

Журнал неорганической химии, 2020, T. 65, № 5, стр. 679-685
Термоаналитическое исследование фазовых превращений метансульфонатов магния и кальция
Д. А. Косова a, *, Д. И. Провоторов a, С. В. Кузовчиков a, И. А. Успенская a
a Московский государственный университет им. М.В. Ломоносова
119991 Москва, Ленинские горы, 1, Россия
* E-mail: dakosova@gmail.com
Поступила в редакцию 15.11.2019
После доработки 04.12.2019
Принята к публикации 24.12.2019
Аннотация
Синтезированы и идентифицированы безводные метансульфонаты магния (Mg(SO3CH3)2) и кальция (Ca(SO3CH3)2), а также гидраты Mg(SO3CH3)2 · 2H2O и Mg(SO3CH3)2 · 12H2O. Методом термогравиметрического анализа изучено термическое разложение солей на воздухе. Методом дифференциальной сканирующей калориметрии установлены параметры фазовых превращений Ca(SO3CH3)2 и Mg(SO3CH3)2 · 2H2O, которые наблюдаются при –62.7 и –119°С соответственно. Определена температура плавления Mg(SO3CH3)2 · 12H2O (45.4°С), синтез которого был проведен непосредственно в приборе ДСК, так как соединение неустойчиво на воздухе. Показано, что Mg(SO3CH3)2 · 12H2O плавится инконгруэнтно.
ВВЕДЕНИЕ
На сегодняшний день объемы производства метансульфоновой кислоты составляют 50 000 т в год, и прогнозируется их дальнейший рост, так как недавно был предложен экологически безопасный и в то же время экономически выгодный способ синтеза метансульфоновой кислоты в одну стадию с выходом >99% [1]. В этой связи изучение свойств солей метансульфоновой кислоты и систем на их основе представляет интерес и для фундаментальной науки, и для практики. Соответствующая информация в литературе представлена фрагментарно [2–5], несмотря на то, что метансульфоновая кислота и ее неорганические производные находят применение в различных областях [6–15]. Так, исследование свойств метансульфонатов магния, кальция и их гидратов интересно для решения задач геохимии и атмосферной химии [16–22]. Метансульфоновая кислота является продуктом окисления диметилсульфида кислородом воздуха, который, в свою очередь, представляет собой продукт жизнедеятельности фитопланктона [23]. Аэрозоли солей метансульфоновой кислоты участвуют в формировании облаков [18, 19]. Попадая с осадками на поверхность Земли, метансульфонаты разлагаются в почве под воздействием различных микроорганизмов [20]. Не так давно исследователи обнаружили [23] метансульфонаты и их гидраты в кернах льда Антарктики, где в заметном количестве представлены метансульфонаты натрия, калия, кальция и магния. В этой связи актуальным является исследование фазовых равновесий в соответствующих водно-солевых системах, так как знание свойств ледяных кернов может быть применено при воссоздании климатических изменений и состава мирового океана в процессе формирования различных пород, т.е. для решения задач геотермобарометрии [24]. Первым этапом изучения фазовых равновесий является оценка диапазонов устойчивости индивидуальных кристаллических фаз, образующихся в системах, и определение температур и энтальпий фазовых переходов веществ.
Объектами настоящего исследования стали гидрат метансульфоната магния и безводные метансульфонаты магния и кальция. Цель работы – изучение термохимических свойств указанных соединений. Для этого предстояло решить следующие задачи: синтезировать объекты исследования; идентифицировать полученные вещества; изучить термическую стабильность безводных и гидратированных соединений и определить параметры фазовых превращений. Основные методы исследования – термогравиметрия (ТГА) и дифференциальная сканирующая калориметрия (ДСК).
Из литературы известно, что метансульфонат кальция не образует гидратов [25], а метансульфонат магния при комнатной температуре кристаллизуется из водных растворов в виде Mg(SO3CH3)2 · 12H2O [26], а при температурах 80–90°С – в форме Mg(SO3CH3)2 · 2H2O [25]. Достоверно установлена только структура Mg(SO3CH3)2 · 12H2O [26, 27] и Ca(CH3SO3)2 [28]. Mg(SO3CH3)2 · 12H2O кристаллизуется в пр. гр. $R\bar {3}$ и имеет тригональную кристаллическую ячейку с параметрами a = 9.27, b = 9.27, c = 21.13 Å, γ = 120°; данные получены при температуре 110 K [26, 27]. Соединение Ca(CH3SO3)2 имеет пр. гр. Pbca, орторомбическую кристаллическую ячейку с параметрами a = 17.235 ± 0.016, b = 10.08 ± 0.01, c = = 9.17 ± 0.01 Å [28].
В работе [25] изучено термическое поведение безводных метансульфонатов магния и кальция в динамической воздушной атмосфере (20 мл/мин) методом ТГА при скорости нагревания 20 град/мин. Температуры начала термического разложения составляют 476 и 474°С, массовая доля сухого остатка – 21.85 и 41.87 мас. % для магниевой и кальциевой солей соответственно. Сухой остаток после разложения солей был исследован методом рентгенофазового анализа. Показано, что он состоит из смеси оксида и сульфата соответствующего металла. В работе [26] представлена кривая ТГА для Mg(SO3CH3)2 · 12H2O, полученная при скорости нагревания 5 град/мин. Дегидратация проходит в три этапа, ступени плохо разделены, однако авторы высказали предположение о том, что Mg(SO3CH3)2 · 12H2O разлагается по следующей схеме:
Информация о плавлении и других фазовых превращениях метансульфонатов кальция, магния и его гидратов в литературе отсутствует.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез. Для синтеза метансульфонатов магния и кальция использовали метансульфоновую кислоту HSO3CH3, смешанный оксид-карбонат магния 4MgCO3 · Mg(OH)2 · nH2O и карбонат кальция CaCO3. Чистота и происхождение реактивов указаны в табл. 1. Вещества получали по следующей методике. К исходным твердым веществам добавляли небольшое количество дистиллированной воды, а затем при активном перемешивании медленно приливали метансульфоновую кислоту в минимальном избытке; pH среды контролировали индикаторной бумагой. В ходе реакции активно выделялся углекислый газ, смесь разогревалась, поэтому во избежание ее вскипания реакционный сосуд помещали в ледяную баню. По окончании реакции смесь упаривали при температуре 60°С, затем оставляли при комнатной температуре. Выпавшие кристаллы дважды перекристаллизовывали из воды и отфильтровывали на пористом фильтре под вакуумом, промывали спиртом и тщательно перетирали. Соли хранили в плотно закрытых тарах, предназначенных для химических реактивов.
Таблица 1.
Чистота и происхождение реактивов, использованных в работе
| Формула | Происхождение | Чистота, мас. % | Метод анализа |
|---|---|---|---|
| HSO3CH3 | Alfa Aesar | 98.0 | Установлено поставщиком |
| 4MgCO3 · Mg(OH)2 · nH2O | Компонент-реактив | >99 | Установлено поставщиком |
| CaCO3 | Компонент-реактив | >99 | Установлено поставщиком |
| Mg(CH3SO3)2 · 12H2O | Синтезирован в настоящей работе | – | ДСК |
| Mg(CH3SO3)2 · 2H2O | Синтезирован в настоящей работе | 99.9 | Термогравиметрия, элементный анализ, комплексонометрическое титрование |
| Mg(CH3SO3)2 | Синтезирован в настоящей работе | 99.9 | Термогравиметрия, комплексонометрическое титрование |
| Ca(CH3SO3)2 | Синтезирован в настоящей работе | 99.9 | Термогравиметрия, элементный анализ, комплексонометрическое титрование |
Идентификацию полученных препаратов проводили методами элементного и термогравиметрического анализа, а также с помощью комплексонометрического титрования с ЭДТА. Результаты исследований представлены в табл. 2–5.
Таблица 2.
Результаты элементного анализа метансульфонатов магния и кальция, мас. %
| Расчет | Mg(CH3SO3)2 · 2H2O | Ca(CH3SO3)2 | ||
|---|---|---|---|---|
| ω(С) | ω(H) | ω(С) | ω(H) | |
| Теория | 9.59 | 4.00 | 10.43 | 2.61 |
| Эксперимент | 9.65 | 4.06 | 10.38 | 2.66 |
Таблица 3.
Содержание кальция и магния в метансульфонатах по результатам комплексонометрического титрования, ω(M2+) – массовая доля катиона магния (кальция)
| Вещество | ω(M2+), мас. % | |
|---|---|---|
| эксперимент | рассчитано по брутто-формуле | |
| Mg(CH3SO3)2 | 11.35 ± 0.02 | 11.34 |
| Mg(CH3SO3)2 · 2H2O | 9.79 ± 0.03 | 9.71 |
| Ca(CH3SO3)2 | 17.19 ± 0.07 | 17.43 |
Таблица 4.
Сравнение экспериментальных и литературных данных [25] по термическому разложению безводных метансульфонатов магния и кальция на воздухе в диапазоне температур 400–600°С. tр – температура начала заметного разложения на кривой ТГА, Δm – потеря массы, мас. % в расчете на безводную соль
Таблица 5.
Результаты термогравиметрического анализа метансульфонатов магния и кальция на содержание воды
| Mg(CH3SO3)2 · 2H2O | Ca(CH3SO3)2 | |
|---|---|---|
| ω(H2O), мас. % | ||
| Рассчитано по брутто-формуле | 14.40 | 0 |
| Эксперимент | 14.48 | 0 |
Элементный анализ проводили на автоматическом CHN-анализаторе CE1106 (навеска образца 0.8–1.0 мг, температура сжигания 1050°С) в ИНЭОС РАН. В момент вспышки в кислороде пробы, которая находилась в оловянной капсуле, температура достигла 1800°С. Процесс сгорания происходил в потоке гелия как транспортного газа, его расход составлял 120 мл/мин. Время полного цикла анализа 10 мин. Разделение продуктов деструкции (азота, углекислоты и воды) происходило газохроматографически на ГХ-колонке прибора в потоке гелия; детектор – катарометр. Расчет содержания C и H производился автоматически с помощью программного обеспечения прибора. При расчете учитывались предварительно установленные по стандартным образцам калибровочные коэффициенты, результаты холостого опыта и величина навески. Ошибка метода составляла 0.5 мас. %. Результаты анализа представлены в табл. 2.
Титрование. Содержание кальция и магния в препаратах определяли методом комплексонометрического титрования 0.005 М раствором ЭДТА с индикатором эриохромовым черным Т по стандартной методике, описанной в [29]. Результаты анализа представлены в табл. 3.
Термогравиметрия. Термогравиметрический анализ выполняли на приборе Netzsch TG 209 F1. Измерения проводили в динамической атмосфере осушенного воздуха, скорость потока газа 40 мл/мин. Скорость нагревания составляла 10 град/мин. Калибровку прибора по температуре осуществляли по температурам плавления стандартных веществ с чистотой не менее 99.999%. В качестве стандартов использовали серебро, алюминий, висмут, индий и олово. Систематическая погрешность потери массы, определенная по разложению моногидрата оксалата кальция, не превышала 1%. Температурный диапазон измерений 30–600°С. Перед измерениями вещества тщательно перетирали и помещали в тигли из корунда. Масса навесок составляла 10–20 мг. Взвешивание проводили на весах A&D GH-202 с точностью 2 × 10–5 г. Результаты анализа представлены в табл. 4 и 5.
Дифференциальная сканирующая калориметрия. Для изучения фазовых переходов индивидуальных кристаллических фаз и фазовых равновесий твердое вещество–жидкость использовали метод дифференциальной сканирующей калориметрии. Измерения проводили на калориметре DSC 204 F1 Phoenix в атмосфере азота марки “ос. ч.” (поток газа через ячейку 40 мл/мин, поток защитного газа 70 мл/мин). Для охлаждения использовали автоматическую систему подачи жидкого азота. Калибровку прибора осуществляли по температуре и чувствительности в соответствии с ASTM E967 и E968. В качестве стандартов использовали циклогексан, ртуть, галлий, бензойную кислоту, воду, галлий, индий, олово, висмут, свинец, хлорид цезия и цинк; чистота стандартов составляла не менее 99.999%. Систематическая ошибка прибора после проведения калибровки составляла 0.1°С по температуре и 5% по теплоте. Исследования проводили в температурном диапазоне 150–60°С.
Вещества помещали в алюминиевый тигель (V = 56 мм3, D = 6 мм) с проколотой крышкой или герметично закрытой в случае работы с жидким образцом. Масса навесок составляла 3–10 мг, точность определения массы 2 × 10–5 г. Твердые образцы тщательно перетирали и распределяли тонким ровным слоем по дну тигля. Жидкие образцы готовили в эппендорфах, а затем с помощью шприца переносили в тигель. Гетерогенные образцы готовили непосредственно в тигле при внесении твердого компонента и добавлении дистиллированной воды из шприца.
Для обработки результатов использовали программное обеспечение Netzsch Proteus Analysis. Температуры фазовых превращений первого рода определяли как tначала (точка пересечения базовой линии, экстраполированной в область пика, и касательной, проведенной через точку перегиба на левом плече пика на кривой ДСК) в соответствии с ASTM E 794. Энтальпии фазовых превращений определяли по площадям соответствующих пиков на кривых ДСК (ASTM E 793). Количественной обработке подвергались только кривые ДСК, полученные в режиме нагревания.
В работе использовали две основные температурные программы ДСК:
первая (I) применялась для поиска и изучения фазовых переходов индивидуальных кристаллических веществ и представляла собой простое сканирование на скорости 10 град/мин; вторая (II) состояла из нескольких этапов и применялась при исследовании составов системы вода–метансульфонат магния:
1) нагревание со скоростью 5 град/мин до 75°С;
2) изотерма при 75°С, 5 мин;
3) охлаждение со скоростью 10 град/мин до ‒150°С;
4) нагревание со скоростью 10 град/мин до ‒15°С;
5) нагревание со скоростью 2 град/мин до 75°С.
После ДСК-экспериментов с жидкостями проводили контрольное взвешивание для проверки герметичности тигля.
ДСК-измерения при повышенном внешнем давлении 107 Па проводили на приборе Netzsch DSC 204 HD, скорость нагревания составляла 10 град/мин, температурный диапазон съемки 25–450°С.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
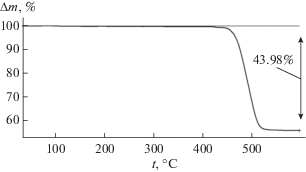
Термогравиметрический анализ солей. На рис. 1 представлена ТГА-кривая образца метансульфоната кальция, полученного по методике, описанной в экспериментальной части. Потеря массы фиксируется только при температурах выше 450°С. Сравнение экспериментальных и литературных данных [25] о термическом разложении метансульфоната кальция на воздухе дано в табл. 4, приведенные величины удовлетворительно согласуются между собой.
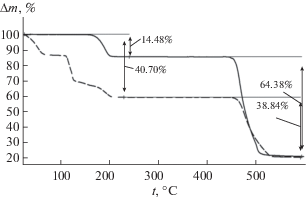
Полученные в ходе синтеза кристаллы магниевой соли метансульфоновой кислоты были охарактеризованы методом ТГА без предварительной пробоподготовки. Результаты эксперимента представлены на рис. 2 пунктирной линией. Видно, что процесс дегидратации начался еще до начала съемки на предшествующем ему изотермическом сегменте при 30°С. В связи с неустойчивостью данного соединения на воздухе не представляется возможным напрямую использовать его для анализа. Перетертые кристаллы были оставлены сушиться на несколько дней на воздухе, после чего провели термоаналитические измерения; полученная кривая ТГА представлена на рис. 2 сплошной линией. Содержание воды соответствует двухводному гидрату Mg(CH3SO3)2 · 2H2O (табл. 5). Характер разложения безводного метансульфоната магния Mg(CH3SO3)2 согласуется с литературными данными (табл. 4). Поскольку безводный метансульфонат магния гигроскопичен на воздухе, все дальнейшие манипуляции проводили с его двухводным гидратом.
Рис. 2.
ТГА гидратов метансульфоната магния Mg(CH3SO3)2. Сплошная линия – Mg(CH3SO3)2 · · 2H2O, штриховая – кристаллы, полученные при синтезе.
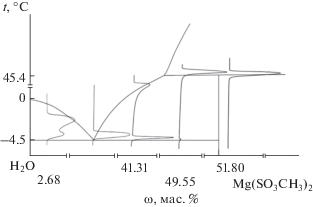
Плавление Mg(CH3SO3)2 · 12H2O. Для термического анализа Mg(CH3SO3)2 · 12H2O его синтезировали непосредственно в приборе ДСК, смешав в тигле соответствующие количества дистиллированной воды и двухводного гидрата Mg(CH3SO3)2 · 2H2O (теоретическое содержание Mg(CH3SO3)2 50.2 мас. %). Смесь исследовали по температурной программе II, описанной в экспериментальной части. На кривой ДСК зафиксирован эндотермический эффект при 45.4°С, соответствующий плавлению образовавшегося двенадцативодного гидрата (рис. 3, табл. 6). Для установления типа плавления соединения было исследовано несколько составов системы вода–метансульфонат магния. Особое внимание уделялось области составов вблизи двенадцативодного гидрата. Для разбавленных растворов фиксировали пики ликвидуса и солидуса (–4.5°С, значение находится в соответствии с данными [26] (–5°C)). Для состава, содержащего 49.55 мас. %, зафиксирован как солидус при –4.5°С, так и плавление двенадцативодного гидрата при 45.4°С (рис. 4). Для составов с бóльшим содержанием соли фиксировались только пики плавления двенадцативодного гидрата. На основании проведенного анализа сделан вывод об инконгруэнтном характере плавления Mg(CH3SO3)2 · 12H2O. Кривые ДСК для исследованных составов схематично нанесены на фазовую диаграмму (рис. 4), а также изображены на рис. 3.
Рис. 3.
Кривые ДСК для различных составов системы H2O–Mg(CH3SO3)2 в области плавления Mg(CH3SO3)2 · · 12H2O.
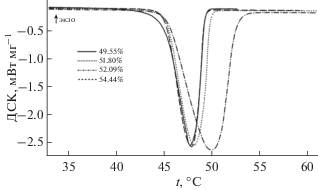
Таблица 6.
Температура инконгруэнтного плавления Mg(CH3SO3)2 · 12H2O, определенная по результатам ДСК-экспериментов смесей разных составов системы H2O–Mg(CH3SO3)2
| Состав смеси, мас. % Mg(CH3SO3)2 | № эксперимента | tпл, °C |
|---|---|---|
| 49.55 | 1 | 45.2 |
| 51.80 | 1 | 45.6 |
| 2 | 45.5 | |
| 3 | 45.3 | |
| 52.09 | 1 | 45.6 |
| 2 | 45.3 | |
| 3 | 45.5 | |
| 54.44 | 1 | 45.4 |
| Среднее значение | 45.4 ± 0.1 | |
Рис. 4.
Схематичное представление фрагмента фазовой диаграммы системы H2O–Mg(CH3SO3)2 с наложенными на нее кривыми ДСК для составов, содержащих 2.68, 41.31, 49.55, 51.80 мас. % Mg(CH3SO3)2.
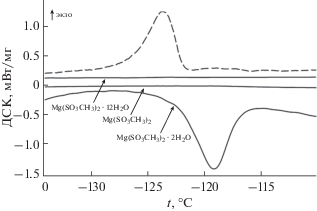
Фазовый переход Mg(CH3SO3)2 · 2H2O. При нагревании и охлаждении со скоростью 10 град/мин обнаружена воспроизводимая аномалия у Mg(CH3SO3)2 · 2H2O с максимумом при –119°С, которая отсутствует у безводной соли и двенадцативодного гидрата (рис. 5). Температурный гистерезис не наблюдается, пик имеет характерную лямбда-форму. Для определения природы наблюдаемого процесса требуются дополнительные структурные исследования при пониженных температурах. При нагревании Mg(CH3SO3)2 · 2H2O в ячейке ДСК вплоть до температуры разложения (115°С) при внешнем давлении 107 Па других фазовых превращений не обнаружено.
Рис. 5.
Кривые ДСК при охлаждении (пунктирная линия) и нагревании (сплошные линии) для Mg(CH3SO3)2 · 2H2O, Mg(CH3SO3)2 · 12H2O и Mg(CH3SO3)2.
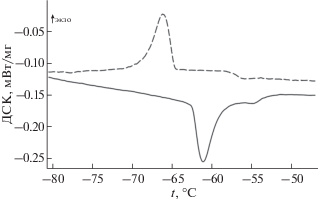
Фазовый переход Ca(CH3SO3)2. У метансульфоната кальция на кривых ДСК обнаружена воспроизводимая при нагревании и охлаждении аномалия (рис. 6), имеющая температурный гистерезис ∼5°C за счет переохлаждения высокотемпературной фазы при охлаждении. В связи с этим аномалия отнесена к фазовому переходу первого рода, температура и энтальпия которого представлены в табл. 7. При нагревании Ca(CH3SO3)2 в ячейке ДСК вплоть до температуры разложения (450°С) при внешнем давлении 107 Па других фазовых превращений не обнаружено.
ЗАКЛЮЧЕНИЕ
Методом термогравиметрии изучено термическое разложение на воздухе следующих соединений: Mg(CH3SO3)2 ∙ 12H2O, Mg(CH3SO3)2 ∙ 2H2O, Mg(CH3SO3)2 и Ca(CH3SO3)2. Показано, что Mg(CH3SO3)2 ∙ 12H2O неустойчив на воздухе и с течением времени превращается в двухводный гидрат Mg(CH3SO3)2 ∙ 2H2O, температура начала разложения которого составляет 115°С. Показано, что Mg(CH3SO3)2 ∙ 2H2O, Mg(CH3SO3)2 и Ca(CH3SO3)2 разлагаются, не достигая плавления, не только при атмосферном давлении, но и при повышении внешнего давления до 107 Па.
Методом дифференциальной сканирующей калориметрии обнаружено, что Mg(CH3SO3)2 ∙ 2H2O и Ca(CH3SO3)2 претерпевают фазовые превращения при –119 и –62.7°С соответственно. На кривых ДСК в области фазового превращения Ca(CH3SO3)2 зафиксирован гистерезис при охлаждении и нагревании, в этой связи достоверно установлено, что фазовый переход относится к первому роду, определена его температура и энтальпия.
Разработана методика синтеза Mg(CH3SO3)2 ∙ ∙ 12H2O непосредственно в приборе ДСК, температура плавления двенадцативодного гидрата Mg(CH3SO3)2 ∙ 12H2O составила 45.4°С. Методом ДСК нескольких составов системы H2O–Mg(CH3SO3)2 установлено, что Mg(CH3SO3)2 ∙ ∙ 12H2O плавится инконгруэнтно.
Список литературы
Diaz-Urrutia C., Ott T. // Science. 2019. V. 363. P. 1326. https://doi.org/10.1126/science.aav0177
Kosova D.A., Navalayeu T.I., Maksimov A.I. et al. // Fluid Phase Equilib. 2017. V. 443. P. 23. https://doi.org/10.1016/j.fluid.2017.04.006
Maxwell R.J., Silbert L.S., Russell J.R. // J. Org. Chem. 1977. V. 42. № 9. P. 1515. https://doi.org/10.1021/jo00429a005
Wei C.H. // Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1986. V. 42. № 12. P. 1839. https://doi.org/10.1107/S0108270186090340
Kokunov Yu.V., Kovalev V.V., Gorbunovaet Yu.E. et al. // Russ. J. Coord. Chem. 2018. V. 44. № 2. P. 103. [Кокунов Ю.В., Ковалев В.В., Горбунова Ю.Е. и др. // Коорд. химия. 2018. Т. 44. № 1. С. 14.]https://doi.org/10.1134/S1070328418020069
Finšgar M., Milošev I. // Corros. Sci. 2010. V. 52. № 7. P. 2430. https://doi.org/10.1016/j.corsci.2010.04.001
Walsh F.C., de León C.P. // Surf. Coat. Technol. 2014. V. 259. P. 676. https://doi.org/10.1016/j.surfcoat.2014.10.010
Tian Y., Meng X., Duan J. // Ind. Eng. Chem. Res. 2012. V. 51. № 42. P. 13627. https://doi.org/10.1021/ie302015v
Luong B.X., Petre A.L., Hoelderich W.F. et al. // J. Catal. 2004. V. 226. № 2. P. 301. https://doi.org/10.1016/j.jcat.2004.05.025
Liu D., Wei Z., Shen Y. et al. // J. Mater. Chem. A. 2015. V. 3. № 40. P. 20322. https://doi.org/10.1039/C5TA05497D
Rackemann D.W., Bartley J.P., Doherty O.S. // Ind. Crops Prod. 2014. V. 52. P. 46. https://doi.org/10.1016/j.indcrop.2013.10.026
Friščić T., Halasz I., Beldon P.J. et al. // Nat. Chem. 2013. V. 5. № 1. P. 66. https://doi.org/10.1038/nchem.1505
Calvin J.J., Asplund M., Akimbekov Z. et al. // J. Chem. Thermodyn. 2018. V. 116. P. 341. https://doi.org/10.1016/j.jct.2017.10.002
Leung P.K., Ponce-de-León C., Low C.T.J. et al. // J. Power Sources. 2011. V. 196. № 11. P. 5174. https://doi.org/10.1016/j.jpowsour.2011.01.095
Nikiforidis G., Daoud W.A. // Electrochim. Acta. 2014. V. 141. P. 255. https://doi.org/10.1016/j.electacta.2014.06.142
Bzdek B.R., Ridge D.P., Johnston M.V. // J. Geophys. Res.: Atmos. 2011. V. 116. P. 1. https://doi.org/10.1029/2010JD015217
Kwong K.C., Chim M.M., Hoffmann E.H. et al. // ACS Earth Space Chem. 2018. V. 2. № 9. P. 895. https://doi.org/10.1021/acsearthspacechem.8b00072
Saltzman E.S., Savoie D.L., Prospero J.M. // J. Atmos. Chem. 1986. V. 4. № 2. P. 227. https://doi.org/10.1007/BF00052002
Pszenny A.A. // J. Atmos. Chem. 1992. V. 14. P. 273. https://doi.org/10.1007/BF00115239
Kelly D.P., Murrell J.C. // Arch. Microbiol. 1999. V. 172. № 6. P. 341. https://doi.org/10.1007/s002030050770
Welch K.A., Mayewski P.A., Whitlow S.I. // Geophys. Res. Lett. 1993. V. 20. № 6. P. 443. https://doi.org/10.1029/93GL00499
Sakurai T., Ohno H., Genceli F.E. et al. // J. Glaciol. 2010. V. 56. № 199. P. 837. https://doi.org/10.3189/002214310794457335
Gernon M.D., Wu M., Buszta T. et al. // Green Chem. 1999. V. 1. № 3. P. 127. https://doi.org/10.1039/A900157C
Воронин Г.Ф. Основы термодинамики. М.: Изд-во Моск. ун-та, 1987. 192 с.
Wang M., Song Z.G., Jiang H. et al. // J. Therm. Anal. Calorim. 2009. V. 98. № 3. P. 801. https://doi.org/10.1007/s10973-009-0119-z
Guner F.E.G., Lutz M., Sakurai T. et al. // Cryst. Growth Des. 2010. V. 10. № 10. P. 4327. https://doi.org/10.1021/cg100234e
Güner F.E.G., Sakurai T., Hondoh T. // Eur. J. Mineral. 2012. V. 25. № 1. P. 79. https://doi.org/10.1127/0935-1221/2013/0025-2257
Charbonnier F., Faure R., Loiseleur H. // J. Appl. Crystallogr. 1975. V. 8. № 6. P. 694. https://doi.org/10.1021/cg100234e
Золотов Ю.А. Основы аналитической химии. М.: Высш. школа, 2001. 432 с.
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии