

Журнал неорганической химии, 2021, T. 66, № 2, стр. 220-228
Комплексный подход к мониторингу летучих органических соединений сенсорными фотонно-кристаллическими матрицами
Е. С. Большаков a, А. В. Иванов a, b, *, А. В. Гармаш a, А. С. Самохин a, А. А. Козлов c, Ю. А. Золотов a, b
a Московский государственный университет имени М.В. Ломоносова
119991 Москва, Ленинские горы, 1, Россия
b Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
c Российский технологический университет МИРЭА, Институт тонких химических технологий
им. М.В. Ломоносова
119454 Москва, пр-т Вернадского, 86, Россия
* E-mail: sandro-i@yandex.ru
Поступила в редакцию 06.04.2020
После доработки 01.06.2020
Принята к публикации 05.06.2020
Аннотация
Предложен комплексный аппаратно-программный подход к регистрации аналитического отклика сенсорных матриц при продолжительном воздействии жидких углеводородов и их паров. Разработана и с помощью 3D-печати изготовлена ячейка для детектирования углеводородов на фотонно-кристаллических сенсорных матрицах с компьютерной записью и накоплением спектров диффузного отражения с последующей метрологической обработкой сигнала. Осуществлен контроль низкого содержания летучих органических соединений в воздухе рабочей зоны, данные хорошо согласуются с этапами рабочего цикла на лакокрасочном производстве. Подтверждена статистическая значимость изменений аналитического сигнала фотонно-кристаллического сенсора, обусловленных меняющейся во времени концентрацией углеводородов.
ВВЕДЕНИЕ
Фотонные кристаллы – это твердотельные функциональные материалы, имеющие упорядоченную структуру с периодически изменяющимся показателем преломления и обладающие структурным цветом [1]. Фотонный кристалл прозрачен в видимой области электромагнитного излучения, за исключением узкой полосы, которая обусловлена наличием стоп-зон, а также полной и псевдозапрещенной зон. Положение максимума полосы отражения фотонного кристалла может быть найдено из закона Брэгга–Снелла:
где m – порядок дифракционного максимума, λ – длина волны максимума отражения, нм, d111 – межплоскостное расстояние между кристаллическими плоскостями, нм (111), neff – эффективный показатель преломления кристаллической структуры (в нашем случае кристаллического коллоидного массива), nair – показатель преломления среды (в нашем случае воздуха), из которой падает свет, θ – угол падения [2, 3]. Из уравнения (1) видно, что положение максимума полосы отражения фотонного кристалла существенно зависит от угла наблюдения. Следовательно, необходим надежный универсальный и унифицированный метод регистрации структурного цвета фотонного кристалла.В последнее время материалы на основе фотонных кристаллов получили распространение в химическом анализе. Разнообразие способов формирования фотонных кристаллов, типов образующихся структур и способов их модифицирования велико [4–10]. К основным методам увеличения селективности фотонных кристаллов можно отнести следующие: а) использование в качестве материала матрицы чувствительных к аналиту веществ (матричное взаимодействие), б) введение в фотонную структуру молекулярного агента распознавания – импрегнирование и иммобилизация реагента, в) формирование в структуре специфических сайтов распознавания для захватывания молекул-мишеней – метод молекулярных отпечатков. Разработка сенсоров, использующих в качестве платформы фотонные кристаллы, сочетается с такими направлениями создания и развития функциональных материалов, как применение полимеризированных ионных жидкостей [11] или токопроводящих полимеров [12] в качестве матрицы для аналитических сенсоров; создание многослойных сборок, состоящих, например, из фотонного гидрогеля и ион-селективной пластифицированной ПВХ-мембраны [13]. Приведем также пример применения композиционных субмикронных частиц: модифицированные суперпарамагнитные наночастицы заключены в полимерную субмикронную капсулу, и непосредственно на ее поверхности отпечатаны молекулы-мишени [14]. Интересным примером является формирование фотонных кристаллов на контактных линзах для непрерывного контроля уровня глюкозы в физиологических жидкостях [15].
Аналитическим сигналом в фотонно-кристаллических (ФК) сенсорах служит смещение максимума полосы отражения, которое регистрируется как гипсохромный или батохромный сдвиг спектра, а также изменение интенсивности отражения (гипер- или гипохромный эффект). Спектрофотометры зеркального отражения позволяют получать спектр с высокой дискретностью (<1 нм), а поскольку при детектировании аналита происходит смещение максимума длины волны на несколько десятков и даже сотен нм, для сканирования полного диапазона видимой области требуется некоторое время. Это обстоятельство ограничивает использование прибора при исследовании кинетики взаимодействия ФК-сенсоров с аналитами. Современные компактные спектрофотометры, оснащенные оптоволоконными волноводами, имеют высокую скорость регистрации сигнала и гибкость установки источника и приемника излучения. Поскольку угол измерения является критическим фактором при регистрации спектров зеркального отражения анизотропных структур, предпочтительным может оказаться метод спектроскопии диффузного отражения, позволяющий получить интегральные характеристики фотонно-кристаллического массива. Серийные настольные спектрофотометры диффузного отражения, используемые в промышленности, неудобны из-за своих больших габаритов, хотя и обладают лучшими аналитическими характеристиками благодаря интегрирующей сфере. Доступный вариант для изучения диффузного отражения ФК-структур – мини-спектрофотометры, входящие в программно-аппаратные комплексы для калибровки принтеров и экранов компьютеров, соответствующие требованиям ISO 13655:2017 и уже использующиеся для колориметрических измерений. Эти устройства хорошо зарекомендовали себя в химическом анализе; оценена их погрешность при определении характеристик цвета (координат цветности) [16–18]. Отметим, что необходимо использовать только спектрофотометры с дифракционной решеткой, обеспечивающие регистрацию спектра диффузного отражения с шагом не больше 10 нм, а не более дешевые варианты, основанные на применении светофильтров [19].
Применение стандартизированного метода измерения цвета ФК-сенсоров позволяет получить цветовые координаты и воспользоваться в химическом анализе цветовыми пространствами RGB, CIE и др. [13, 20]. Такой подход дает возможность построить в цветовом пространстве треки цвета в зависимости от концентрации аналита, а также в дальнейшем проводить определение веществ при помощи простых спектрофотометров или цифровых бытовых устройств типа сканеров и фотоаппаратов.
Надежно и гибко объединить различные узлы экспериментальных установок позволяет технология 3D-печати. Ее применяют для создания самодельных устройств: корпусов, держателей и других вспомогательных элементов [21–23]. Одно из основных преимуществ технологии 3D-печати заключается в возможности быстрого и относительно недорогого производства уникальных деталей или даже конструктивно-законченных устройств, предназначенных для решения специализированных задач [24], что открывает дополнительные возможности в области химического анализа. Мы применили технологию 3D-печати для стандартизации нанесения проб на ФК-сенсоры и для совершенствования системы регистрации сигнала.
Цель настоящей работы – разработка комплексного подхода к исследованию отклика сенсорных устройств на основе фотонных кристаллов при воздействии жидких углеводородов и их паров, включающего ячейку, подготовленную с помощью 3D-печати, а также компьютерный сбор результатов с их последующей метрологической обработкой. Функциональные материалы на основе фотонных кристаллов представляют собой хорошую платформу для создания тест-систем.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Экспериментальная установка. Серии спектров диффузного отражения получали с использованием спектрофотометра eye-one Pro (X-Rite, Inc., USA), имеющего голографическую дифракционную решетку с 128-пиксельной диодной матрицей. Спектрофотометр располагали на разработанных нами приставках, фиксирующих прибор в двух положениях – апертурой вверх или вниз. Расположение апертурой вниз на специальной приставке позволяло детектировать пары химических веществ в замкнутом пространстве – насыщенные пары (рис. 1а). Приставка имеет гнездо для кюветы и оснащена датчиком влажности, температуры и давления BMP280 (Robert Bosch GmbH, Germany). Сенсор размещали на кювете и прижимали кольцом, герметично закрывая ее, что позволяло создавать насыщенные пары. Измерения спектров диффузного отражения проводили с обратной стороны сенсора через оптически прозрачную подложку (поликарбонат, полиэтилентерефталат, стекло) в непрерывном режиме. Установку использовали на этапе модельных экспериментов с насыщенными парами растворителей.
Рис. 1.
Схема экспериментальных установок для детектирования углеводородов с использованием фотонных кристаллов: а – детектирование насыщенных паров: 1 – сенсор на основе фотонного кристалла, 2 – спектрофотометр eye-one Pro, 3 – фиксирующая приставка к спектрофотометру, 4 – кювета с аналитом, 5 – датчик влажности, температуры и давления; б – детектирование паров и аликвот: 1 – сенсор на основе фотонного кристалла, 2 – спектрофотометр eye-one Pro, 3 – фиксирующая приставка к спектрофотометру, 4 – микрошприц Hamilton Microliter 705 N, 5 – шприцевой насос.
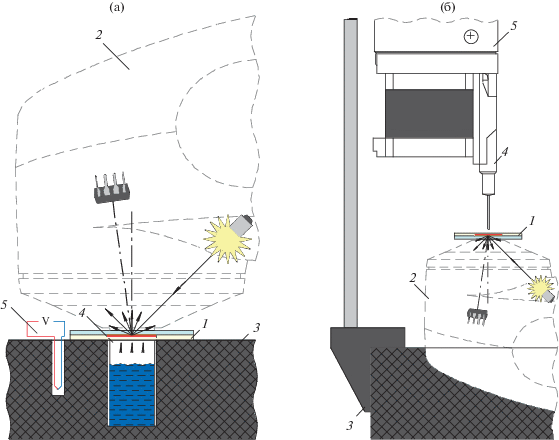
При фиксации прибора апертурой вверх (рис. 1б) можно работать как с газообразными аналитами в окружающем воздухе, так и с аликвотами жидкостей, которые наносят с помощью шприцевого насоса (дозатора) с использованием микрошприца Hamilton Microliter 705 N непосредственно на поверхность сенсора. Микрошприц устанавливали в шприцевой насос, который фиксировали в приставке спектрофотометра. В работе использовали самодельный шприцевой насос, управляемый при помощи платы Arduino UNO. Большинство деталей шприцевого насоса и корпус блока управления изготавливали методом 3D-печати. Конструкцию шприцевого насоса, описанную в работе [24], изменили с целью вертикального расположения шприца. Основание установки отлили из эпоксидной смолы, что, с одной стороны, обеспечивало надежную фиксацию спектрофотометра, а с другой – увеличивало массу и устойчивость всей конструкции. Насос устанавливали на штативе, что позволяло точно выставлять зазор между кончиком иглы шприца и поверхностью сенсора. Крепление штатива к основанию спроектировано таким образом, чтобы игла шприца располагалась перпендикулярно плоскости пластины с сенсором над центром измерительного окна прибора. Данные получали с помощью стандартного программного обеспечения i1Share. Автоматизацию измерений осуществляли на сценарном языке, который представляет собой последовательность команд. Температуру и влажность записывали в файл формата .csv на протяжении всего эксперимента. Результаты обрабатывали в программе MS Excel 2019.
Материалы. ФК-сенсоры изготовлены из субмикронных полистирольных частиц (≈200 нм), упакованных в гранецентрированную кристаллическую решетку и закрытых по типу “сэндвича” чувствительной гидрофобной матрицей (полидиметилсилоксан Sylgard 184 silicone elastomer, Dow Corning, USA). Сэндвичевую структуру собирали на стеклянной либо полимерной подложке [25].
В предварительных модельных экспериментах использовали пара-ксилол квалификации “ч.” (Реахим, Россия) и толуол “ч.д.а.” (Реахим, Россия).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
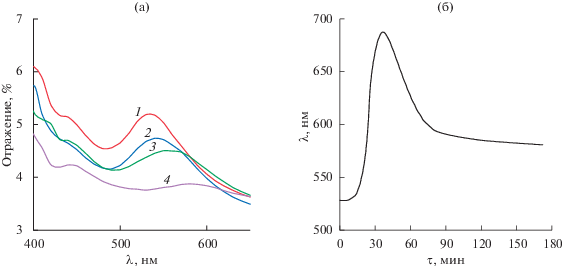
В наших предыдущих работах мы исследовали сенсоры на основе фотонных кристаллов на соответствие закону Брэгга–Снелла, определили основные характеристики (средний размер частиц, межплоскостное расстояние, показатели преломления) и установили кинетические зависимости при воздействии летучих органических соединений на примере паров ароматических и алифатических углеводородов [25]. Использованные нами фотонные кристаллы представляли собой кристаллические коллоидные массивы из полистирольных субмикронных частиц, закрытых матрицей из полидиметилсилоксана; структура собрана по способу самоорганизации, хорошо известному для формирования неорганических, в том числе наноразмерных, структур (например, из оксидов металлов и др.) [26, 27]. В случае нанесения капли пара-ксилола непосредственно на поверхность кристалла наблюдается градиент цвета. Если превысить допустимый объем пробы, полоса отражения ФК-сенсора уширяется, а кристалл приобретает белый цвет. Этот эффект сводит на нет возможность регистрации смещения максимума полосы отражения без фиксации образца на апертуре прибора, поскольку в область измерения могут попадать различные обработанные участки сенсора (рис. 2а). На рис. 2б представлена кинетическая кривая воздействия аликвоты пара-ксилола (20 мкл), полученная с применением нашего подхода со стандартизованным нанесением проб с использованием дозатора. Подход позволяет избежать этого влияния и получить кинетические кривые практически без выбросов экспериментальных точек.
Рис. 2.
Воздействие жидкого пара-ксилола на ФК-сенсор: а – изменение спектра диффузного отражения по радиусу обработанного пятна, наблюдается три области изменения: 1 – необработанный участок; 2, 3 – участок, подвергшийся воздействию; 4 – деградировавший участок в центре пятна; б – кинетическая кривая отклика (положение максимума полосы).
Воздействие концентраций углеводородов, близких к насыщенным парам, приводит к сильному батохромному сдвигу, гиперхромному и гипохромному эффекту, уширению и раздвоению пика в спектре за счет неравномерного изменения периода структуры по слоям (рис. 3а). При длительном воздействии аналитов, особенно жидких проб ароматических углеводородов, может происходить деградация обработанного участка сенсора за счет размягчения и слипания полистирольных частиц [28], что выражается в визуальном помутнении ФК-массива и вырождении максимумов в спектрах.
Рис. 3.
Принцип обработки данных (пик на 425 нм обусловлен отражением поликарбоната): а – серия спектров при воздействии паров углеводородов с концентрацией, близкой к насыщенной (батохромный сдвиг λmax на сотни нанометров); б – серия спектров при воздействии паров углеводородов с концентрацией на уровне ПДК (наблюдается незначительное смещение спектров); в – выбор точек для дальнейшей обработки данных, три точки с максимальной интенсивностью на полосе отражения фотонного кристалла (отмечены плюсом) и пять точек с максимальной интенсивностью (две дополнительные точки отмечены крестом); г – нахождение максимума на полосе отражения фотонного кристалла интерполяцией (сплошная линия) или аппроксимацией (пунктирная линия) выбранных данных.
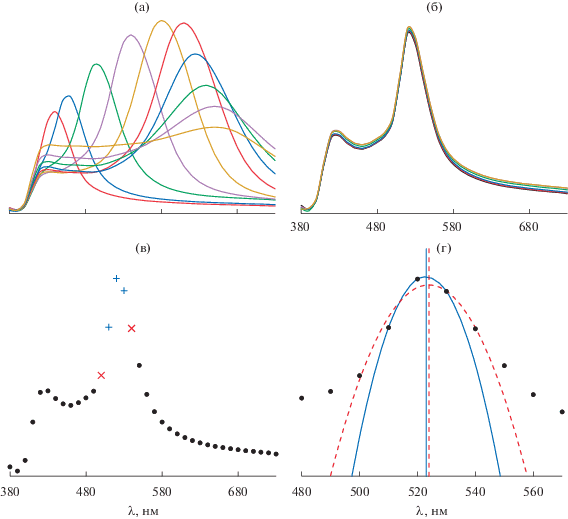
Наибольший интерес представляет регистрация низкой концентрации паров летучих органических соединений в динамике (on-line), так как в этом случае не наблюдается значительной деградации сенсора за счет растворения полимерных частиц (рис. 3б). Интенсивность излучения на полосе отражения имеет по длине волны распределение, подобное гауссову, что может расширить возможности подхода.
При регистрации спектра с низкой дискретностью (большим шагом длин волн) возникает проблема точного определения положения максимума. Для этого мы применили описание участка спектра вблизи максимума квадратичной функцией R = aλ2 + bλ + c, где λ – длина волны, R – сигнал (коэффициент отражения). Параметры a, b, c находили двумя способами: интерполяцией по трем точкам спектра (использовали участок спектра протяженностью 20 нм) или аппроксимацией по пяти точкам (участок в 40 нм) с помощью регрессионного анализа (рис. 3в). После нахождения этих параметров положение максимума полосы находили как λmax = –b/2a. Оказалось, что в большинстве случаев значения λmax, рассчитанные этими двумя способами, различаются между собой на 1–2 нм и более (рис. 3г). Это свидетельствует о существенной асимметрии спектральной полосы, в силу которой ее описание квадратичной зависимостью на значительном протяжении недостаточно адекватно. Кроме того, на значительном удалении от максимума (±20 нм) величины сигналов достаточно малы, что также снижает точность оценки. Поэтому в дальнейшем для нахождения максимума применяли интерполяцию по трем точкам спектра.
Предложенную методику измерения и обработки данных применили на производстве лакокрасочных материалов, где различные летучие органические соединения используются и в качестве основного растворителя в рецептурах материалов, и для очистки оборудования. Мини-спектрофотометр, помещенный в приставку (рис. 1б), с зафиксированным на апертуре фотонным кристаллом оставляли в производственном цехе. Эксперименты проводили в рабочие дни (не менее 5 раз), когда концентрация паров углеводородов в рабочей зоне была максимальной; в выходные дни, когда концентрация паров была равновесной (2 раза), и в “чистом” помещении, где заведомо отсутствовали пары углеводородов (2 раза) (рис. 4). Показано резкое смещение положения вычисленного максимума полосы отражения фотонного кристалла, которое хорошо согласуется со стадиями производства (табл. 1).
Рис. 4.
Регистрация паров углеводородов с использованием ФК-сенсора: 1 – в рабочий день, когда концентрация паров углеводородов изменяется в зависимости от стадии производства; штриховая линия – предел обнаружения; 2 – в выходной день, когда в помещении устанавливается равновесная концентрация паров углеводородов; 3 – контрольный эксперимент в помещении, где заведомо отсутствовали пары углеводородов.
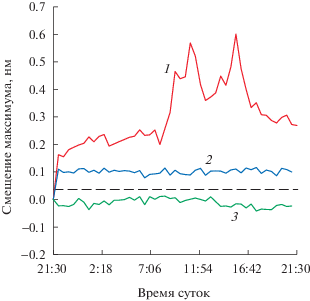
Таблица 1.
Смещение максимума длины волны отражения (λ) от изначального положения в зависимости от этапа эксперимента
| Этап | Время суток | λ, нм |
|---|---|---|
| На сенсор не оказывают воздействия пары углеводородов | 21:30 | 0.00 |
| В производственном помещении не проводятся работы | 22:00–7:30 | 0.16–0.25 |
| Включение приточно-вытяжной вентиляции | 8:00 | 0.20 |
| Загрузка сырьевых компонентов | 8:30 | 0.26 |
| Первый максимум на графике | 11:00 | 0.57 |
| Обеденный перерыв | 12:00 | 0.42 |
| Фасовка готового материала | 15:00 | 0.48 |
| Второй максимум на графике | 15:30 | 0.60 |
| Завершение рабочего дня | 16:30 | 0.40 |
| Выключение приточно-вытяжной вентиляции | 17:00 | 0.33 |
| В производственном помещении не проводятся работы | 17:30–22:00 | 0.33–0.27 |
При перемещении сенсора в производственный цех практически сразу на графике наблюдали смещение рассчитанного максимума полосы в длинноволновую область на 0.2 нм, что может быть обусловлено наличием паров углеводородов. Перед началом смены происходит включение вентиляции, на графике наблюдается практически незначимый гипсохромный сдвиг (ниже 0.08 нм). Первый этап производственного цикла – загрузка сырья, включающего углеводородные растворители, что проявляется в существенном для данных условий батохромном сдвиге на 0.57 нм. Далее происходит диспергирование материала, в этот момент наблюдается спад на графике, который совпадает с обеденным перерывом. Заключительным этапом является фасовка готового продукта и мытье оборудования, когда снова можно видеть значительное смещение (0.60 нм) максимума длины волны.
Шум сигнала на графике в целом объясняется колебанием температуры в помещении, работой вентиляции, изменением внешнего освещения в рабочее и нерабочее время и вибрациями, вызванными работающим оборудованием. Отключение освещения и вентиляции в нерабочее время, а также дрейф температуры в рабочие часы не приводят к значительным отклонениям. Вероятно, наибольший вклад в уровень шума могут вносить вибрации, приводящие к смещению сенсора на апертуре спектрофотометра, поскольку поверхность фотонного кристалла имеет анизотропную структуру, а изменяющийся вследствие этого угол измерения, согласно закону Брэгга–Снелла, приводит к изменению максимума длины волны на полосе отражения.
Отметим дополнительные факторы, которые могли исказить аналитический сигнал сенсора. Температура в ходе всех экспериментов колебалась в пределах 22–25°С, ее влияние оценено далее. Внешнее освещение изменялось в зависимости от времени суток и от дня проведения эксперимента, но природа ФК-сенсора и особенности конструкции ячейки, а также режима измерения позволяют избежать этого влияния. Непосредственно сенсор выступает в качестве “светофильтра”, что не приводит к искажению спектра диффузного отражения, а лишь незначительно изменяет интенсивность отражения максимума длины волны на полосе отражения. В результатах экспериментов не наблюдалось корреляции между изменениями в спектрах диффузного отражения и интенсивностью внешнего освещения. Мы не приводим статистических данных по измерениям в производственном цехе, поскольку каждый день выпускалась продукция разного объема и на разных растворителях (смесях углеводородов). Замечено, что при увеличении производимых объемов сдвиг максимума полосы имел большую амплитуду.
Из этих экспериментов видно, что изменения положения максимума полосы отражения хорошо воспроизводятся и соотносятся со стадиями производственного процесса. Однако величина этих изменений – десятые доли нанометра – значительно (на 2–3 порядка) меньше, чем шаг регистрации спектра (10 нм). Поэтому необходимо было установить, являются ли эти изменения статистически значимыми. Для этого был проведен ряд контрольных экспериментов в условиях, исключающих возможные колебания концентрации паров углеводородов в атмосфере: в производственном помещении в нерабочее время и в “чистом помещении”, где заведомо отсутствуют пары углеводородов. Сигнал, регистрируемый в таких условиях, принимали за фоновый. В каждом эксперименте регистрировали спектры диффузного отражения с интервалом 30 мин (20–48 спектров на протяжении всего эксперимента), для каждого спектра рассчитывали положение максимума λmax и величину батохромного сдвига Δλmax (по отношению к исходному положению максимума). Для данных, полученных в каждом эксперименте, рассчитывали s(λmax) – стандартное отклонение значений λmax. Для учета возможного влияния температуры рассчитывали коэффициент корреляции rT между величиной λmax и температурой окружающей среды T, а в случае статистической значимости коэффициента корреляции rT – также температурный коэффициент aT линейной регрессионной зависимости λmax = aTT + bT.
Значения коэффициента корреляции rT во всех экспериментах, кроме одного, составили от 0.11 до 0.32 и оказались статистически незначимыми при уровне значимости 0.05. В одном эксперименте он составил –0.68 и оказался статистически значимым, в том числе и после введения поправки на множественную проверку гипотез методом Холма–Бонферрони [29], а соответствующий температурный коэффициент aT составил –0.0245. Для этой серии данных все величины λmax были пересчитаны – приведены к температуре начала эксперимента.
Значения стандартных отклонений s(λmax) составили в отдельных экспериментах от 0.009 до 0.039 нм, средневзвешенная величина составила 0.024 нм. При расчете использовали данные пяти экспериментов в рабочей зоне. Отметим, что в эксперименте, в котором наблюдалась значимая корреляция между λmax и температурой, учет влияния температуры не привел к существенному изменению величины стандартного отклонения: значения s(λmax), рассчитанные для данных без коррекции и с коррекцией на температуру, составили 0.015 и 0.023 нм соответственно. Таким образом, если принять средневзвешенное значение s(λmax) = 0.024 нм за уровень колебаний фонового сигнала (шум), то по критерию 3s батохромный сдвиг, превышающий 0.08–0.10 нм, следует считать статистически значимым.
Поскольку такая величина более чем на два порядка ниже шага регистрации спектра, была проведена дополнительная оценка возможных колебаний величины λmax непосредственно из статистических характеристик исходных спектральных данных – коэффициентов диффузного отражения.
Пусть x1, x2, x3 – значения трех длин волн, по которым вычисляется величина λmax, а y1, y2, y3 – соответствующие значения измеренных коэффициентов диффузного отражения. Обозначим x1 = = x – Δx, x2 = x, x3 = x + Δx, где x – длина волны, соответствующая наибольшему зарегистрированному значению коэффициента диффузного отражения, а Δx – шаг регистрации спектра. Тогда положение максимума спектра, вычисленное интерполяцией квадратичной зависимостью по трем точкам, составит
(2)
${{\lambda }_{{\max }}} = x + 0.5\Delta x\frac{{{{y}_{1}} - {{y}_{3}}}}{{{{y}_{1}} - 2{{y}_{2}} + {{y}_{3}}}}.$Стандартное отклонение s(λmax) можно оценить по закону суммирования неопределенностей с учетом корреляции аргументов [30] из величин si (стандартных отклонений значений yi) и ri,j (коэффициентов корреляции между yi и yj (i, j = 1, 2, 3)) как
(3)
$\begin{gathered} {{s}^{2}}({{\lambda }_{{\max }}}) = \sum\limits_i {{{{\left( {\frac{{\partial {{\lambda }_{{\max }}}}}{{\partial {{y}_{i}}}}} \right)}}^{2}}s_{i}^{2}} + \\ + \,\,2\sum\limits_{j < i} {\left( {\frac{{\partial {{\lambda }_{{\max }}}}}{{\partial {{y}_{i}}}}} \right)} \left( {\frac{{\partial {{\lambda }_{{\max }}}}}{{\partial {{y}_{j}}}}} \right){{r}_{{i,j}}}{{s}_{i}}{{s}_{j}}. \\ \end{gathered} $Подставив выражение (2) в формулу (3), полагая все si = s и все ri,j = r и проведя преобразования, получаем:
(4)
$s({{\lambda }_{{\max }}}) = \sqrt 2 \Delta x\frac{{\sqrt {{{u}^{2}} + {{{v}}^{2}} - u{v}} }}{{{{{(u + {v})}}^{2}}}}s\sqrt {1 - r} ,$Для серии спектров, полученных в каждом контрольном эксперименте, были рассчитаны и усреднены значения si и ri,j. В отдельных экспериментах они составили от 1 × 10–4 до 7 × 10–4 и от 0.93 до 0.98 соответственно. Оценки s(λmax), рассчитанные из этих величин, составили от 0.010 до 0.044 нм, средневзвешенное значение равно 0.028 нм. Эта величина хорошо согласуется со значением 0.024 нм, рассчитанным ранее из значений спектральных максимумов, и подтверждает вывод о статистической значимости батохромного сдвига на 0.08–0.10 нм. Столь высокая точность нахождения положения спектрального максимума объясняется малыми колебаниями измеряемых величин коэффициентов диффузного отражения R, обусловленными конструкцией измерительной установки, а также высокой коррелированностью этих колебаний при трех соседних длинах волн, используемых для расчета максимума (симбатные изменения значений R не приводят к изменению положения максимума).
ЗАКЛЮЧЕНИЕ
Предложен комплексный подход к контролю воздуха рабочей зоны на присутствие летучих органических соединений. Подход включает разработанное устройство для детектирования углеводородов с помощью фотонно-кристаллических сенсорных матриц и для регистрации спектров диффузного отражения с последующей метрологической обработкой сигнала. Полученные данные по изменению содержания углеводородов хорошо согласуются с этапами рабочего цикла на лакокрасочном производстве.
Список литературы
Елисеев А.А., Лукашин А.В. Функциональные наноматериалы. М.: Физматлит, 2010. 456 с.
Sato A., Ikeda Y., Yamaguchi K., Vohra V. // Nanomaterials. 2018. V. 8. № 3. P. 169. https://doi.org/10.3390/nano8030169
Burratti L., De Matteis F., Casalboni M. et al. // Mater. Chem. Phys. 2018. V. 212. P. 274. https://doi.org/10.3390/ma11091547
Nair R.V., Vijaya R. // Prog. Quantum Electron. 2010. V. 34. № 3. P. 89. https://doi.org/10.1016/j.pquantelec.2010.01.001
Fenzl C., Hirsch T., Wolfbeis O.S. // Angew. Chem., Int. Ed. 2014. V. 53. № 13. P. 3318. https://doi.org/10.1002/anie.201307828
Inan H., Poyraz M., Inci F. et al. // Chem. Soc. Rev. 2017. V. 46. № 2. P. 366. https://doi.org/10.1039/c6cs00206d
Kuang M., Wang J., Jiang L. // Chem. Soc. Rev. 2016. V. 45. № 24. P. 6833. https://doi.org/10.1039/C6CS00562D
Men D., Liu D., Li Y. // Sci. Bull. 2016. V. 61. № 17. P. 1358. https://doi.org/10.1007/s11434-016-1134-7
Phillips K.R., England G.T., Sunny S. et al. // Chem. Soc. Rev. 2016. V. 45. № 2. P. 281. https://doi.org/10.1039/C5CS00533G
Tseng R.C., Chen C.C., Hsu S.M., Chuang H.S. // Sensors. 2018. V. 18. № 8. P. 2651. https://doi.org/10.3390/s18082651
Cui J., Zhu W., Gao N. et al. // Angew. Chem., Int. Ed. 2014. V. 53. № 15. P. 3844. https://doi.org/10.1002/anie.201308959
Zhong Q., Xu H., Ding H. et al. // Colloids Surf. A. 2013. V. 433. P. 59. https://doi.org/10.1016/j.colsurfa.2013.04.053
Fenzl C., Kirchinger M., Hirsch T., Wolfbeis O.S. // Chemosensors. 2014. V. 2. № 3. P. 207. https://doi.org/10.3390/chemosensors2030207
You A.M., Ni X.J., Cao Y.H., Cao G.Q. // J. Chin. Chem. Soc. (Weinheim, Germany). 2017. V. 64. № 10. P. 1235. https://doi.org/10.1002/jccs.201700126
Elsherif M., Hassan M.U., Yetisen A.K., Butt H. // ACS nano. 2018. V. 12. № 6. P. 5452. https://doi.org/10.1021/acsnano.8b00829
Apyari V.V., Dmitrienko S.G., Batov I.V., Zolotov Yu.A. // J. Anal. Chem. 2011. V. 66. № 2. P. 144. [Апяри В.В., Дмитриенко С.Г., Батов И.В., Золотов Ю.А. // Журн. аналит. химии. 2011. Т. 66. № 2. С. 148.]https://doi.org/10.1134/s1061934811020043
Milić N., Novaković D., Kašiković N. // J. Graphic Engineer. Design. 2011. V. 2. № 2. P. 16.
Monogarova O.V., Oskolok K.V., Apyari V.V. // J. Anal. Chem. 2018. V. 73. № 11. P. 1076. [Моногарова О.В., Осколок К.В., Апяри В.В. // Журн. аналит. химии. 2018. Т. 73. № 11. С. 857.]https://doi.org/10.1134/S1061934818110060
Ivanov A.V., Bolshakov E.S., Apyari V.V. et al. // J. Anal. Chim. 2019. V. 74. № 2. P. 198. [Иванов А.В., Большаков Е.С., Апяри В.В. и др. // Журн. аналит. химии. 2019. Т. 74. № 2. С. 154.]https://doi.org/10.1134/S1061934819020072
Kuo W.K., Weng H.P., Hsu J.J., Yu H.H. // Mater. Chem. Phys. 2016. V. 173. P. 285. https://doi.org/10.1016/j.matchemphys.2016.02.014
Chen Y., Fu Q., Li D. et al. // Anal. Bioanal. Chem. 2017. V. 409. P. 6567. https://doi.org/10.1007/s00216-017-0605-2
Chen C.-L., Chen T.-R., Chiu S.-H., Urban P.L. // Sens. Actuators, B: Chem. 2017. V. 239. P. 608. https://doi.org/10.1016/j.snb.2016.08.031
Carvalho M.C., Murray R.H. // HardwareX. 2018. V. 3. P. 10. https://doi.org/10.1016/j.ohx.2018.01.001
Samokhin A.S. // J. Anal. Chem. 2011. V. 75. № 3. P. 416. [Самохин А.С. // Журн. аналит. химии. 2020. Т. 75. № 3. С. 281.]https://doi.org/10.1134/S1061934820030156
Bol'shakov E.S., Ivanov A.V., Kozlov A.A., Abdullaev S.D. // Russ. J. Phys. Chem. A. 2018. V. 92. № 8. P. 1530. [Большаков Е.С., Иванов А.В., Козлов А.А., Абдуллаев С.Д. // Журн. физ. химии. 2018. Т. 92. № 8. С. 1283.]https://doi.org/10.1134/S0036024418080083
Yamanovskaya I.A., Gerasimova T.V., Agafonov A.V. // Russ. J. Inorg. Chem. 2018. V. 63. № 9. P. 1096. [Ямановская И.А., Герасимова Т.В., Агафонов А.В. // Журн. неорган. химии. 2018. Т. 63. № 9. С. 1574.]https://doi.org/10.1134/S0036023618090218
Zaytseva M.P., Muradova A.G., Sharapaev A.I. et al. // Russ. J. Inorg. Chem. 2018. V. 63. P. 1684. https://doi.org/10.1134/S0036023618120239
Kozlov A.A., Abdullaev S.D., Aksenov A.S. et al. // J. Intern. Scient. Publica. Materials, Methods and Technol. 2018. V. 12. P. 64.
Holm S. // Scand. J. Stat. 1979. V. 6. № 2. P. 65.
Руководство ЕВРАХИМ/СИТАК. Количественное описание неопределенности в аналитических измерениях. СПб.: ВНИИМ им. Д.И. Менделеева, 2002. 149 с.
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии