

Журнал неорганической химии, 2021, T. 66, № 2, стр. 141-148
Селективный синтез полиморфных модификаций диоксида марганца гидротермальной обработкой водных растворов KMnO4
А. А. Егорова a, Т. М. Бушкова a, И. В. Колесник b, А. Д. Япрынцев a, С. Ю. Котцов a, b, А. Е. Баранчиков a, *
a Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
b Московский государственный университет им. М.В. Ломоносова
119991 Москва, Ленинские горы, 1, Россия
* E-mail: a.baranchikov@yandex.ru
Поступила в редакцию 20.04.2020
После доработки 29.04.2020
Принята к публикации 30.04.2020
Аннотация
Показано, что гидротермальная обработка нейтральных и подкисленных растворов KMnO4 c концентрацией 0.037–0.093 моль/л при температурах 120, 170, 220°С в течение 24 ч позволяет осуществлять селективный синтез трех различных полиморфных модификаций диоксида марганца: α-MnO2, δ-MnO2, β-MnO2. Показано, что температура гидротермальной обработки и кислотность среды оказывают существенное влияние на фазовый состав продуктов восстановления KMnO4 и выход реакции. Полученные образцы MnO2 охарактеризованы методами рентгеновской дифракции, растровой электронной микроскопии, спектроскопии комбинационного рассеяния, спектроскопии диффузного отражения.
ВВЕДЕНИЕ
Диоксид марганца, благодаря своим уникальным физическим и физико-химическим свойствам, является основой для создания широкого круга функциональных материалов: компонентов источников постоянного тока, суперконденсаторов, катализаторов и др. [1–6]. Одной из ярких особенностей диоксида марганца является его структурное разнообразие, это соединение образует целый спектр полиморфных модификаций, различающихся способом сочленения элементарных структурных единиц, октаэдров [MnO6] [7]. Так, δ-MnO2 (бирнессит) представляет собой слоистое соединение, структура α-MnO2 (криптомелан) и β-MnO2 (пиролюзит) включает в себя туннели различного размера [8].
Фазовый состав материалов на основе диоксида марганца в значительной степени определяет их функциональные свойства и область практического применения. В частности, δ-MnO2 проявляет высокую каталитическую активность в реакциях гетерогенного каталитического озонирования [9]. α-MnO2 рассматривается в качестве перспективного материала катодов литий-ионных источников тока [10], а также молекулярных сит [11]. Структура β-MnO2 характеризуется наличием каналов наименьшего размера среди других полиморфных модификаций диоксида марганца, что обусловливает возможность получения данного соединения в высокочистом состоянии. В связи с этим β-MnO2 может быть использован для создания люминофоров [12, 13]. Кроме того, данное соединение применяется для эффективной очистки сточных вод и воздуха от техногенных отходов (в частности SO2) и загрязнений органической природы [14], для создания источников тока [15], каталитических систем [16, 17] и т.д.
Для направленного синтеза диоксида марганца(IV) с контролируемым фазовым составом используют различные методы, в том числе электроосаждение, гидротермальный синтез, методы растворной химии и др. [18–22]. Гидротермальный синтез является одним из наиболее гибких подходов мягкой химии, поскольку обеспечивает возможность получения веществ в высокодисперсном и нанокристаллическом состоянии с заданными химическим и фазовым составом, размером частиц и морфологией, определяющими их функциональные характеристики [23–28]. Синтез диоксида марганца с использованием гидротермальной обработки, как правило, проводят восстановлением перманганата калия в присутствии органических (гексаметилентетрамин [29], этанол [30]) или неорганических (нитрит натрия [31], сульфат марганца [32], соляная кислота [33]) восстановителей. В качестве восстановителя перманганата калия может выступать и вода. Известно, что в водном растворе перманганат калия постепенно разлагается с образованием оксидов марганца(IV) [34]. В гидротермальных условиях скорость восстановления перманганат-ионов значительно увеличивается. Показано, что при температуре >170°С формируются твердофазные марганецсодержащие продукты состава KxMnO2 ⋅ yH2O со слоистой структурой (бирнесситоподобные фазы) [35]. Длительная гидротермальная обработка растворов перманганата калия в присутствии азотной кислоты при 170°C в течение 4 сут приводит к практически количественному превращению KMnO4 в δ-MnO2 [35]. Гидротермальная обработка водных растворов KMnO4 в течение нескольких часов при более низких температурах (120–160°С) может быть использована для синтеза бирнесситоподобных фаз [9] и α-MnO2 [36]. Отметим, что преимуществом синтеза MnO2 гидротермальной обработкой водных растворов перманганата калия является присутствие в реакционной системе только двух компонентов (перманганата калия и воды), что может обеспечить высокую воспроизводимость химического и фазового состава получаемых твердофазных продуктов. Анализ существующих публикаций показывает, что сведения об условиях формирования различных полиморфных модификаций диоксида марганца при гидротермальной обработке водных растворов KMnO4, не содержащих дополнительных восстановителей, достаточно противоречивы и к настоящему времени их нельзя считать достоверно установленными.
Целью данной работы является анализ влияния условий проведения гидротермальной обработки (температура, концентрация и кислотность среды) водных растворов перманганата калия на фазовый состав получаемого при этом диоксида марганца.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве исходных соединений использовали перманганат калия “ос. ч.” (Реахим), азотную кислоту “ос. ч.” (Химмед), дистиллированную воду.
Для синтеза диоксида марганца навеску KMnO4 (от 0.280 до 1.806 г) растворяли в 70 мл дистиллированной воды (pH растворов составлял ~8). Полученные растворы (табл. 1) перемешивали в течение 10 мин, затем помещали в тефлоновые автоклавы емкостью 100 мл. Автоклавы нагревали в сушильном шкафу при 120, 170 или 220°С в течение 24 ч. Затем автоклавы извлекали и охлаждали до комнатной температуры. Осадки отфильтровывали, несколько раз промывали дистиллированной водой и высушивали при 80°С в течение 12 ч на воздухе.
Таблица 1.
Условия гидротермальной обработки водных растворов KMnO4 и выход продукта реакции
| t, °С | рН 8.1 | рН 1.3 | |||
|---|---|---|---|---|---|
| m(KMnO4), г | выход*, % | m(KMnO4), г | HNO3, мл | выход*, % | |
| 120 | 1.088 | 0.7 | 0.400 | 0.250 | 46 |
| 1.508 | 0.9 | 0.600 | 0.376 | 48 | |
| 1.806 | 0.8 | 1.000 | 0.626 | 44 | |
| 170 | 1.060 | 5.0 | 0.300 | 0.188 | 45 |
| 1.500 | 5.0 | 0.500 | 0.313 | 66 | |
| 1.800 | 6.0 | 1.000 | 0.626 | 72 | |
| 220 | 0.280 | 98.0 | 0.400 | 0.250 | 45 |
| 0.400 | 99.0 | 0.600 | 0.376 | 86 | |
| 0.600 | 96.0 | 1.000 | 0.626 | 67 | |
Аналогичным способом проводили гидротермальную обработку водных растворов KMnO4, подкисленных добавлением концентрированной азотной кислоты (табл. 1). Величина pH растворов перед проведением гидротермальной обработки составлялa 1.3.
Рентгенофазовый анализ (РФА) образцов проводили на дифрактометре Bruker D8 Advance (CuKα-излучение) в диапазоне углов 2θ 10°–80° с шагом 0.02° и выдержкой не менее 0.5 с на шаг.
Спектры комбинационного рассеяния (КР) регистрировали с использованием спектрометра Renishaw inVia Reflex (длина волны лазерного излучения 531 нм).
Растровую электронную микроскопию (РЭМ) и рентгеноспектральный микроанализ (РСМА) образцов проводили с использованием микроскопа Carl Zeiss NVision 40, оснащенного анализатором Oxford Instruments X-Max (80 мм2). Ускоряющее напряжение при проведении анализа методом РЭМ составляло 7 кВ, методом РСМА – 20 кВ.
Анализ образцов методом спектроскопии диффузного отражения проводили с использованием модульного спектрометра Ocean Optics QE65000, снабженного интегрирующей сферой Ocean Optics ISP-50-8-R-GT и источником излучения Ocean Optics HPX-2000.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
В результате гидротермальной обработки водных растворов перманганата калия при температурах 120, 170, 220°С во всех случаях наблюдали образование осадков черного или темно-коричневого цвета. В результате гидротермальной обработки неподкисленных растворов (стартовое значение pH ~ 8) происходило значительное изменение кислотности маточных растворов (до pH ~ 12). Напротив, pH подкисленных растворов в ходе синтеза оставался практически неизменным – по окончании синтеза он составлял во всех случаях ~1.9. Эти наблюдения находятся в соответствии с уравнениями реакций формирования диоксида марганца при взаимодействии перманганата калия и воды в нейтральной среде:
(1)
${\text{KMn}}{{{\text{O}}}_{4}} + 1/2{{{\text{H}}}_{{\text{2}}}}{\text{O}} \to {\text{Mn}}{{{\text{O}}}_{2}} + {\text{KOH}} + 3/2{{{\text{O}}}_{2}}$(2)
$\begin{gathered} {\text{KMn}}{{{\text{O}}}_{4}} + {\text{HN}}{{{\text{O}}}_{3}} \to {\text{Mn}}{{{\text{O}}}_{2}} + 1/2{{{\text{H}}}_{{\text{2}}}}{\text{O}} + \\ {\text{ + }}\,\,{\text{KN}}{{{\text{O}}}_{3}} + 3/2{{{\text{O}}}_{2}}. \\ \end{gathered} $Оценка величины окислительно-восстановительного потенциала по уравнению Нернста привела к значениям 0.64–0.68 и 1.69 для реакций (1) и (2) соответственно, это свидетельствует о том, что в обоих случаях равновесие в указанных системах сдвинуто в сторону образования продуктов реакции.
На рис. 1 приведены дифрактограммы образцов, полученных гидротермальной обработкой неподкисленных растворов KMnO4 при 120, 170, 220°С. Образец, полученный при наименьшей температуре, является рентгеноаморфным, на его дифрактограмме присутствует гало с максимумами в районе 30° и 37° 2θ. Отметим, что положение максимумов гало близко к положению рефлексов для пиролюзита. Данное наблюдение может свидетельствовать о том, что у рентгеноаморфной фазы MnO2, полученной в ходе гидротермальной обработки при низкой температуре, есть ближний порядок, характерный для β-MnO2.
Рис. 1.
Дифрактограммы образцов MnO2, полученных гидротермальной обработкой неподкисленных растворов KMnO4 при 120 (1), 170 (2), 220°С (3) и подкисленных HNO3 растворов KMnO4 при 120 (6), 170 (5), 220°С (4).
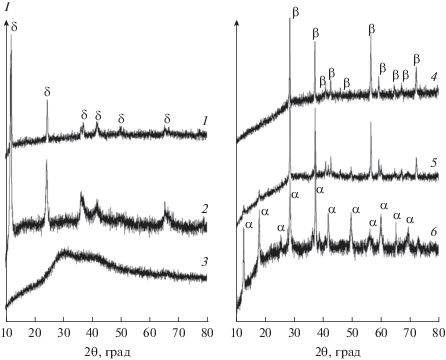
Синтез при более высоких температурах (170 и 220°С) приводит к получению хорошо закристаллизованного δ-MnO2, бирнессита (PDF2 #80-1098). Отметим, что уширение рефлексов на дифрактограмме образца δ-MnO2, полученного при температуре 220°С, значительно меньше, чем на дифракограмме аналогичного образца, полученного при температуре 170°С. Оценка размеров областей когерентного рассеяния для этих образцов по уравнению Шеррера привела к значениям 15 и 29 нм (для рефлекса с индексами Миллера 001) соответственно. Полученные данные удовлетворительно согласуются с результатами РЭМ (рис. 3а–3в), согласно которым микроструктура образца δ-MnO2, полученного при более высокой температуре, характеризуется наличием пластинчатых частиц большего размера и толщины (рис. 3в) по сравнению с образцами, полученными при более низких температурах (рис. 3а, 3б). Увеличение размеров кристаллитов δ-MnO2 с ростом температуры гидротермальной обработки соответствует представлениям о том, что в гидротермальных условиях рост частиц нанокристаллического бирнессита реализуется по механизму растворения–кристаллизации (оствальдова созревания [37]).
На рис. 1 (дифрактограммы 4–6) приведены данные РФА образцов, полученных путем гидротермальной обработки раствора KMnO4, подкисленного азотной кислотой, при температурах 120, 170, 220°С. В этом случае выход реакции в меньшей степени зависит от температуры обработки и во всех случаях превышаeт 40%, достигая 70–80%. Отметим, что количественного превращения KMnO4 в MnO2 при гидротермальной обработке подкисленных растворов мы не наблюдали. Увеличение концентрации перманганата калия в исходном растворе приводит к некоторому увеличению выхода MnO2 при проведении синтеза при относительно высоких температурах (170 и 220°С).
Фазовый состав продукта, полученного гидротермальной обработкой подкисленного раствора KMnO4 при 120°С, отвечает α-MnO2 (PDF2 #44-0141), при этом размер областей когерентного рассеяния составляет 26 и 20 нм для рефлексов 110 и 200 соответственно. Увеличение температуры гидротермальной обработки до 170°С дополнительно приводит к формированию β-MnO2 (PDF2 #24-0735), при этом размер областей когерентного рассеяния α-MnO2 закономерно возрастает до 33 и 23 нм для рефлексов 110 и 200 соответственно.
В свою очередь, в результате гидротермальной обработки при 220°С происходит формирование практически однофазного β-MnO2, пиролюзита, рефлексы примесной фазы α-MnO2 имеют крайне малую интенсивность. Формирование β-MnO2 в ходе гидротермальной обработки подкисленных растворов перманганата калия является достаточно необычным результатом, если принять во внимание известный анализ маршрутов кристаллизации диоксида марганца в аналогичной системе, выполненный Ченом с соавт. [38]. Согласно результатам in situ и ex situ рентгеновской дифракции [38], формирование β-MnO2 наблюдалось только в результате гидротермальной обработки растворов марганцовой кислоты, полученных удалением ионов калия из растворов KMnO4 путем его пропускания через колонку, заполненную катионитом в H-форме. Гидротермальная обработка растворов KMnO4, содержавших эквимолярное количество калия и марганца, в зависимости от концентрации KMnO4, приводила к формированию либо δ-MnO2 (в области высоких концентраций KMnO4), либо α-MnO2 (в области низких концентраций KMnO4). Отметим, что концентрации растворов KMnO4, применявшиеся Ченом с соавт. [38], составляли 0.2–0.6 моль/л, тогда как в наших экспериментах – 0.026–0.163 моль/л.
С другой стороны, формирование β-MnO2 в выбранных нами условиях может происходить вследствие фазового превращения α-MnO2 в β-MnO2, происходящего при продолжительной гидротермальной обработке [39, 40]. Принято считать, что такое превращение реализуется по механизму растворения–кристаллизации, в результате чего формируется кристаллический диоксид марганца с наименьшим размером структурных каналов (1 × 1 в единицах октаэдров [MnO6]) [39, 40]. Аналогичным образом реализуется и фазовое превращение γ-MnO2 в β-MnO2 [41, 42].
В то же время Чен с соавт. [35] не наблюдали образования β-MnO2 в результате гидротермальной обработки подкисленного азотной кислотой водного раствора KMnO4 при 170°С в течение 4 сут. Продуктом такого синтеза являлся δ-MnO2 (вернее, гидратированный манганит калия) со слоистой структурой, при этом превращение происходило количественно. В проведенных нами экспериментах наблюдали образование δ-MnO2 только при гидротермальной обработке неподкисленных растворов KMnO4. Существенные различия в фазовом составе продуктов гидротермальной обработки, полученных Ченом с соавт. [35] и нами в настоящей работе, по-видимому, связаны с различным стартовым значением pH реакционных смесей (3.5 и 1.3 соответственно). По завершении гидротермальной обработки растворов со стартовым pH 3.5 происходило существенное увеличение водородного показателя до 12–13 [35]. В нашем эксперименте, как было отмечено выше, значение pH изменялось незначительно (от 1.3 до 1.9).
Фазовый состав образцов диоксида марганца, полученных в ходе гидротермальной обработки в различных условиях, определенный методом РФА, находится в удовлетворительном соответствии с результатами спектроскопии комбинационного рассеяния света (рис. 2).
Рис. 2.
Спектры КР образцов диоксида марганца, полученных гидротермальной обработкой неподкисленных водных растворов KMnO4 при 120 (1) и 220°С (2) и подкисленных HNO3 растворов KMnO4 при 120 (3) и 220°С (4).
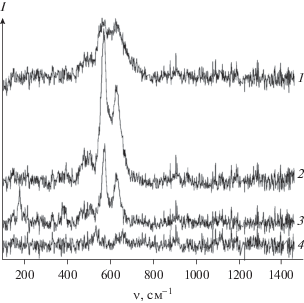
В спектрах КР всех образцов наблюдаются полосы при 634 см–1, отвечающие симметричным валентным колебаниям Mn–O в октаэдрах [MnO6], и при 575 см–1, отвечающие валентным колебаниям Mn–O в плоскости слоев [43, 44]. В спектрах КР образцов, отвечающих кристаллическому α-MnO2, дополнительно присутствуют полосы при 183 и 389 см–1, отвечающие трансляционным смещениям октаэдров [MnO6] и деформационным колебаниям связей Mn–O соответственно. Положение указанных полос удовлетворительно соответствует литературным данным [44–46].
Фазовый состав образцов диоксида марганца, полученных гидротермальной обработкой водных растворов перманганата калия в различных условиях, косвенно подтверждается результатами их рентгеноспектрального микроанализа. Действительно, в составе β-MnO2, структура которого характеризуется наличием каналов наименьшего размера, мольное соотношение Mn/K равно нулю, для образца α-MnO2 значение данной величины составляет 0.12, а для образцов δ-MnO2 мольное соотношение Mn/K варьирует в диапазоне 0.26–0.30.
Рис. 3.
РЭМ-изображения образцов MnO2, полученных гидротермальной обработкой неподкисленных (а–в) и подкисленных растворов (г–е) KMnO4 при 120 (а, г); 170 (б, д); 220°С (в, е).

Данные РЭМ, приведенные на рис. 3, для образцов, содержащих α-MnO2 и β-MnO2, также удовлетворительно согласуются с результатами РФА. α-MnO2 кристаллизуется в виде нитевидных или игольчатых кристаллов длиной около 100–200 нм и толщиной около 18–28 нм. β-MnO2 кристаллизуется в виде крупных кристаллов размером 200–1000 нм, некоторые из которых имеют характерную огранку, образованную плоскостями (110), (100) и (101) [38].
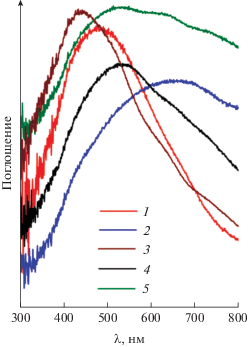
Выше было отмечено, что порошки MnO2, в зависимости от условий их синтеза, характеризовались различной окраской – черной или темно-коричневой. Из данных, приведенных на рис. 4, следует, что вид спектров оптического поглощения порошков значительно различается, при этом фазовый состав порошков в значительной степени определяет их окраску. Темно-коричневая окраска характерна для порошков α-MnO2, а также рентгеноаморфного диоксида марганца. Порошки β-MnO2 и δ-MnO2 обладают черной окраской. В целом полученные оптические спектры соответствуют литературным данным [47, 48]. Интересно, что оптические характеристики δ-MnO2 зависят от условий его получения: порошки, синтезированные при более высокой температуре и характеризующиеся большим размером частиц, обладают более высоким светопоглощением в области больших длин волн.
Рис. 4.
Оптические спектры поглощения порошков диоксида марганца, полученных гидротермальной обработкой подкисленных (1, 2) и неподкисленных (3, 4, 5) водных растворов перманганата калия при 120 (1, 3); 170 (4); 220°С (2, 5). Фазовый состав порошков соответствует α-MnO2 (1); β-MnO2 (2); рентгеноаморфному MnO2 (3); δ-MnO2 (4, 5).
ЗАКЛЮЧЕНИЕ
Проанализировано влияние температуры и кислотности среды на фазовый состав твердофазных продуктов гидротермальной обработки водных растворов перманганата калия, не содержащих дополнительных восстановителей. Выявлены условия формирования однофазных порошков δ-MnO2, α-MnO2 и β-MnO2 с высоким выходом (от 50 до 100%).
Список литературы
Parker D., Webb S.M., Verity R. et al. // Annu. Rev. Earth Planet. Sci. 2004. V. 32. P. 287. https://doi.org/10.1146/annurev.earth.32.101802.120213
Tretyakov Y.D. // Russ. Chem. Rev. 2004. V. 73. P. 831. https://doi.org/10.1070/rc2004v073n09abeh000914
Manickam M., Singh P., Issa T.B. et al. // J. Power Sources. 2004. V. 130. P. 254. https://doi.org/10.1016/j.jpowsour.2003.12.018
Gellings P.J., Bouwmeester H.J.M. // Catal. Today. 1992. V. 12. P. 1. https://doi.org/10.1016/0920-5861(92)80046-P
Hu J., Odom T.W., Lieber C.M. // Acc. Chem. Res. 1999. V. 32. P. 435. https://doi.org/10.1021/ar9700365
Ivanova A.G., Karasev L.V., Masalovich M.S. et al. // Glass Phys. Chem. 2020. V. 46. P. 96. https://doi.org/10.1134/S1087659620010101
Post J.E. // Proc. Natl. Acad. Sci. 1999. V. 96. P. 3447. https://doi.org/10.1073/pnas.96.7.3447
Thackeray M.M. // Prog. Solid State Chem. 1997. V. 25. P. 1. https://doi.org/10.1016/S0079-6786(97)81003-5
Luo K., Zhao S.X., Wang Y.F. // New J. Chem. 2018. V. 42. P. 6770. https://doi.org/10.1039/c8nj00240a
Hill L.I., Verbaere A., Guyomard D. // J. Power Sources. 2003. V. 119–121. P. 226. https://doi.org/10.1016/S0378-7753(03)00238-6
Li W.N., Yuan J., Shen X.F. et al. // Adv. Funct. Mater. 2006. V. 16. P. 1247. https://doi.org/10.1002/adfm.200500504
Brekhovskikh M.N., Batygov S.K., Moiseeva L.V. et al. // Inorg. Mater. 2018. V. 54. P. 1157. https://doi.org/10.1134/S0020168518110031
Brekhovskikh M.N., Solodovnikov S.P., Moiseeva L.V. et al. // Inorg. Mater. 2019. V. 55. P. 710. https://doi.org/10.1134/S0020168519070045
Sun W., Ding S., Zeng S. et al. // J. Hazard. Mater. 2011. V. 192. P. 124. https://doi.org/10.1016/j.jhazmat.2011.04.104
Wang D., Liu L.M., Zhao S.J. et al. // Phys. Chem. Chem. Phys. 2013. V. 15. P. 9075. https://doi.org/10.1039/c3cp50392e
Buzanov G.A., Simonenko N.P., Zhizhin K.Y. et al. // Russ. J. Inorg. Chem. 2019. V. 64. P. 1335. https://doi.org/10.1134/S0036023619110044
Buzanov G.A., Simonenko N.P., Zhizhin K.Y. et al. // Russ. J. Inorg. Chem. 2019. V. 64. P. 1482. https://doi.org/10.1134/S0036023619120040
Song J., Li H., Li S. et al. // New J. Chem. 2017. V. 41. P. 3750. https://doi.org/10.1039/c6nj04118c
Subramanian V., Zhu H., Wei B. // J. Power Sources. 2006. V. 159. P. 361. https://doi.org/10.1016/j.jpowsour.2006.04.012
Reddy R.N., Reddy R.G. // J. Power Sources. 2003. V. 124. P. 330. https://doi.org/10.1016/S0378-7753(03)00600-1
Fedosova S.P. // Growth of Crystals. 1975. V. 9. P. 62.
Sharipov K.B., Yapryntsev A.D., Baranchikov A.E. et al. // Russ. J. Inorg. Chem. 2017. V. 62. P. 139. https://doi.org/10.1134/S0036023617020164
Byrappa K., Yoshimura M. // Handbook of Hydrothermal Technology. Elsevier, 2013. P. 615.
Komarneni S., Li Q., Stefansson K.M., Roy R. // J. Mater. Res. 1993. V. 8. P. 3176. https://doi.org/10.1557/JMR.1993.3176
Meskin P.E., Baranchikov A.E., Ivanov V.K. et al. // Inorg. Mater. 2004. V. 40. P. 1058. https://doi.org/10.1023/b:inma.0000046468.73127.f5
Meskin P.E., Baranchikov A.E., Ivanov V.K. et al. // Dokl. Chem. 2003. V. 389. P. 62. https://doi.org/10.1023/A:1022978107113
Meskin P.E., Gavrilov A.I., Maksimov V.D. et al. // Russ. J. Inorg. Chem. 2007. V. 52. P. 1648. https://doi.org/10.1134/S0036023607110022
Ivanov V.K., Maksimov V.D., Shaporev A.S. // Russ. J. Inorg. Chem. 2010. V. 55. P. 150. https://doi.org/10.1134/S0036023610020026
Korotkov R.F., Baranchikov A.E., Boytsova O.V. // Russ. J. Inorg. Chem. 2016. V. 61. P. 129. https://doi.org/10.1134/S0036023616020091
Pan H., Chen X., Sanz O. et al. // Chin. J. Catal. 2019. V. 40. P. 940. https://doi.org/10.1016/S1872-2067(19)63339-5
Korotkov R.F., Baranchikov A.E., Boytsova O.V., Ivanov V.K. // Russ. J. Inorg. Chem. 2015. V. 60. P. 1299. https://doi.org/10.1134/s0036023615110108
Birgisson S., Saha D., Iversen B.B. // Cryst. Growth Des. 2018. V. 18. P. 827. https://doi.org/10.1021/acs.cgd.7b01304
Zhang X., Li B., Liu C. et al. // Mater. Res. Bull. 2013. V. 48. P. 2696. https://doi.org/10.1016/j.materresbull.2013.03.025
Parsons R. // J. Electroanal. Chem. Interfacial Electrochem. 1967. V. 13. P. 471. https://doi.org/10.1016/0022-0728(67)80059-7
Cao H., Wei B., Yu B. et al. // ICAFPM 2009 – Proc. 2009 Int. Conf. Adv. Fibers Polym. Mater. 2009. V. 2. P. 1302. https://doi.org/10.1002/chin.200523238
Tang N., Tian X., Yang C. et al. // Mater. Res. Bull. 2009. V. 44. P. 2062. https://doi.org/10.1016/j.materresbull.2009.07.012
Xu N., Liu Z.H., Ma X. et al. // J. Nanoparticle Res. 2009. V. 11. P. 1107. https://doi.org/10.1007/s11051-008-9517-6
Chen B.R., Sun W., Kitchaev D.A. et al. // Nat. Commun. 2018. V. 9. P. 2553. https://doi.org/10.1038/s41467-018-04917-y
Zhang X., Yang W., Yang J., Evans D.G. // J. Cryst. Growth. 2008. V. 310. P. 716. https://doi.org/10.1016/j.jcrysgro.2007.11.113
Zhang X., Yu P., Wang D., Ma Y. // J. Nanosci. Nanotechnol. 2010. V. 10. P. 898. https://doi.org/10.1166/jnn.2010.1893
Gao T., Fjellvåg H., Norby P. // Nanotechnology. 2009. V. 20. P. 055610. https://doi.org/10.1088/0957-4484/20/5/055610
Kao W.-H. // J. Electrochem. Soc. 2006. V. 136. P. 13. https://doi.org/10.1149/1.2096574
Julien C., Massot M., Baddour-Hadjean R. et al. // Solid State Ionics. 2003. V. 159. P. 345. https://doi.org/10.1016/S0167-2738(03)00035-3
Julien C., Massot M., Poinsignon C. // Spectrochim. Acta, Part A. 2003. V. 60. P. 689. https://doi.org/10.1016/s1386-1425(03)00279-8
Gao T., Fjellvåg H., Norby P. // Anal. Chim. Acta. 2009. V. 648. P. 235. https://doi.org/10.1016/j.aca.2009.06.059
Buciuman F., Patcas F., Craciun R., Zahn D.R.T. // Phys. Chem. Chem. Phys. 1999. V. 1. P. 185. https://doi.org/10.1039/a807821a
Li W., Cui X., Zeng R. et al. // Sci. Rep. 2015. V. 5. P. 8987. https://doi.org/10.1038/srep08987
Toufiq A.M., Wang F., Javed Q.U.A. // J. Nanosci. Nanotechnol. 2013. V. 13. P. 2948. https://doi.org/10.1166/jnn.2013.7408
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии