

Журнал неорганической химии, 2020, T. 65, № 9, стр. 1198-1207
Пербромированные сульфонил-клозо-декабораты с экзополиэдрическими аминогруппами [2-B10Br9S((CH2)nNH2)2]– (n = 1–3)
А. В. Голубев a, А. С. Кубасов a, *, Е. С. Турышев a, А. Ю. Быков a, К. Ю. Жижин a, b, Н. Т. Кузнецов a
a Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
b Российский технологический университет
119571 Москва, пр-т Вернадского, 86, Россия
* E-mail: fobosax@mail.ru
Поступила в редакцию 15.04.2020
После доработки 28.04.2020
Принята к публикации 30.04.2020
Аннотация
Разработан метод получения полностью бромированных сульфониевых производных клозо-декаборатного аниона с функциональными аминогруппами (n-Bu4N)[2-B10Br9S((CH2)nNH2)2]– (n = 1–3). Метод основан на взаимодействии сульфониевых производных клозо-декаборатного аниона с фталимидными группами (n-Bu4N)[2-B10Br9S((CH2)npht)2]– (n = 1–3) с элементарным бромом в ацетонитриле в инертной атмосфере с последующим снятием фталимидной защиты метиламином.
ВВЕДЕНИЕ
Кластерные анионы бора представляют собой необычный класс соединений, которые могут выступать как слабокоординирующиеся лиганды и образовывать различные комплексы металлов с “мягкими кислотами” Льюиса [1–3]. Кроме того, благодаря замещению экзополиэдрических атомов водорода борного кластера возможно образование большого числа производных с различными типами экзополиэдрических заместителей [4–8], что, в свою очередь, позволяет существенно изменить свойства борного остова. Следует отметить, что введение экзополиэдрического заместителя позволяет понизить общий заряд молекулы, что также существенно влияет на координационную способность соединений этого класса [9–12]. Синтез комплексов на основе производных клозо-декаборатного аниона является актуальной задачей, решение которой позволит существенно расширить области практического применения производных кластерных анионов бора благодаря возможности получать вещества, сочетающие свойства катионной и анионной частей соединения [13–16].
Другим способом изменения физико-химических свойств кластерных анионов бора, таких как растворимость, полярность, электрохимические потенциалы и др., является галогенирование борного остова [17, 18]. При этом полное замещение атомов водорода в борном кластере можно проводить различными галогенами, что также позволяет существенно варьировать свойства кластерных соединений бора [19–23].
Среди большого разнообразия производных клозо-декаборатного аниона особо можно отметить производные со связью бор–сера [24–26]. Данные анионы обладают высокой химической устойчивостью [27], что очень важно для дальнейшей модификации этих соединений, особенно для полного замещения атомов водорода в борном остове.
Ранее в работе [28] мы представили методы получения сульфониевых производных клозо-декаборатного аниона с экзополиэдрическими аминогруппами, а также их перхлорированных аналогов. В настоящей работе мы исследовали и описали методы получения пербромированных аналогов этих соединений.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
(n-Bu4N)[2-B10H9S(R)2] (R = CH2pht, (CH2)2pht, (CH2)3pht) получали по методике [28]. Элементарный бром использовали без дополнительной очистки.
Ацетонитрил кипятили с гидридом кальция в течение 2 ч и затем перегоняли. Полученный ацетонитрил хранили над молекулярными ситами 4 Å.
Элементный анализ на углерод, азот, водород и серу выполняли на CHNS-анализаторе Eurovector EuroEA 3000.
ИК-спектры соединений записывали на ИК-фурье-спектрофотометре Инфралюм ФТ-08 (НПФ АП “Люмекс”) в области 4000–400 см–1 с разрешением 1 см–1. Образцы готовили в виде суспензии исследуемого вещества в тетрахлорметане.
Спектры ЯМР 1Н, 11B, 13C растворов исследуемых веществ в DMSO-D6 записывали на импульсном фурье-спектрометре Bruker MSL-300 (ФРГ) на частотах 300.3, 96.32 и 75.49 МГц соответственно с внутренней стабилизацией по дейтерию. В качестве внешних стандартов использовали тетраметилсилан.
РСА. Пригодные для РСА монокристаллы соединения [Ag(PPh3)4][2-B10H9S(CH2N(CO)2C6H4)2] (I) были получены в результате смешивания растворов [Ag(PPh3)4]NO3 в дихлорметане и (n‑Bu4N)[2-B10H9S(CH2N(CO)2C6H4)2] в ацетонитриле в молярном соотношении 1 : 1. После этого раствор оставляли медленно упариваться на несколько дней в холодильнике.
Наборы дифракционных отражений для кристаллов I и IV получены в Центре коллективного пользования ИОНХ РАН на автоматическом дифрактометре Bruker SMART APEX2 (λMoKα, графитовый монохроматор, ω–ϕ-сканирование). Структуры расшифрованы прямым методом с последующим расчетом разностных синтезов Фурье. Все неводородные атомы уточнены в анизотропном приближении. Все атомы водорода уточнены по модели “наездника” с тепловыми параметрами U(H) = 1.2Uэкв (1.5Uэкв для СН3-групп) соответствующих неводородных атомов. Одна из фталимидных групп в структуре I разупорядочена по трем позициям с заселенностью 0.4 : 0.4 : 0.2 с тремя общими атомами: N1, C3 и O2.
При сборе и обработке массива отражений использованы программы APEX2, SAINT и SADABS [29]. Структура расшифрована и уточнена с помощью комплекса программ OLEX2 [30].
Основные кристаллографические данные, параметры эксперимента и характеристики уточнения структуры приведены в табл. 1.
Таблица 1.
Основные кристаллографические данные, параметры эксперимента и уточнения структур для I и IV
| Соединение | I | IV |
|---|---|---|
| Брутто-формула | C90H81AgB10N2O4P4S | C39.5H59.5B10Br9N3.5O4.5S |
| М | 1626.47 | 1514.75 |
| T, K | 150 | 296.0 |
| Сингония | Триклинная | Триклинная |
| Пр. гр. | P$\bar {1}$ | P$\bar {1}$ |
| a, Å | 13.8921(6) | 12.4399(5) |
| b, Å | 14.4311(7) | 12.7882(8) |
| c, Å | 23.5375(11) | 22.4401(8) |
| α, град | 91.759(2) | 99.485(2) |
| β, град | 106.042(2) | 90.6220(10) |
| γ, град | 90.135(2) | 119.1000(10) |
| V, Å3 | 4532.6(4) | 3059.4(3) |
| Z | 2 | 2 |
| ρрасч, г/см3 | 1.192 | 1.644 |
| μ, мм–1 | 0.364 | 5.969 |
| F(000) | 1680.0 | 1476.0 |
| Размеры кристалла, мм | 0.4 × 0.3 × 0.05 | 0.8 × 0.15 × 0.02 |
| Излучение, λ, Å | 0.71073 | 0.71073) |
| Интервал углов 2θ, град | 3.084–52 | 3.702–54 |
| Число отражений: | ||
| измеренных независимых (N) |
45 109 16 643 [Rint = 0.0345] |
25 485 13 047 [Rint = 0.0490] |
| R1, wR2 по No | 0.0853, 0.2442 | 0.0489, 0.1200 |
| R1, wR2 по N | 0.1015, 0.2636 | 0.1143, 0.1454 |
Кристаллографические данные депонированы в Кембриджском банке структурных данных (№ 1996448, 1996449).
Синтез (n-Bu4N)[2-B10Br9S(CH2N(CO)2C6H4)2] (II). Соль (n-Bu4N)[2-B10H9S(CH2N(CO)2C6H4)2] (0.5 г, 0.7 ммоль) помещали в круглодонную колбу на 25 мл и приливали 5 мл ацетонитрила. Полученный раствор охлаждали до 0°С на ледяной бане и прикапывали к нему 10-кратный избыток брома (360 мл, 7 ммоль) в атмосфере аргона при постоянном перемешивании. Затем реакционную смесь медленно нагревали до комнатной температуры и оставляли перемешиваться в течение 24 ч. Далее раствор упаривали на пластинчато-роторном насосе до полного удаления летучих продуктов реакции. К полученному твердому остатку приливали 30 мл гексана и обрабатывали на ультразвуковой бане в течение 10 мин. Полученный мелкокристаллический порошок отфильтровывали и промывали дистиллированной водой (2 × 30 мл) и диэтиловым эфиром (2 × 30 мл). Выход 88%.
| C | H | N | S | |
|---|---|---|---|---|
| Найдено, %: | 28.64; | 3.45; | 2.92; | 2.23. |
| Для C34H48B10Br9N3O4S1 | ||||
| вычислено, %: | 28.71; | 3.40; | 2.95; | 2.25. |
11B ЯМР (DMSO-D6, δ, м.д.): –0.5 (s, 1B, B1), –4.7 (s, 1B, B10), –13.3 (s, 8B, B2–B9).
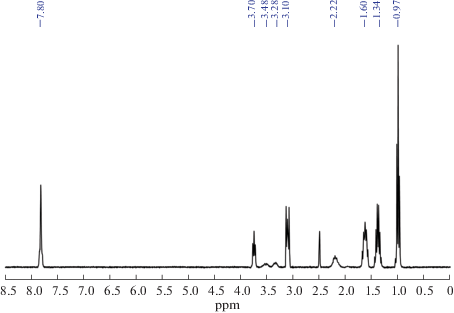
1H ЯМР (DMSO-D6, δ, м.д.): 7.80 (m, 8H, Ph), 5.21 (s, 4H, SCH2), 3.10 (m, 8H, n-Bu4N+), 1.60 (m, 8H, n-Bu4N+), 1.34 (m, 8H, n-Bu4N+), 0.97 (m, 8H, n‑Bu4N+).
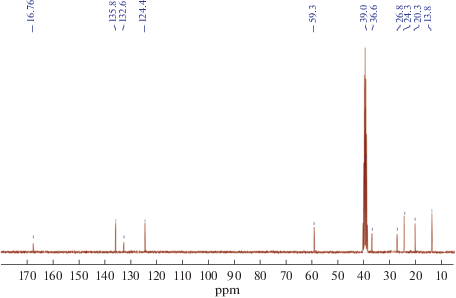
13C ЯМР (DMSO-D6, δ, м.д.): 167.6 (CO), 135.8, 132.6, 124.4 (Ph), 59.3 (n-Bu4N+), 57.6 (SCH2), 24.3 (n-Bu4N+), 20.3 (n-Bu4N+), 13.8 (n-Bu4N+).
ИК-спектр (CCl4, ν, см–1): 3374, 3309, 3241, 3179, 2950, 2908, 2845, 1764, 1734, 1615, 1541, 1460, 1421, 1369, 1180, 1153, 1110, 1071, 1010, 830, 841, 784, 760, 724, 624, 665, 650, 580, 524.
(n-Bu4N)[2-B10Br9S(CH2CH2N(CO)2C6H4)2] (III) получали аналогично II. Выход 86%.
| C | H | N | S | |
|---|---|---|---|---|
| Найдено, %: | 29.76; | 3.70; | 2.87; | 2.17. |
| Для C36H52B10Br9N3O4S1 | ||||
| вычислено, %: | 29.81; | 3.61; | 2.90; | 2.21. |
11B ЯМР (DMSO-D6, δ, м.д.): –0.5 (s, 1B, B1), ‒4.6 (s, 1B, B10), –13.4 (s, 8B, B2–B9).
1H ЯМР (DMSO-D6, δ, м.д.): 7.80 (m, 8H, Ph), 4.13 (t, 4H, CH2N), 3,96 (t, 2H, SCHaHb), 3.80 (t, 2H, SCHaHb), 3.10 (m, 8H, n-Bu4N+), 1.60 (m, 8H, n‑Bu4N+), 1.34 (m, 8H, n-Bu4N+), 0.97 (m, 8H, n‑Bu4N+).
13C ЯМР (DMSO-D6, δ, м.д.): 167.6 (CO), 135.8, 132.6, 124.4 (Ph), 59.3 (n-Bu4N+), 40.5 (SCH2), 35.4 (CH2N), 24.3 (n-Bu4N+), 20.3 (n-Bu4N+), 13.8 (n-Bu4N+).
ИК-спектр (CCl4, см–1): 3367, 3304, 3245, 3182, 2945, 2907, 2845, 1760, 1732, 1621, 1545, 1460, 1419, 1370, 1185, 1147, 1104, 1068, 1003, 834, 780, 761, 730, 624, 648,579, 526.
(n-Bu4N)[2-B10Br9S(CH2CH2CH2N(CO)2C6H4)2] (IV) получали аналогично II. Выход 90%.
| C | H | N | S | |
|---|---|---|---|---|
| Найдено, %: | 30.84; | 3.79; | 2.85; | 2.16. |
| Для C38H56B10Br9N3O4S1 | ||||
| вычислено, %: | 30.87; | 3.81; | 2.84; | 2.17. |
11B ЯМР (DMSO-D6, δ, м.д.): –0.5 (s, 1B, B1), –4.5 (s, 1B, B10), –13.2 (s, 8B, B2–B9).
1H ЯМР (DMSO-D6, δ, м.д.): 7.80 (m, 8H, Ph), 3.70 (t, 4H, CH2N), 3.48 (t, 2H, SCHaHb), 3.28 (t, 2H, SCHaHb), 3.10 (m, 8H, n-Bu4N+), 2.22 (m, 4H, SCH2CH2), 1.60 (m, 8H, n-Bu4N+), 1.34 (m, 8H, n‑Bu4N+), 0.97 (m, 8H, n-Bu4N+).
13C ЯМР (DMSO-D6, δ, м.д.): 167.6 (CO), 135.8, 132.6, 124.4 (Ph), 59.3 (n-Bu4N+), 39.0 (SCH2), 36.6 (CH2N), 26.8 (SCH2CH2), 24.3 (n-Bu4N+), 20.3 (n‑Bu4N+), 13.8 (n-Bu4N+).
ИК-спектр (CCl4, ν, см–1): 3360, 3315, 3240, 3183, 2951, 2903, 2846, 1761, 1728, 1629, 1547, 1458, 1423, 1370, 1181, 1150, 1107, 1068, 1006, 830, 780, 758, 734, 621, 647, 571, 531.
Монокристаллы IV, пригодные для РСА, получали путем насыщения раствора данного соединения в диметилформамиде водой в эксикаторе.
Синтез (n-Bu4N)[2-B10Br9S(CH2NH2)2] (V). Соль II (0.2 г, 0.28 ммоль) помещали в круглодонную колбу на 50 мл и приливали 20 мл абсолютного этанола и MeNH2 (40%, 65.23 мкл, 0.84 ммоль). Реакционную смесь нагревали в течение 4 ч при 70°С, затем добавляли эквимолярное количество H2O, подкисляли соляной кислотой до pH < 2 и нагревали еще 1 ч при 70°С. Далее реакционную смесь охлаждали до комнатной температуры, к суспензии приливали раствор KOH до pH > 10 и помещали на ультразвуковую баню на 10 мин. Полученный осадок отфильтровывали и промывали дистиллированной водой (2 × 10 мл). Выход 82%.
| C | H | N | S | |
|---|---|---|---|---|
| Найдено, %: | 18.68; | 3.94; | 3.57; | 2.72. |
| Для C18H44B10Br9N3S1 | ||||
| вычислено, %: | 18.61; | 3.81; | 3.61; | 2.76. |
11B ЯМР (DMSO-D6, δ, м.д.): –0.5 (s, 1B, B1), –4.4 (s, 1B, B10), –13.3 (s, 8B, B2–B9).
1H ЯМР (DMSO-D6, δ, м.д.): 3.50 (s, 4H, SCH2), 3.10 (m, 8H, n-Bu4N+), 2.17 (brs, 4H, NH2), 1.60 (m, 8H, n-Bu4N+), 1.34 (m, 8H, n-Bu4N+), 0.97 (m, 8H, n-Bu4N+).
13C ЯМР (DMSO-D6, δ, м.д.): 59.3 (n-Bu4N+), 43.2 (SCH2), 24.3 (n-Bu4N+), 20.3 (n-Bu4N+), 13.8 (n‑Bu4N+).
ИК-спектр (CCl4, ν, см–1): 3587, 3366, 3294, 2965, 2928, 2882, 1651, 1588, 1520,1464, 1389, 1302, 1154, 998, 881, 850, 740, 725, 526.
(n-Bu4N)[2-B10Br9S(CH2CH2NH2)2] (VI) получали аналогично V. Выход 81%.
| C | H | N | S | |
|---|---|---|---|---|
| Найдено, %: | 20.25; | 4.16; | 3.52; | 2.67. |
| Для C20H48B10Br9N3S1 | ||||
| вычислено, %: | 20.19; | 4.06; | 3.53; | 2.69. |
11B ЯМР (DMSO-D6, δ, м.д.): –0.5 (s, 1B, B1), –4.5 (s, 1B, B10), –13.3 (s, 8B, B2–B9).
1H ЯМР (DMSO-D6, δ, м.д.): 3.45 (s, 4H, CH2N), 3.10 (m, 8H, n-Bu4N+), 2.68 (t, 4H, SCH2) 2.18 (brs, 4H, NH2), 1.60 (m, 8H, n-Bu4N+), 1.34 (m, 8H, n‑Bu4N+), 0.97 (m, 8H, n-Bu4N+).
13C ЯМР (DMSO-D6, δ, м.д.): 59.3 (n-Bu4N+), 42.5 (SCH2), 36.9 (CH2N), 24.3 (n-Bu4N+), 20.3 (n‑Bu4N+), 13.8 (n-Bu4N+).
ИК-спектр (CCl4, ν, см–1): 3584, 3359, 3300, 2964, 2921, 2879, 1641, 1589, 1518,1460, 1395, 1299, 1150, 1001, 880, 847, 742, 720, 531.
(n-Bu4N)[2-B10Br9S(CH2CH2CH2NH2)2] (VII) получали аналогично V. Выход 83%
| C | H | N | S | |
|---|---|---|---|---|
| Найдено, %: | 21.72; | 4.40; | 3.50; | 2.61. |
| Для C22H52B10Br9N3S1 | ||||
| вычислено, %: | 21.69; | 4. 30; | 3.45; | 2.63. |
11B ЯМР (DMSO-D6, δ, м.д.): –0.5 (s, 1B, B1), –4.4 (s, 1B, B10), –13.4 (s, 8B, B2–B9).
1H ЯМР (DMSO-D6, δ, м.д.): 3.35 (t, 4H, CH2N), 3.10 (m, 8H, n-Bu4N+), 2.60 (t, 4H, SCH2) 2.17 (brs, 4H, NH2), 1.88 (m, 4H, SCH2CH2), 1.60 (m, 8H, n‑Bu4N+), 1.34 (m, 8H, n-Bu4N+), 0.97 (m, 8H, n‑Bu4N+).
13C ЯМР (DMSO-D6, δ, м.д.): 59.3 (n-Bu4N+), 41.5 (SCH2), 36.0 (CH2N), 29.4 (SCH2CH2), 24.3 (n‑Bu4N+), 20.3 (n-Bu4N+), 13.8 (n-Bu4N+).
ИК-спектр (CCl4, ν, см–1): 3580, 3361, 3301, 2959, 2923, 2881, 1638, 1593, 1524,1454, 1383, 1304, 1147, 1004, 879, 852, 738, 718, 521.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
В работе [28] описан метод получения перхлорированных производных клозо-декаборатного аниона с экзополиэдрическими аминогруппами с использованием сульфурилхлорида в качестве хлорирующего агента.
Получение пербромированных аналогов сульфониевых производных с N-алкилфталимидными группами [2-B10Br9S((CH2)nN(CO)2C6H4)2]– (n = 1, 2, 3) протекает в схожих условиях: в ацетонитриле в инертной атмосфере с использованием Br2 в качестве галогенирующего агента. Реакция протекает при комнатной температуре, и полное замещение атомов водорода на бром происходит примерно за 24 ч (рис. 1).
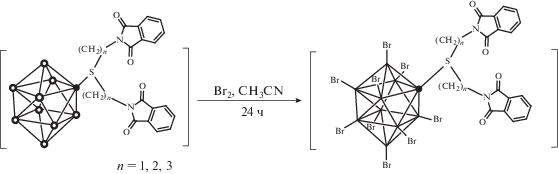
В отличие от исходных соединений и хлорированных производных, их бромированные аналоги гораздо хуже растворяются в ацетонитриле и дихлорметане.
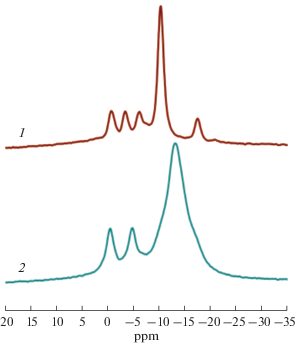
Сигналы в спектрах 11B ЯМР соединений II–IV существенно вырождены по сравнению с хлорированными аналогами (рис. 2), поэтому мы наблюдаем только два сигнала от апикальных вершин при –0.5 и –4.7 м.д. и один широкий сигнал от экваториальных атомов бора при –13.2 м.д.
В спектрах 1H ЯМР наблюдается картина, аналогичная и для хлорированных производных: сигналы от α-метиленовых групп в соединениях II–IV смещаются на 0.8 м.д. в слабое поле по сравнению с исходным небромированным производным клозо-декаборатного аниона, сигналы от остальных групп существенно не меняются (рис. 3).
В спектрах 13С ЯМР наблюдается смещение сигналов от α-метиленовых групп на 2 м.д. в область слабого поля, смещения остальных сигналов практически не происходит (рис. 4).
Аналогично хлорированным производным (n-Bu4N)[2-B10Cl9S((CH2)nPht)2] снять защитную группу можно метиламином (рис. 5).
В спектрах 11В ЯМР соединений V–VII существенных изменений не происходит, в 1H, 13С ЯМР и ИК-спектрах исчезают сигналы от фталимидных групп и появляются сигналы от аминогруппы.
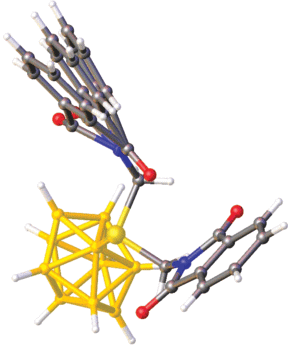
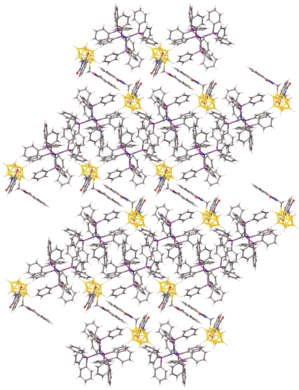
Кристаллографически независимая часть триклинной элементарной ячейки (P1) I включает комплексный катион [Ag(PPPh3)4]+ и анион [2‑B10H9S(CH2N(CO)2C6H4)2]– (рис. 6). Одна из фталимидных групп экзополиэдрического заместителя I разупорядочена и уточнена в трех позициях с заселенностью 0.4 : 0.4 : 0.2 (рис. 7). При этом разупорядоченная фталимидная группа развернута относительно второй таким образом, что углы между плоскостями, в которых лежат эти группы, находятся в диапазоне от 77.4° до 87.6°. Следует отметить, что данный угол увеличивается с уменьшением спейсерной цепи. Так, в анионе [2-B10H9S(CH2CH2N(CO)2C6H4)2]– этот угол составляет 57.0°, в анионе [2‑B10 Cl9S(CH2CH2CH2N(CO)2C6H4)2]– – 34.9° [26], а в соединении IV – 41.2°. Расстояние от плоскости неразупорядоченной группы до ближайшего атома кислорода O2 второй группы составляет 2.944 Å.
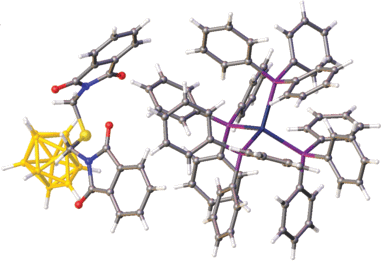
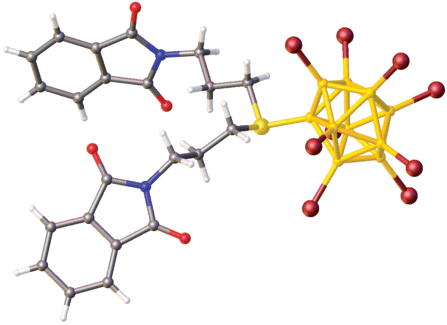
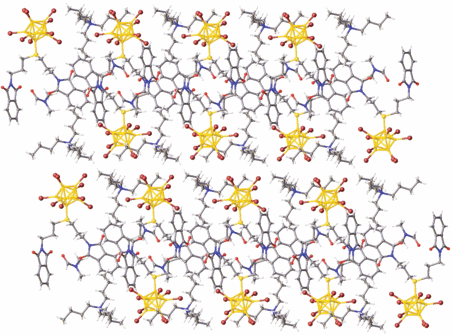
Кристаллографически независимая часть триклинной элементарной ячейки (P$\bar {1}$) IV состоит из катиона (Bu4N)+, аниона [2‑B10Br9S(CH2CH2CH2N(CO)2C6H4)2]– и молекулы DMF (рис. 8). При этом молекула растворителя находится между двумя фталимидными группами и разупорядочена, молекулу удалось локализовать только с заселенностью 0.5.
Длины связей B–S в соединениях I и IV составляют 1.891(7) и 1.894(6) Å (табл. 2) соответственно, а в хлорированном анионе [2‑B10 Cl9S(CH2CH2CH2N(CO)2C6H4)2]– – 1.890(3) Å. Таким образом, замена атомов водорода на хлор и бром практически не влияет на эту величину и согласуется с длинами связей в других сульфониевых производных [24, 26–28].
Таблица 2.
Длины связей (Å) и валентные углы (град) для соединений I и IV
| Параметр | I | IV |
|---|---|---|
| B–S, Å | 1.891(7) | 1.894(6) |
| S–C, Å | 1.834(7) 1.841(7) |
1.812(7) 1.815(6) |
| C–N, Å | 1.430(8) 1.44(1) |
1.451(8) 1.463(8) |
| BSC, град | 104.3(3) 103.0(3) |
105.0(3) 103.8(3) |
| CSC, град | 100.2(4) | 105.3(3) |
В кристалле I (рис. 9) катионы [Ag(PPPh3)4]+ и анионы [2-B10H9S(CH2N(CO)2C6H4)2]– образуют катионно-анионные слои, параллельные плоскости ab. Атомы H клозо-декаборатного аниона образуют короткие контакты с протонами [Ag(PPPh3)4]+, которые лежат в диапазоне 2.29–2.69 Å.
В структуре IV (рис. 10) анионы [2-B10Br9S(CH2CH2CH2N(CO)2C6H4)2]– обращены друг к другу экзополиэдрическими заместителями S(CH2CH2CH2N(CO)2C6H4)2, образуя слой, параллельный плоскости ab, из фталимидных фрагментов. При этом анионы [2‑B10Br9S(CH2CH2CH2N(CO)2C6H4)2]– располагаются между катионами Bu4N+ в одном слое.
ЗАКЛЮЧЕНИЕ
В результате данного исследования разработан метод получения пербромированных сульфониевых производных клозо-декаборатного аниона с экзополиэдрическими аминогруппами [2‑B10Br9S((CH2)nNH2)2]– (n = 1–3). Метод заключается в бромировании соответствующих фталимидов [2-B10Br9S((CH2)nN(CO)2C6H4)2]– (n = 1–3) элементарным бромом в ацетонитриле и последующем снятии фталимидной защиты метиламином.
Список литературы
Avdeeva V.V., Polyakova I.N., Churakov A.V. et al. // Polyhedron. 2019. V. 162. P. 65. https://doi.org/10.1016/j.poly.2019.01.051
Avdeeva V.V., Malinina E.A., Kuznetsov N.T. // Russ. J. Inorg. Chem. 2020. V. 65. № 3. P. 335. [Авдеева В.В., Малинина Е.А., Кузнецов Н.Т. // Журн. неорган. химии. 2020. Т. 65. № 3. С. 334.]https://doi.org/10.1134/S003602362003002X
Avdeeva V.V., Malinina E.A., Kuznetsov N.T. // Polyhedron. 2016. V. 105. P. 205. https://doi.org/10.1016/j.poly.2015.11.049
Sivaev I.B., Prikaznov A.V., Naoufal D. // Collect. Czech. Commun. 2010. V. 75. P. 1149. https://doi.org/10.1135/cccc2010054
Matveev E.Yu., Akimov S.S., Kubasov A.S. et al. // Russ. J. Inorg. Chem. 2019. V. 64. P. 1513. https://doi.org/10.1134/S003602361912009X
Avdeeva V.V., Polyakova I.N., Vologzhanina A.V. et al. // Polyhedron. 2017. V. 123. P. 396. https://doi.org/10.1016/j.poly.2016.12.009
Spokoyny A.M., Machan C.W., Clingerman D.J. et al. // Nature Chem. 2011. V. 3. P. 590. https://doi.org/10.1038/nchem.1088
Kubasov A.S., Matveev E.Yu., Turyshev E.S. et al. // Dokl. Chem. 2017. V. 477. P. 257. https://doi.org/10.1134/S0012500817110088
Zhang X., Dai H., Yan H. et al. // J. Am. Chem. Soc. 2016. V. 138. № 13. P. 4334. https://doi.org/10.1021/jacs.6b01249
Avdeeva V.V., Malinina E.A., Kuznetsov N.T. // Russ. J. Inorg. Chem. 2017. V. 62. P. 1673. https://doi.org/10.1134/S0036023617130022
Zhizhin K.Yu., Zhdanov A.P., Kuznetsov N.T. // Russ. J. Inorg. Chem. 2010. V. 55. P. 2089. https://doi.org/10.1134/S0036023610140019
Kubasov A.S., Matveev E.Yu., Retivov V.M. et al. // Russ. Chem. Bull. 2014. V. 63. P. 187. https://doi.org/10.1007/s11172-014-0412-2
Drozdova V.V., Lisovskii M.V., Polyakova I.N. et al. // Russ. J. Inorg. Chem. 2006. V. 51. P. 1716. https://doi.org/10.1134/S00360236061100664
Avdeeva V.V., Malinina E.A., Zhizhin K.Yu. et al. // Russ. J. Inorg. Chem. 2020. V. 65. № 4. P. 514.
Zhdanov A.P., Voinova V.V., Klyukin I.N. et al. // Russ. J. Coord. Chem. 2019. V. 45. P. 563. https://doi.org/10.1134/S1070328419080098
Malinina E.A., Kochneva I.K., Avdeeva V.V. et al. // Russ. J. Inorg. Chem. 2019. V. 64. P. 1210. https://doi.org/10.1134/S0036023619100085
Shmal’ko A.V., Sivaev I.B. // Russ. J. Inorg. Chem. 2019. V. 64. № 14. P. 1726. https://doi.org/10.1134/S0036023619140067
Gu W., Ozerov O.V. // Inorg. Chem. 2011. V. 50. P. 2726. https://doi.org/10.1021/ic200024u
Geis V., Guttsche K., Knapp C. et al. // Dalton Trans. 2009. V. 15. P. 2649. https://doi.org/10.1039/B821030F
Warneke J., Konieczka S.Z., Hou G.-L. et al. // Phys. Chem. Chem. Phys. 2019. V. 21. № 11. P. 5903. https://doi.org/10.1039/c8cp05313h
Holub J., Anwar S.El., Jelínek T. et al. // J. Inorg. Chem. 2017. V. 38. P. 4499. https://doi.org/10.1002/ejic.201700651
Kravchenko E.A., Gippius A.A., Korlyukov A.A. et al. // Inorg. Chim. Acta. 2016. V. 447. P. 22. https://doi.org/10.1016/j.ica.2016.03.025
Sharma M., Sethio D., Lawson Daku L.M. et al. // J. Phys. Chem. A. 2019. V. 123. № 9. P. 1807. https://doi.org/10.1021/acs.jpca.8b11638
Kubasov A.S., Turishev E.S., Polyakova I.N. et al. // J. Organomet. Chem. 2017. V. 828. P. 106. https://doi.org/10.1016/j.jorganchem.2016.11.035
Kubasov A.S., Matveev E.Yu., Turyshev E.S. et al. // Dokl. Chem. 2018. V. 483. P. 263. https://doi.org/10.1134/S001250081811006X
Kubasov A.S., Matveev E.Yu., Polyakova I.N. et al. // Russ. J. Inorg. Chem. 2015. V. 60. P. 198. https://doi.org/10.1134/S0036023615020084
Kubasov A.S., Matveev E.Yu., Turyshev E.S. et al. // Inorg. Chim. Acta. 2018. V. 477. P. 277. https://doi.org/10.1016/j.ica.2018.03.013
Kubasov A.S., Turishev E.S., Golubev A.V. et al. // Inorg. Chim. Acta. 2020. V. 507. P. 119589. https://doi.org/10.1016/j.ica.2020.119589
APEX2 (V. 2009, 5-1), SAINT (V7.60A), SADABS (2008/1). Bruker AXS Inc., Madison, Wisconsin, USA, 2008-2009.
Dolomanov O.V., Bourhis L.J., Gildea R.J. et al. // J. Appl. Crystallogr. 2009. V. 42. P. 339. https://doi.org/10.1107/S0021889808042726
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии