

Журнал неорганической химии, 2020, T. 65, № 9, стр. 1283-1291
Особенности поведения ионов Mg2+ и их влияние на структуру и морфологию наногидроксиапатита при адсорбционном способе создания композиционного материала ГА-Mg
А. В. Северин a, *, В. Н. Рудин a, М. Э. Пауль a
a Московский государственный университет им. М.В. Ломоносова
119991 Москва, Ленинские горы, 1, Россия
* E-mail: severin@radio.chem.msu.ru
Поступила в редакцию 12.03.2020
После доработки 12.04.2020
Принята к публикации 27.04.2020
Аннотация
Выявлены особенности процессов сорбционного взаимодействия ионов магния(II) в водном растворе с наночастицами гидроксиапатита в качестве сорбента. Показано, что кинетика процесса хорошо описывается моделью псевдовторого порядка. Изотерма сорбции отражает последовательное изменение механизмов взаимодействия сорбата и сорбента при увеличении концентрации последнего. Выявлено два основных участка на изотерме сорбции. Основным механизмом на первом участке является ионный обмен, который дополняется физической адсорбцией на втором участке. Каждый участок по отдельности формально может быть описан моделью Ленгмюра со следующими параметрами: коэффициент адсорбции K = 33.7, 32.1 л/моль и предельная адсорбция Г∞ = 2.2, 8.5 моль Mg/моль гидроксиапатита соответственно для первого и второго участков. При высоких концентрациях ионов магния обнаружено морфологическое и структурное изменение сорбента, которое может быть связано с возможностью протекания химической реакции между компонентами с образованием новой фазы.
ВВЕДЕНИЕ
Гидроксиапатит (ГА) является минеральной составляющей костной ткани животных и человека, причем находится он не только в ткани, но и в кровотоке виде нанокристаллов, связанных с коллагеном [1]. Синтетический ГА, в том числе в наноформе, давно и активно используется в мировой медицинской практике и как непосредственно лекарственный препарат, как материал для трансплантатов и имплантатов, а также в качестве носителя других лекарственных форм или композитов [2–6]. Это определяется тем, что среди всех видов биокерамики фосфаты кальция, в частности ГА, являются наиболее биосовместимыми материалами для реконструкции дефектов костной ткани [5], поскольку их химический состав подобен минеральной фазе костной ткани. Для лечения костных травм широкое применение нашла паста ГА, полученная в лаборатории гетерогенных процессов кафедры радиохимии химического факультета МГУ [7–9], которая представляет собой высокодисперсный наногидроксиапатит с удельной поверхностью ~170 м2/г со средней длиной единичного кристалла ~60 нм и шириной ~15 нм. Лечение с использованием нанодисперсного ГА [10, 11] показало высокие результаты в репарации костной ткани.
Добиться более выраженного результата можно, создавая биокомпозиты с природными биополимерами или биоактивными металлами, например с магнием. Хорошо известно, что у млекопитающих ионы магния являются четвертыми по количеству катионами после натрия, калия и кальция [12, 13]. Кроме того, магний является вторым наиболее распространенным внутриклеточным катионом, активность которого связана с регулированием ионных каналов кальция и натрия, стабилизацией ДНК. Он является кофактором и катализатором для многих ферментов и стимулирует клеточный рост и пролиферацию [14, 15]. Кости являются “главным депо магния” в человеческом организме. Как показано в работе [16], необеспеченность организма магнием влияет на состояние соединительной ткани и приводит к ее дисплазии (нарушение структуры ткани). Это связано с кальциевым обменом, так как магний является его синергистом в метаболическом процессе. Клиническое исследование [17] показывает, что в странах, где встречается более высокое употребление магния по сравнению с кальцием, обнаруживается меньшее количество больных остеопорозом. Чтобы оценить перспективы использования кальций-магниевых фосфатов в составе композита в медицине необходимо понимание как существенной физиологической роли магния в организме человека, так и особенностей его взаимодействия с ГА.
Большинство литературных данных по взаимодействию ионов магния с ГА касается введения магния в решетку ГА с целью получения различных материалов медицинского назначения, например посредством сокристаллизации Mg2+ при получении ГА [18–21]. Вместе с тем доступность иона Mg для организма при его инкорпорации в решетку ГА весьма ограничена и связана с его резорбцией. Поэтому если поставлена задача создать относительно высокую концентрацию ионов магния в области функционирования, необходим сорбционный способ связывания ГА и целевого катиона. Данных по изучению адсорбции ионов Mg2+ на синтетическом ГА крайне мало [22, 23], и исследована она при очень малых концентрациях (2–10 ммоль/л) и на гидроксиапатите с малой удельной поверхностью (25–30 м2/г). Вследствие этого при изучении адсорбции исследователям было достаточно низких концентраций магния, чтобы выйти на предельную величину адсорбции, и дальнейшее увеличение концентрации ионов магния представлялось им нецелесообразным. В связи с этим актуально выявление закономерностей адсорбционного взаимодействия в водных и физиологических растворах ионов Mg2+ с нанокристаллами ГА в широком диапазоне концентраций катиона.
Цель настоящей работы – выявление особенностей механизма и кинетики адсорбции и десорбции ионов магния на ГА во всем возможном диапазоне концентраций Mg2+, получение количественных характеристик данных процессов, а также определение степени влияния сорбирующихся ионов на структуру и морфологию самого сорбента. Данная информация необходима для создания медицинских препаратов на основе ГА с различным содержанием магния и применения их для лечения костных травм и остеопороза.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве сорбента использовали водную суспензию нанокристаллов ГА. Синтез сорбента проводили путем введения раствора ортофосфорной кислоты с контролируемой скоростью в водную суспензию СаО (в процессе гидролиза образуется Са(ОН)2) при интенсивном перемешивании и постоянном контроле рН реакционной смеси. Более подробно методика описана в работе [24], физико-химические механизмы, приводящие к получению наночастиц ГА, рассмотрены в работе [7]. Изучение полученного материала заключалось в определении содержания твердой фазы в суспензии (5.73 ± ± 0.20% по массе) и определении морфологических и поверхностных характеристик – средние размеры кристаллов и их удельная поверхность близки к значениям, приведенным во введении. Кроме того, методами химического и рентгенофазового (РФА) анализа твердой фазы показано, что в системе присутствует только ГА.
Для проведения сорбционных экспериментов готовили 0.1 и 0.3 М водные растворы хлорида магния путем растворения точных навесок MgCl2 · 6H2O квалификации “ч. д. а.” в 1 л дистиллированной воды. Концентрацию ионов магния контролировали спектрофотометрически с помощью специального аналитического набора реагентов Магний-Ольвекс (Ольвекс-диагностикум, Россия) по методике, указанной производителем, на спектрофотометре Shimadzu UV-1280 (Япония) при длине волны 575 нм. Поскольку данный метод позволяет с хорошей точностью (~7%) определять концентрацию магния в диапазоне от 0.3 до 2.0 ммоль/л, бóльшую часть образцов растворов (как исходного, так и после адсорбции) разбавляли дистиллированной водой (в 10–100 раз). Помимо концентрации ионов магния в растворах контролировали содержание ионов Са2+ с помощью реагента о-крезолфталеин комплексона (КФК) на спектрофотометре Shimadzu UV-1280 при длине волны 575 нм по методике, указанной производителем (Ольвекс-диагностикум, Россия). При проведении экспериментов по десорбции готовили ряд растворов CaCl2 различной концентрации. Для этого точную навеску CaCl2 квалификации “ч. д. а.” растворяли в дистиллированной воде. Точную концентрацию раствора устанавливали спектрофотометрически. Затем полученный раствор разбавляли дистиллированной водой до необходимых концентраций.
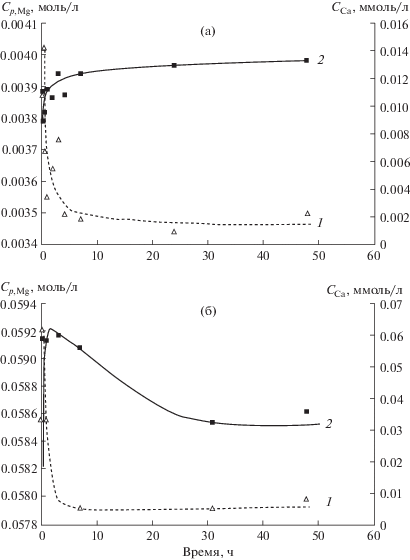
При исследовании кинетики сорбции точную навеску суспензии ГА с известным содержанием твердой фазы помещали во флакон емкостью 12 мл, добавляли точный объем раствора хлорида магния и воды. Массу флакона постоянно контролировали весовым методом на аналитических весах Shimadzu AX-200 (Япония). Во всех образцах было одинаковое соотношение ГА и хлорида магния. Кинетику изучали при двух исходных концентрациях магния (5 и 62 ммоль/л). Образцы помещали на орбитальный шейкер SkyLine Shaker S-3.08L (Elmi, Латвия) и перемешивали в идентичных условиях при комнатной температуре. Через определенные промежутки времени образцы центрифугировали (центрифуга MLW-T51.1, 3000 g, ГДР) для отделения твердой фазы и отбирали пробы растворов (по 1 мл в пробирки эппендорф на 1.5 мл) с помощью автоматического дозатора Eppendorf Research (Нидерланды) для определения содержания ионов магния и кальция по методике, указанной выше. Выявлено, что стационарное состояние системы в процессе сорбции в среднем достигается в течение 7–10 ч. Результаты экспериментов представлены на рис. 1.
Рис. 1.
Кинетика сорбции ионов магния (1) на ГА и изменение содержания ионов кальция (2) в маточном растворе при исходной концентрации ионов магния 5 (а) и 62 ммоль/л (б), точность измерений 7%.
В эксперименте по изучению изотермы адсорбции точную навеску суспензии ГА помещали во флакон емкостью 12 мл, добавляли точный объем раствора хлорида магния и воды. Как и в случае экспериментов по кинетике адсорбции, контролировали массу каждого компонента. В данном эксперименте варьировали соотношение массы суспензии ГА и концентрацию раствора хлорида магния. Образцы помещали на орбитальный шейкер и перемешивали в идентичных условиях. Минимальное время проведения эксперимента, предварительно определенное по кинетике адсорбции (время выхода кинетической кривой на стационар), было примерно удвоено для надежности и достоверного выхода системы на стационар и составляло 24 ч. По истечении данного времени образцы центрифугировали в течение 10 мин, отбирали пробы раствора и раздельно определяли в них концентрации ионов кальция и магния. Экспериментальная изотерма адсорбции в избранном диапазоне равновесных концентраций ионов магния представлена на рис. 2 (кривая 1).
Рис. 2.
Изотермы сорбции (кривая 1) и десорбции (кривая 2) ионов магния из водного раствора MgCl2 на высокодисперсном ГА. На графике обозначены точки отбора проб на физико-химический анализ.
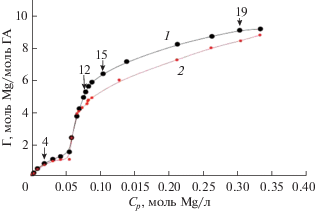
Для определения времени и степени десорбции была проведена серия идентичных экспериментов по десорбции (в точке с концентрацией магния 31 ммоль/л). Предварительную сорбцию проводили по методике, описанной выше. После центрифугирования образцов (центрифуга MLW T 51.1, 3000 g) маточный раствор удаляли и заменяли его таким же количеством воды. Образцы интенсивно встряхивали для гомогенизации суспензии и продолжали перемешивать на орбитальном шейкере. Далее через разные промежутки времени пробы центрифугировали и отбирали раствор для определения содержания ионов магния спектрофотометрическим методом. По результатам эксперимента выявлено, что при данной концентрации десорбция незначительна даже через 48 ч.
Изотерму десорбции исследовали в экспериментальных точках с соотношением исходных компонентов, как в экспериментах по изучению изотермы сорбции. После ее проведения и анализа маточного раствора на содержание ионов кальция и магния его удаляли и заменяли раствором с концентрацией ионов кальция, определенной для предыдущей точки изотермы адсорбции, приготовленным, как было указано выше. После этого пробы гомогенизировали и перемешивали в течение 2 сут. По истечении времени отбирали аликвоты маточного раствора для спектрофотометрического определения содержания ионов магния. Полученные данные представлены на рис. 2 (кривая 2).
Все сорбционные эксперименты проводили при температуре 22 ± 0.1°С, каждый опыт повторяли не менее трех раз.
Для выявления влияния адсорбции ионов магния на структуру и морфологию ГА был проведен комплекс физико-химических методов анализа твердой фазы после адсорбции при соотношении компонентов, соответствующих точкам 4, 12, 15 и 19 на изотерме адсорбции (рис. 2).
Фазовый состав образцов исследовали с помощью автоматического рентгеновского дифрактометра ДР-01 РАДИАН (λ(СuKα) = 1.54178 Å) c шагом 0.05° в диапазоне углов 2θ от 10° до 70° и временем экспозиции 12 с/град.
Морфологию нанокристаллов изучали методом трансмиссионной электронной микроскопии (ТЭМ) на просвечивающем электронном микроскопе JEM-1011B (Япония) с разрешением 0.3 нм. Образцы для ТЭМ готовили нанесением на специальную медную сеточку с формваровой пленкой капли суспензии изучаемого препарата, разбавленной дистиллированной водой в соотношении 100 : 1. Далее образцы высушивали на воздухе и хранили в специальных пеналах.
Предварительно был исследован состав и степени окисления элементов поверхности сорбента методом рентгеновской фотоэлектронной спектроскопии (РФЭС). Результаты этих экспериментов подробно описаны в работе [25] и использованы для обсуждения результатов данного исследования.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Кинетика адсорбции ионов магния на гидроксиапатите (при двух исходных концентрациях магния в растворе) и соответствующая кинетика выхода ионов кальция в раствор (рис. 1) показывают, что среднее время выхода на стационарный режим составляет (независимо от начальной концентрации магния) 7–10 ч. Из данных рис. 1 можно сделать вывод об изменении динамики поступления ионов кальция в раствор в зависимости от исходной концентрации магния.
На экспериментальной изотерме адсорбции наблюдаются две четко выраженные области в диапазонах равновесных концентраций ионов магния 0–55 и 55–350 ммоль/л (рис. 2, кривая 1).
Аналогично можно выделить два участка и на изотерме десорбции (рис. 2, кривая 2).
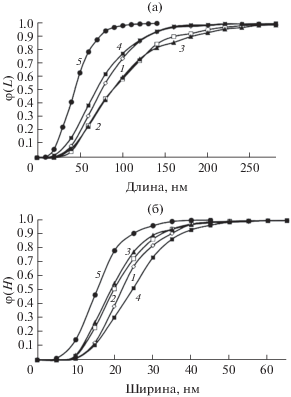
Данные электронной микроскопии кристаллов ГА до и после адсорбции ионов магния представлены на рис. 3. Приведены результаты морфологического анализа экспериментальных фотографий в виде рассчитанных интегральных функций распределения кристаллов по длине (φ(L)) и ширине (φ(H)) (рис. 4).
Рис. 3.
Данные трансмиссионной электронной микроскопии чистого ГА (а) и образцов ГА после адсорбции ионов магния, соответствующие точкам 4 (б), 12 (в), 15 (г) и 19 (д) (рис. 2).

Рис. 4.
Интегральные функции распределения нанокристаллов чистого ГА (1) и ГА после сорбции ионов Mg в различных точках экспериментальной изотермы (2–5, соответствующие образцам 4, 12, 15 и 19 соответственно, рис. 2) по длине φ(L) (а) и ширине φ(Н) (б).
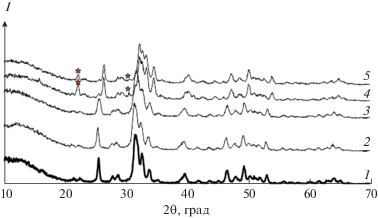
Дифрактограммы исходного ГА и образцов, полученных после сорбции ионов магния, приведены на рис. 5.
Рис. 5.
Рентгеновские дифрактограммы образцов чистого ГА (1) и ГА после адсорбции ионов магния (2–5, дифрактограммы образцов 4, 12, 15 и 19 соответственно, рис. 2). Звездочками обозначены пики, относящиеся к фазе Ca9Mg(HPO4)(PO4)6 (витлокит).
Анализ кинетических кривых показал, что основное их различие при больших и малых концентрациях ионов магния заключается в динамике выхода ионов кальция в раствор. При малых начальных концентрациях ионов магния ионы кальция симбатно выходят в раствор и их концентрация достигает стационарного значения 13 ммоль/л, а при больших концентрациях сначала происходит выброс ионов кальция до концентрации 60 ммоль/л, а затем плавное снижение до концентрации 30–40 ммоль/л. Скорее всего, при низких концентрациях происходит частичный обмен ионов Mg2+ с наиболее подвижными поверхностными ионами кальция ГА. Такие ионы находятся в положении Са(I) (девятивершинники СаО9, расположенные вдоль тройной оси) и, по различным литературным данным, более склонны к обмену с ионами Mg [26]. При более высоких концентрациях может протекать обратная сорбция излишнего количества ионов кальция на нанокристаллы ГА или возможно превышение в растворе произведения растворимости по одному из фосфатов кальция, скорее всего самого гидроксиапатита или октакальцийфосфата, и уход его обратно в твердую фазу. Для анализа кинетических кривых были применены две кинетические модели: псевдопервого и псевдовторого порядка [27]. Установлено, что обсчет экспериментальных данных по модели псевдопервого порядка применим только для начального участка кинетической кривой, соответствующей низким концентрациям сорбата. Гораздо лучше кинетические данные описываются моделью псевдовторого порядка. Уравнение реакции обратимой адсорбции псевдовторого порядка выводится [27] из уравнения:
а в линеаризованном виде: где qt также обозначает адсорбцию в данный момент времени; qe – равновесную адсорбцию; k1 – константу скорости адсорбции, а также из соотношения: где v0 – скорость реакции в начальный момент времени.В результате были рассчитаны параметры кинетики псведовторого порядка для разных концентраций ионов магния, представленных в табл. 1.
Таблица 1.
Параметры кинетики сорбции по модели псевдовторого порядка
| Концентрация ионов магния, моль/л | k1, г/мг/мин | v0, г/мг/мин |
|---|---|---|
| 0.0053 | 0.009 ± 0.001 | 3.31 ± 0.23 |
| 0.061 | 0.16 ± 0.01 | 39 ± 3 |
Различия в константах k1 и скоростях v0 практически пропорциональны изменению исходной концентрации ионов магния в растворе.
По данным ТЭМ, увеличение концентрации ионов магния сопровождается изменением характеристик кристаллов ГА – уменьшением длины и увеличением ширины (рис. 4). Кроме того, известно [28], что наличие в системе хлорид-иона способствует образованию более вытянутых и узких кристаллов. При высоких концентрациях ионов магния (точка 19) наблюдается частичный распад наночастиц сорбента, сопровождаемый существенным уменьшением размеров индивидуальных нанокристаллов ГА. Более наглядно это видно на микрофотографии на рис. 6.
Рис. 6.
Дезагрегирование индивидуальных наночастиц ГА в процессе сорбции ионов магния при высоких концентрациях сорбата.

Как указано выше, на экспериментальной изотерме сорбции наблюдаются две четко выраженные области. Начальный участок изотермы в данной работе можно формально описать уравнением типа Ленгмюра. Полученные параметры сорбции приведены в табл. 2 в сравнении с литературными данными [29].
Таблица 2.
Параметры адсорбции, рассчитанные при линеаризации начального участка изотермы в сравнении с литературными данными
| Тип сорбата, среда | Коэффициент корреляции, R2 | Коэффициент адсорбции K, л/моль | Предел адсорбции, Г∞, моль Mg/моль ГА |
|---|---|---|---|
| Mg(NO3)2, вода [29] | 0.99 | 532 ± 26 | 0.14 ± 0.01 |
| MgCl2, вода | 0.98 | 33.7 ± 2.4 | 2.18 ± 0.15 |
Предполагаем, что основным механизмом сорбции магния на начальном участке является ионообменный механизм, что подтверждается данными по кинетике сорбции ионов магния и выходу ионов кальция (рис. 1), а также данными [22]. При этом сорбция происходит преимущественно на боковых гранях нанокристаллов ГА, где сосредоточены наиболее подвижные ионы кальция. Подобный процесс наблюдался нами в предшествующих экспериментах по сорбции из растворов нитрата магния [29].
На втором участке, по нашему мнению, действует смешанный механизм сорбции – ионообменный по ионам кальция и поверхностная координация хлорида магния с фосфат-ионами. Кроме того, резкое увеличение концентрации ушедшего в твердую фазу катиона связано, скорее всего, с частичной дезагрегацией наночастиц в суспензии (рис. 6). Увеличение концентрации хлорид-ионов также приводит к уменьшению слоя гидратированной воды на поверхности нанокристаллов. В результате для сорбционного (ионообменного или поверхностного) взаимодействия становится доступной почти вся поверхность наночастиц.
Формально второй участок (по аналогии с начальным) также можно описать моделью, аналогичной модели Ленгмюра. При этом из результирующего значения сорбции необходимо вычесть значение максимальной адсорбции, найденной для начального участка (табл. 2). Рассчитанные параметры равны: коэффициент адсорбции K = = 32.05 ± 2.24 л/моль; предельная адсорбция Г∞ = = 8.48 ± 0.59 моль Mg/моль ГА (R2 = 0.98).
Расчет общего количества ионов магния, способных сорбироваться на поверхности ГА, исходя из радиуса иона Mg2+ (0.74 Å) и величины удельной свободной поверхности ГА в суспензии (~170 м2/г) составляет величину ~8.2 моль Mg2+ на моль ГА. Однако максимальная сорбция по изотерме составляет ~9.3 моль Mg/моль ГА (рис. 2). Возможно, часть ионов магния после обмена на ионы Са2+ диффундирует в глубь приповерхностного слоя ГА. Такое предположение подтверждается данными РФЭС, приведенными нами в работе [25] и представленными в табл. 3.
Таблица 3.
Атомный состав поверхности образцов по отношению к атому фосфора Р для различных равновесных концентраций ионов магния Ср,Mg, моль/л [25]
| Образец | Ср,Mg, моль/л | Г, моль Mg/моль ГА | Состав поверхности |
|---|---|---|---|
| ГА-0 | – | – | Ca1.00P0.60O2.40 + 0.20H2 (расчет) |
| ГА-0 | – | – | Ca0.95P0.60O2.51 + 0.34С0.12 (эксперимент) |
| ГА-4 | 0.019 | 0.84 | Ca0.92P0.60O2.55 + 0.50C0.06 Mg0.15 Cl0.03 |
| ГА-12 | 0.078 | 5.31 | Ca0.88P0.60O2.53 + 0.22C0.01 Mg0.31 Cl0.09 |
| ГА-15 | 0.103 | 6.43 | Ca0.92P0.60O2.60 + 0.90C0.14 Mg0.33 Cl0.3 |
| ГА-19 | 0.310 | 9.19 | Ca0.87P0.60O2.60 + 1.18C0.15 Mg0.30 Cl0.26 |
Как при ионообменном, так и при смешанном механизме сорбции, ионы магния должны занимать только поверхностные позиции на нанокристалле ГА, не влияя на его структуру. Это подтверждают данные РФА (рис. 5). Анализ экспериментальных дифрактограмм в сравнении с чистым ГА показал отсутствие в образцах других фаз, кроме ГА, для точек 4 и 12. Однако уже в точках 15 и 19 возможна диффузия ионов магния в глубь кристалла с заменой ионов кальция в основных кристаллографических позициях (Са(I)) и образование посторонней фазы, по-видимому, витлокита.
Из приведенных выше данных РФЭС [25], РФА и морфологического анализа видно, что при больших концентрациях сорбата (окончание второго участка изотермы), возможно, начинает проявляться третий механизм взаимодействия хлорида магния и ГА. При этом происходит процесс, связанный, по нашему мнению, с реакцией между этими веществами в приповерхностном слое сорбента. В результате возможно образование новой фазы и разрушение частиц ГА, что мы и наблюдаем в эксперименте (рис. 5, 6). Кроме того, по данным РФЭС, в твердую фазу входит и хлорид-ион (табл. 3).
Предлагаемый выше механизм сорбционного взаимодействия ионов магния с ГА косвенно подтверждают результаты экспериментов по десорбции. На экспериментальной кривой изотермы десорбции (рис. 2, кривая 2) можно также выделить две основные области. На начальном участке, когда превалирует преимущественно ионообменный механизм взаимодействия, десорбция незначительная, что дополнительно подтверждают данные по ее кинетике. Она протекает медленно, и количество ушедших в маточный раствор ионов магния не превышает 2–3%. На втором участке, когда в системе реализуется смешанный механизм взаимодействия, десорбция более заметна и ее максимальная величина достигает 16%. По-нашему мнению, это связано с более легким уходом в раствор хлорида магния, который сорбировался за счет координации с фосфат-ионами ГА. И, наконец, в самом конце второго участка, когда начинает сказываться образование смешанных кальций-магниевых твердых фаз, за счет протекания химических реакций десорбция вновь резко уменьшается.
Как видно из данных табл. 2, 3, а также морфологического анализа, немаловажным является поведение противоиона в процессе сорбции. Это особенно необходимо учитывать в случае хлорид-ионов, поскольку предполагается функционирование будущего препарата в физиологической среде.
ЗАКЛЮЧЕНИЕ
Таким образом, можно представить себе следующую картину сорбционного взаимодействия ионов магния с ГА в выбранном экспериментальном диапазоне концентраций сорбата. На первом этапе в диапазоне концентраций хлорида магния 0–55 ммоль/л сорбция протекает по ионообменному механизму, при котором ионы магния заполняют все боковые грани нанокристаллов ГА. Этот процесс сопровождается выходом ионов кальция в маточный раствор и характеризуется затрудненным процессом десорбции. Изотерму адсорбции на этом участке формально можно описать моделью Ленгмюра с параметрами, указанными выше. На втором этапе (55–350 ммоль/л) происходит дезагрегация частиц ГА, при которой становятся доступными для сорбции остальные грани нанокристаллов. Здесь реализуется смешанный механизм сорбционного взаимодействия, при котором помимо ионного обмена протекает поверхностная сорбция хлорида магния с координацией через фосфатные группы ГА. Второй участок формально может быть описан уравнением типа Ленгмюра с параметрами, указанными выше. В конце второго этапа возможно протекание химических реакций, при которых возникает посторонняя фаза (витлокит или смешанные Са-Mg апатиты с частичной заменой ОН на Cl), резко меняется морфология частиц ГА. В результате для создания композиционных материалов ГА-Mg медицинского назначения сорбционным способом в дальнейшем необходимо ограничиться концентрационным диапазоном – до 0.3 моль/л (по магнию). При этом для пролонгированного действия препарата с небольшим выходом ионов магния необходимо использовать соотношения компонентов в пределах первого участка изотермы. Если по медицинским показаниям необходим первоначальный выброс стимулирующего компонента, концентрации должны соответствовать второму участку.
Список литературы
Титов А.Т., Ларионов П.М., Щукин B.C., Зайковскии В.И. // Докл. АН. 2000. Т. 373. № 3. С. 257.
Yu Ya-Dong, Zhu Ying-Jie, Qi Chao et al. // J. Colloid Interface Sci. 2017. V. 496. P. 416. https://doi.org/10.1016/j.jcis.2017.02.041
Ezhova Z.A., Zakharov N.A., Koval E.M. // Russ. J. Inorg. Chem. 2018. V. 63. № 8. P. 1001. https://doi.org/10.1134/S0036023618080065
Forte L., Sarda S., Combes C. et al. // Colloids Surf. B: Biointerfaces. 2017. V. 160. P. 493. https://doi.org/10.1016/j.colsurfb.2017.09.055
Szcześ A., Hołysz L., Chibowski E. // Adv. Colloid Interface Sci. 2017. V. 249. P. 321. https://doi.org/10.1016/j.cis.2017.04.007
Orlovskii V.P., Komlev V.S., Barinov S.M. // Inorg. Mater. 2002. V. 38. № 10. P. 973. [Орловский В.П., Комлев В.С., Баринов С.М. // Неорган. материалы. 2002. Т. 38. № 10. С. 1159.]
Melikhov I.V., Komarov V.F., Severin A.V. et al. // Dokl. Phys. Chem. 2000. V. 373. P. 125. [Мелихов И.В., Комаров В.Ф., Северин А.В. и др. // Докл. АН. 2000. Т. 373. С. 355.]
Domínguez-Trujilloa C., Peónb E., Chicardia E. et al. // Surf. Coat. Technol. 2018. V. 333. P. 158. https://doi.org/10.1016/j.surfcoat.2017.10.079
Medvecky L., Sopcak T., Durisin J. et al. // Mater. Lett. 2011. V. 65. P. 3566. https://doi.org/10.1016/j.matlet.2011.07.096
Зуев В.П., Сергеев П.В., Мелихов И.В. // Химико-фармацевтический журнал. 1994. № 2. С. 10.
Зуев В.П., Панкратов А.С. // Стоматология. 1999. Т. 1. С. 37.
Weisinger J.R., Bellorín-Font E. // Lancet. 1998. V. 9125. P. 391.
Wolf F.I., Cittadini A. // Mol. Aspects Med. 2003. V. 24. № 1–3. P. 3.
Sethua S.N., Namashivayama S., Devendrand S. // Int. J. Biol. Macromol. 2017. V. 98. P. 67. https://doi.org/10.1016/j.ijbiomac.2017.01.089
Wu L., Luthringer B.J.C., Feyerabend F. et al. // Acta Biomaterialia. 2014. V. 10. P. 2843. https://doi.org/10.1016/j.actbio.2014.02.010
Torshin I.Yu., Gromova O.A. // Russ. Med. J. 2008. № 4. P. 230. [Торшин И.Ю., Громова О.А. // Рус. мед. журн. 2008. № 4. С. 230.]
Schlingmann K.P., Weber S., Peters M. et al. // Nat. Genet. 2002. V. 31. № 2. P. 166.
Suchanek W.L., Byrapp S.K., Shuk P. et al. // Biomaterials. 2004. V. 25. P. 4647. https://doi.org/10.1016/j.biomaterials.2003.12.008
Lala S., Ghosh M., Das P.K. et al. // Mater. Chem. Phys. 2016. V. 170. P. 319. https://doi.org/10.1016/j.matchemphys.2015.12.058
Akemi Yasukawa, Takashi Yokoyama, Kazuhiko Kandori, Tatsuo Ishikawa // Colloids Surf. A. 2004. V. 238. № 1–3. P. 133. https://doi.org/10.1016/j.colsurfa.2004.03.001
Rasskazova L.A., Zhuk I.V., Korotchenko N.M. et al. // Sci. Rep. 2019. V. 9. № 1. P. 14836.
Shimabayashi S., Tamura Ch., Nakagaki M. // Chem. Pharm. Bull. 1981. V. 29. № 8. P. 2116.
Aoba T., Moreno E.C., Shimoda S. // Calcif. Tissue. Int. 1992. V. 51. P. 143.
Severin A.V., Pankratov D.A. // Russ. J. Inorg. Chem. 2016. V. 61. № 3. P. 265. [Северин А.В., Панкратов Д.А. // Журн. неорган. химии. 2016. Т. 61. № 3. С. 279.]https://doi.org/10.1134/S0036023616030190
Тетерин Ю.А., Рудин В.Н., Северин А.В. и др. // Неорган. материалы. 2020. Т. 56. № 5. С. 507–518. https://doi.org/10.31857/S0002337X20050139
Вересов А.Г., Третьяков Ю.Д., Путляев В.И. // Рос. хим. журн. 2004. Т. 48. № 4. С. 52.
Prasad M., Saxena S. // Indust. Eng. Chem. Res. 2004. V. 43. № 8. P. 1512. https://doi.org/10.1021/ie030152d
Северин А.В., Васильев А.Н., Остапенко В.С. и др. // IX Междунар. научн. конф. “Кинетика и механизм кристаллизации. Кристаллизация и материалы будущего”. Тез. докл. Иваново, 2016. С. 171.
Северин А.В., Рудин В.Н. // IX Междунар. научн. конф. “Кинетика и механизм кристаллизации. Кристаллизация и материалы будущего”. Тез. докл. Иваново, 2016. С. 92.
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии