

Журнал неорганической химии, 2020, T. 65, № 9, стр. 1216-1221
Теоретические модели химической связи в расплавах бинарных антимонидов кадмия и цинка в полупроводниках группы АIIВV
А. А. Ащеулов a, О. Н. Маник a, *, Т. О. Маник b, В. Р. Билинский-Слотыло a, А. Д. Изотов c, И. В. Федорченко c
a Черновицкий национальный университет им. Ю. Федьковича
58000 Черновцы, ул. Коцюбинского, 2, Украина
b Военно-техническая академия им. Ярослава Домбровского
00-908 Варшава-49, ул. Ген. Сильвестра Калиского, 2, Польша
c Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
* E-mail: slotulo@gmail.com
Поступила в редакцию 20.03.2020
После доработки 27.04.2020
Принята к публикации 30.04.2020
Аннотация
Предложен и рассмотрен комплексный подход к формированию ближнего порядка расплавов бинарных антимонидов кадмия и цинка с учетом особенностей тонкой структуры химической связи и межатомного взаимодействия в полупроводниковых соединениях группы АIIВV. Разработана методика и проведены расчеты энергии разрыва неэквивалентных химических связей в антимонидах кадмия и цинка в зависимости от межатомных расстояний и атомных характеристик исходных компонентов.
ВВЕДЕНИЕ
Полупроводниковые соединения группы АIIВV широко используются в электронной технике [1–10]. В связи с этим следует выделить два наиболее важных направления работ. Первое исходит из задачи совершенствования технологии существующих приборов. Второе включает в себя разработку новых материалов [1, 11–17].
Оба направления связаны с изучением диаграмм состояния, фазовых превращений как между твердыми фазами, так и в области плавления, природы химической связи [18–24]. Следует отметить, что последовательной теории фазовых превращений с позиций химической связи пока еще нет. Поэтому особо важным и актуальным становится проведение исследований взаимозависимости макроскопических свойств исследуемых материалов с их микроскопическими характеристиками с позиций химической связи. Решение задачи создания новых материалов с необходимыми параметрами требует сочетания эмпирического и квантово-теоретического подходов к взаимосвязи свойств элементов и образуемых ими соединений.
Теоретический анализ многочисленных эмпирических зависимостей связан с пересмотром системы сложившихся взглядов на проблему межатомного взаимодействия, а также с появлением качественно новых представлений, которые не являются результатом развития существующих теорий, а зачастую отрицают некоторые из них.
Квантовые модели электронного строения и межатомного взаимодействия соединений группы АIIВV
На основе результатов работ [15, 16, 20] о термической перегруппировке атомов, способах оптимизации материалов, взаимосвязи термодинамических характеристик ионов с параметрами их электронного строения нами построены уравнения, связывающие величины ионных радиусов с числом электронов на орбитах атомов.
Наиболее простые соотношения получены путем постулирования линейной зависимости числа электронов во внешней оболочке атома от логарифма его радиуса Ферми. Взаимосвязь тангенса угла наклона ${\text{tg}}\alpha = \frac{{\Delta \lg {{R}_{u}}}}{{\Delta n}}$ и электроотрицательностей исключает возможность произвольного изменения сопоставляемых величин.
Хорошее согласование комплекса опытных данных дает постулируемая в [20] зависимость:
где $R_{{uA}}^{0}$ – радиус атома в невозбужденном состоянии, а х – валентность.Поскольку уравнение (1) описывает изменение Ru атомов А и В при изменении числа электронов на орбитах каждого, то допуская равенство абсолютных значений зарядов взаимодействующих атомов, зависимость (1) принимает вид системы уравнений:
d – сумма ионных радиусов, равная межатомному расстоянию.С позиций квантового подхода формально система уравнений (2)–(4) рассматривает геометрические условия контакта сферических электронных облаков с разным уровнем плотности на границе.
Поэтому необходимы дополнительные критерии, позволяющие перевести систему (2)–(4) на формализм квантовой химии с учетом тонкой структуры химической связи. Для этого необходимо проанализировать зависимость межатомных расстояний от эффективных зарядов. Результаты анализов показали, что в любой точке, кроме d1= dmin, плотность заряда на границе ионов различна.
Для определения эффективных зарядов и эффективных радиусов в связях с d1< dmin образование связи А–В сопровождается уходом электронов на другие направления межатомного взаимодействия, т.е. связь становится донорной. При этом удаление электронов ($ + \Delta q$) или их локализация ($ - \Delta q$) на данном направлении связи одинаково изменяет значения зарядов, которые имеет данная пара при d1= dmin. При таком подходе система (2)–(4) переходит в систему:
(5)
$\lg R_{{uA}}^{{ZA}} = \lg R_{{uA}}^{0} - \left( {{{z}_{{\min }}} + \frac{{\Delta q}}{2}} \right){\text{tg}}{{\alpha }_{A}},$(6)
$\lg R_{{uB}}^{{ZB}} = \lg R_{{uB}}^{0} - \left( {{{z}_{{\min }}} + \frac{{\Delta q}}{2}} \right)~{\text{tg}}{{\alpha }_{B}},$Плотность заряда, рассчитанная в кристаллохимическом подходе (xA = –xB), корректна с квантовой точки зрения только при d1= dmin, но этого достаточно, чтобы система (5)–(7) решалась при известном d1 [20].
Таким образом, в результате учета квантовой интерпретации эмпирического материала путем сочетания принципов теоретического и экспериментального подходов в едином количественном методе расчета электронного строения вещества получена зависимость энергии связей неэквивалентных орбиталей от их длины и электронных конфигураций атомов в соединениях АIIВV.
В отличие от результатов, полученных в [20], построение рабочей формулы нами выполнено с учетом требований теории подобия, и выражение для энергии связей приняло вид:
(8)
$D_{{A - B}}^{i} = \frac{{{{C}_{1}}\left( {R_{{uA}}^{0} + R_{{uB}}^{0}} \right)}}{{\left( {{\text{tg}}{{\alpha }_{A}} + {\text{tg}}{{\alpha }_{B}}} \right)}}\left( {\frac{{{{C}_{2}}{{d}_{i}}}}{{d_{i}^{2} - {{R}_{{uA}}}{{R}_{{uB}}}}} - \frac{1}{{{{d}_{i}}}}} \right),$С1 – коэффициент, отражающий взаимосвязь размерных и энергетических характеристик межатомного взаимодействия, таких как потенциалы ионизации, эффекты экранирования, электроотрицательность с эффективными радиусами и межатомными расстояниями. В случае использования несистемных единиц, когда расстояние измеряется в Å, С1 измеряется в эВ.
С2 – коэффициент, зависящий от типа кристаллической структуры, химической связи и отражающий количественную связь между коэффициентами ${\text{tg}}{{\alpha }_{A}}$ и ${\text{tg}}{{\alpha }_{B}}$ из уравнений (2)–(4) с величинами ${{d}_{i}}$ и (${{{{R}_{{uA}}}} \mathord{\left/ {\vphantom {{{{R}_{{uA}}}} {{{R}_{{uB}}}}}} \right. \kern-0em} {{{R}_{{uB}}}}}$). С учетом требований теории подобия коэффициент С2 выбирается безразмерным. Апробация полученных уравнений проведена путем расчетов эффективных зарядов, эффективных радиусов и энергии диссоциации неэквивалентных гибридных орбиталей (НГО) в расплавах антимонидов кадмия и цинка.
ТЕОРЕТИЧЕСКИЕ РАСЧЕТЫ
В качестве объекта исследования выбраны антимониды кадмия и цинка [21–24]. Особенностью химической связи в кристаллах ZnSb является то, что каждый атом Zn в своем ближайшем окружении имеет три атома Sb и один атом Zn, а каждый атом Sb имеет три ближайших атома Zn и один атом Sb. Всего имеется пять семейств НГО, различных как по межатомным расстояниям, так и по составу компонентов (φ1, φ2, φ3 – соответствуют связям Zn–Sb различной длины, а также φ4(Sb–Sb) и φ5(Zn–Zn)). Аналогичную структуру имеет CdSb: φ6, φ7, φ8 – соответствуют связям Cd–Sb различной длины, а также φ9(Sb–Sb) и φ10(Cd–Cd) [21, 22].
Для решения поставленной задачи необходимо записать систему уравнений (5)–(7) для каждой i-НГО. Затем путем решения обратной задачи по известным межатомным расстояниям di (1 ≤ i ≤ 10) найти ${{R}_{{u{\text{Cd}}}}}$, ${{R}_{{u{\text{Sb}}}}}$ в кристаллах CdSb и ${{R}_{{u{\text{Zn}}}}}$, ${{R}_{{u{\text{Sb}}}}}$ в кристаллах ZnSb.
Решение системы логарифмических трансцендентных уравнений проводили численными методами. Задавали погрешность $\varepsilon \geqslant d_{i}^{{\left( {{\text{эксп}}} \right)}} - d_{i}^{{\left( {{\text{теор}}} \right)}}$, которая в нашем случае для всех i не превышала 0.1%. Точность расчетов ограничивалась точностью экспериментальных методов нахождения межатомных расстояний.
Необходимые для составления систем уравнений (5)–(7) коэффициенты $R_{u}^{0}$ и ${\text{tg}}\alpha $ находили по методике [20]. Результаты проведенных расчетов для всех компонентов соединений АIIВV приведены в табл. 1.
Таблица 1.
Коэффициенты уравнений (2)–(4) исходных компонентов соединений группы АIIВV
| z | Элемент | $R_{u}^{0}$, Å | ${\text{tg}}\alpha $ | z | Элемент | $R_{u}^{0}$, Å | ${\text{tg}}\alpha $ |
|---|---|---|---|---|---|---|---|
| 4 | Be | 1.130 | 0.2650 | 41 | Nb | 1.465 | 0.063 |
| 12 | Mg | 1.600 | 0.1960 | 48 | Cd | 1.510 | 0.097 |
| 15 | P | 1.040 | 0.0950 | 51 | Sb | 1.450 | 0.074 |
| 20 | Ca | 2.020 | 0.1510 | 56 | Ba | 2.240 | 0.115 |
| 23 | V | 1.468 | 0.0750 | 73 | Ta | 1.469 | 0.060 |
| 30 | Zn | 1.370 | 0.1350 | 80 | Hg | 1.550 | 0.068 |
| 33 | As | 1.220 | 0.0835 | 83 | Bi | 1.650 | 0.068 |
| 38 | Sr | 2.200 | 0.1450 |
Эти результаты согласуются с данными [20] и позволяют провести расчеты межатомного взаимодействия в соединениях группы АIIВV.
С целью корректного использования формализма НГО в численных расчетах в настоящей работе построена зависимость ${{d}_{i}}~\,\,{\text{от}}~\,\,~z_{{}}^{{{\text{эф}}}}$, из которой следует, что для CdSb dmin = 2.9271 Å, а в случае ZnSb dmin = 2.7233 Å. В обоих случаях dmin превышает реальные межатомные расстояния вдоль соответствующих связей. Все это привело к необходимости перерасчета эффективных зарядов для каждой связи.
Далее с учетом казанных выше замечаний рассчитаны ионные радиусы $R_{{u{\text{Cd}}}}^{i}$, $R_{{u{\text{Sb}}}}^{i}$ и $\Delta {{q}_{i}}$ для соединений Cd–Sb, а также $R_{{u{\text{Zn}}}}^{i}$, $R_{{u{\text{Sb}}}}^{i}$ и $\Delta {{q}_{i}}$ для соединений Zn–Sb.
Результаты расчетов эффективных зарядов, эффективных радиусов и энергии диссоциации НГО для кристаллов ZnSb приведены в табл. 2, а для кристаллов CdSb – в табл. 3 .
Таблица 2.
Эффективные заряды, эффективные радиусы и энергии диссоциации НГО в кристаллах ZnSb
| Параметр | Zn–Sb | Sb–Sb | Zn–Zn | ||
|---|---|---|---|---|---|
| φ1 | φ2 | φ3 | φ4 | φ5 | |
| $d_{i}^{{\left( {{\text{эксп}}} \right)}}$ , Å | 2.6400 | 2.6400 | 2.7600 | 2.8000 | 2.8200 |
| $d_{i}^{{\left( {{\text{теор}}} \right)}}$, Å | 2.6405 | 2.6440 | 2.7640 | 2.7943 | 2.8203 |
| $R_{u}^{{{\text{Zn}}}}$, Å | 0.9800 | 0.9750 | 1.0210 | – | 1.4100 |
| $R_{u}^{{{\text{Sb}}}}$, Å | 1.6640 | 1.6690 | 1.7450 | 1.3975 | – |
| ∆q связи φj | 0.0874 | 0.0279 | 0.0169 | 0.0345 | 0.0290 |
| Di, эВ | 1.5544 | 1.5526 | 1.4808 | 2.3327 | 1.1955 |
Таблица 3.
Эффективные заряды, эффективные радиусы и энергии диссоциации НГО в кристаллах CdSb
| Параметр | Cd–Sb | Sb–Sb | Cd–Cd | ||
|---|---|---|---|---|---|
| φ6 | φ7 | φ8 | φ9 | φ10 | |
| $d_{i}^{{\left( {{\text{эксп}}} \right)}}$, Å | 2.8400 | 2.9100 | 2.8100 | 2.810 | 2.9900 |
| $d_{i}^{{\left( {{\text{теор}}} \right)}}$, Å | 2.8398 | 2.9102 | 2.8102 | 2.810 | 2.9894 |
| $R_{u}^{{{\text{Cd}}}}$, Å | 1.4408 | 1.4813 | 1.4239 | – | 1.4947 |
| $R_{u}^{{{\text{Sb}}}}$, Å | 1.3990 | 1.4289 | 1.3865 | 1.405 | – |
| ∆q связи φi | 0.2100 | 0.0860 | 0.2630 | 0.185 | 0.0500 |
| Di, эВ | 2.0300 | 1.9800 | 2.0500 | 2.318 | 1.7340 |
Анализ приведенных результатов показал, что вывод полуэмпирических зависимостей, используемых для вычисления эффективных радиусов, при образовании химических связей, эффективных зарядов при расчетах перераспределения электронной плотности между неэквивалентными химическими связями, энергии взаимодействия (энергии связей) между неоднородными и однородными атомами в различных молекулярных группировках, которые имеют место при синтезе соединений АIIВV, различающихся по стехиометрии, структуре, типу химической связи и физико-химическим свойствам, дает основание утверждать, что использование вышеуказанных методов и подходов позволяет перейти к решению задач прогнозирования физико-химических свойств в материаловедении.
На рис. 1 приведена зависимость межатомных расстояний неэквивалентных орбиталей от перераспределения электронной плотности на них. С ростом межатомных расстояний энергия взаимодействия атомов должна уменьшаться. В то же время, изменение электронной плотности на связях может как усиливать этот эффект, так и ослаблять.
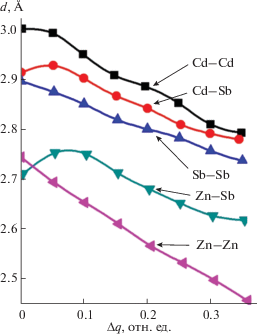
На рис. 2 приведены зависимости энергии диссоциации НГО в соединениях Zn–Sb от коэффициентов C1 и C2, а на рис. 3 – аналогичные зависимости для соединений антимонидов кадмия. Представленные зависимости значительно расширяют технологические возможности получения новых материалов путем учета формализма тонкой структуры химической связи, особенности эвтектико-перитектических реакций [1], образование метастабильных и стабильных фаз [16], уточняют протекание эндотермических и экзотермических реакций синтеза и термической перегруппировки атомов [15] при формировании ближнего порядка в расплавах антимонидов кадмия и цинка.
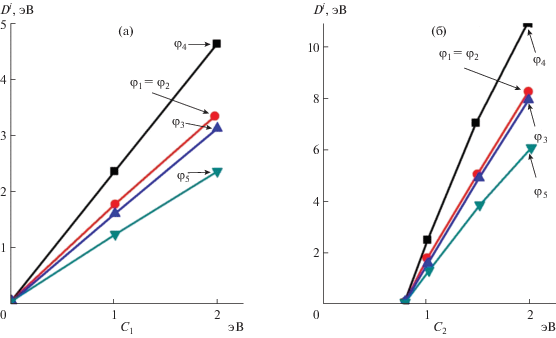
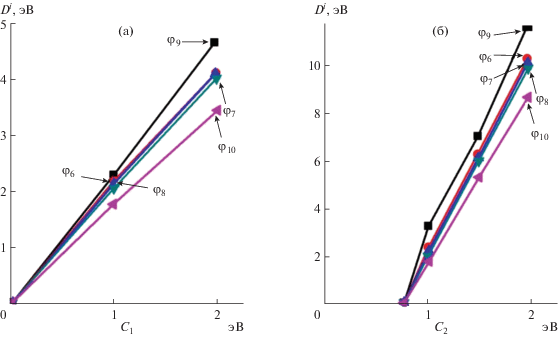
ЗАКЛЮЧЕНИЕ
Предложен метод построения теоретических моделей ближнего порядка расплавов антимонидов кадмия и цинка с учетом их тонкой структуры химической связи и впервые показано, что перераспределение электронной плотности ведет к образованию донорной и акцепторной химических связей. Это позволило разработать методику и провести расчеты энергии разрыва неэквивалентных химических связей в антимонидах кадмия и цинка в зависимости от межатомных расстояний и атомных характеристик исходных компонентов. Отмечено, что полученные результаты хорошо согласуются с расчетами параметров химической связи методами микроскопической теории стабильных и метастабильных фаз, что существенно оптимизирует технологические возможности синтеза материалов на основе антимонидов кадмия и цинка с прогнозируемыми параметрами.
Список литературы
Лазарев В.Б., Шевченко В.Я., Гринберг Я.Х., Соболев В.В. Полупроводниковые соединения группы АIIВV. М.: Наука, 1978. 256 с.
Маренкин С.Ф., Трухан В.М. Фосфиды, арсениды цинка и кадмия. Минск, 2010. 220 с.
Chai R.G., Lou Z., Shen G.Z. // J. Mater. Chem. C. 2019. V. 7. № 5. P. 4581. https://doi.org/10.1039/c8tc06383d
Zhou B.Q., Sun C., Wang X. et al. // ACS Appl. Mater. Interfaces. 2019. V. 11. № 30. P. 27098. https://doi.org/10.1021/acsami.9b10042
Trukhan V.M., Izotov A.D., Shoukavaya T.V. // Inorg. Mater. 2014. V. 50. № 9. P. 868. [Трухан В.М., Изотов А.Д., Шёлковая Т.В. // Неорган. материалы. 2014. Т. 50. № 9. С. 941.]https://doi.org/10.1134/S0020168514090143
Fischer A., Scheidt E.-W., Scherer W. et al. // Phys. Rev. B. 2015. V. 91. № 22. P. 224309. https://doi.org/10.1103/PhysRevB.91.224309
Hermet P., Koza M.M., Ritter C. et al. // RSC Adv. 2015. V. 5. № 106. P. 87118. https://doi.org/10.1039/c5ra16956a
Fischer A., Eklof D., Benson D.E. et al. // Inorg. Chem. 2014. V. 53. № 16. P. 8691. https://doi.org/10.1021/ic501308q
Popov P.A., Oleinik E.A., Trukhan V.M. et al. // Inorg. Mater. 2018. V. 54. № 3. P. 237. [Попов П.А., Олейник Е.А., Трухан В.М. и др. // Неорган. материалы. 2018. Т. 54. № 3. С. 249.].https://doi.org/10.1134/S0020168518030123
Trukhan V.M., Marenkin S.F., Shoukavaya T.V. // Crystallogr. Reports. 2014. V. 59. № 1. P. 53. https://doi.org/10.1134/S1063774514010180
Marenkin S.F., Izotov A.D., Fedorchenko I.V., Novotortsev V.M. // Russ. J. Inorg. Chem. 2015. V. 60. № 3. P. 295. [Маренкин С.Ф., Изотов А.Д., Федорченко И.В., Новоторцев В.М. // Журн. неорган. химии. 2015. Т. 60. № 3. С. 343.]https://doi.org/10.1134/S0036023615030146
Nipan G.D., Aronov A.N. // Russ. J. Inorg. Chem. 2019. V. 64. № 12. P. 1565. [Нипан Г.Д., Аронов А.Н. // Журн. неорган. химии. 2019. Т. 64. № 12. С. 1312.]https://doi.org/10.1051/epjconf/201818501018
Dzhamamedov R.G., Arslanov T.R., Mollaev A.Yu., Kochura A.V. // J. Alloys Compd. 2017. V. 699. P. 1104. https://doi.org/10.1016/j.jallcom.2017.01.014
Kim K., Kaviany M. // Phys. Rev. B. 2016. V. 94. № 15. P. 155 203. https://doi.org/10.1103/PhysRevB.94.155203
Псарев В.И. // Журн. физ. химии. 1997. Т. 77. № 6. С. 1055.
Ащеулов А.А., Воронка Н.К., Маренкин С.Ф., Раренко И.М. // Неорган. материалы. 1996. Т. 32. № 9. С. 1049.
Mekhiya A.B., Kazakov A.A., Oveshnikov L.N. et al. // Semiconductors 2019. V. 53. № 11. P. 1439. [Мехия А.Б., Казаков А.А., Овешников Л.Н. и др. // Физика и техника полупроводников. 2019. Т. 53. № 11. С. 1479.]https://doi.org/10.1134/S1063782619110137
Marenkin S.F., Novodvorsky O.A., Shorokhova A.V. et al. // Inorg. Mater. 2014. V. 50. № 9. P. 897. [Маренкин С.Ф., Новодворский О.А., Шорохова А.В. и др. // Неорган. материалы. 2014. Т. 50. № 9. С. 973.]https://doi.org/10.1134/S0020168514090076
Trukhan V.M., Soshnikov L.E., Marenkin S.F., Haliakevich T.V. // Inorg. Mater. 2005. V. 41. № 9. P. 901. [Трухан В.М., Сошников Л.Е., Маренкин С.Ф., Голякевич Т.В. // Неорган. материалы. 2005. Т. 41. № 9. С. 1031.]https://doi.org/10.1007/s10789-005-0233-7
Приходько Э.В. // Известия ВУЗ. Черная металлургия. 1991. № 2. С. 1.
Manik O.N., Manik T.O., Bilinsky-Slotylo V.R. // Thermoelectricity. 2016. № 5. P. 57.
Маник О.Н., Маник Т.О., Билинский-Слотыло В.Р. // Термоэлектричество. 2018. № 4. С. 33.
Benson D., Sankey O.F., Haeussermann U. // Phys. Rev. B. 2011. V. 84. № 12. P. 125211. https://doi.org/10.1103/PhysRevB.84.125211
Wang S.Y., Yang J., Wu L.H. et al. // Chem. Mater. 2015. V. 27. № 3. P. 1071. https://doi.org/10.1021/cm504398d
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии