

Журнал неорганической химии, 2021, T. 66, № 5, стр. 601-609
Исследование влияния мольных отношений в кристаллохимической структуре биомиметического наноструктурного гидроксиапатита на характеристики синтезированного продукта
М. А. Трубицын a, Хоанг Вьет Хунг a, Л. В. Фурда a, Нгуен Тхи Тхам Хонг b, c, *
a Белгородский государственный национальный исследовательский университет
308015 Белгород, ул. Победы, 85, Россия
b Отдел вычислительной физики, Институт вычислительных наук, Университет Тон Дык Тханг
758307 Хошимин, ул. Нгуен Хыу Тхо, Вьетнам
c Факультет электротехники и электроники, Университет Тон Дык Тханг
758307 Хошимин, ул. Нгуен Хыу Тхо, Вьетнам
* E-mail: nguyenthithamhong@tdtu.edu.vn
Поступила в редакцию 19.10.2020
После доработки 29.12.2020
Принята к публикации 30.12.2020
Аннотация
Изложены результаты исследования влияния мольных отношений в кристаллохимической структуре биомиметического гидроксиапатита (БМГАП) на физико-химические характеристики полученного продукта. Показано, что при увеличении мольного отношения в интервале 1.50–1.67 возрастают параметры решетки а, с и средний размер кристаллитов (от 7.52 до 70.30 нм). Установлено, что в водной суспензии (рН 7) частицы всех образцов несут отрицательный заряд. Выявлена закономерность изменения величины ζ-потенциала синтезированных порошков в исследованном диапазоне мольных отношений. Проведена оценка биоактивности полученных образцов. Установлено, что по сравнению со стехиометрическим немодифицированным гидроксиапатитом все образцы БМГАП обладают более высокой биорезорбируемостью, которая хорошо коррелирует с величиной мольного отношения в структуре синтезированных продуктов.
ВВЕДЕНИЕ
Разработка новых синтетических биоматериалов, предназначенных для реставрации костных дефектов, является одним из актуальных направлений медицинского материаловедения [1–4]. Материалы на основе гидроксиапатита ([Са10(PO4)6(ОН)2], ГАП) на протяжении ряда лет успешно используются в качестве имплантатов в реконструктивной хирургии, ортопедии и стоматологии, поскольку являются химическими аналогами биоапатита костной ткани человека и животных [5–8]. Однако имплантаты на основе керамических ГАП имеют неудовлетворительную биорезорбируемость и плохо индуцируют образование новой костной ткани, что является их существенным недостатком [9, 10]. Перспективным подходом к решению данной проблемы может стать разработка биомиметического ГАП путем химического модифицирования биосовместимыми анионами, что позволяет целенаправленно регулировать биоактивность и остеоиндукцию таких материалов.
Согласно литературным данным [11–13], биогенный апатит – это наноструктурный кальций – дефицитный гидроксиапатит с соответствующими катионными (Na+, Mg2+, K+, Zn2+, Sr2+) и анионными (F–, Cl–, ${\text{SO}}_{{\text{4}}}^{{{\text{2}}--}}$ и т.д.) замещениями. Наличие данных анионов в структуре ГАП оказывает существенное влияние на живой организм и стимулирует процессы регенерации полноценной костной ткани.
Среди используемых на настоящий момент при синтезе ГАП ионных замещений наиболее важными являются силикат- и карбонат-анионы [14–20]. Силикат-анионы $\left( {{\text{SiO}}_{{\text{4}}}^{{4--}}} \right)$ представляют особый интерес из-за их определяющей роли в процессе реставрации костной ткани, а внедрение карбонат-анионов $\left( {{\text{CO}}_{{\text{3}}}^{{{\text{2}}--}}} \right)$ в структуру ГАП улучшает резорбцию и увеличивает скорость остеоинтеграции.
Также сообщалось [21, 22], что введение при синтезе ГАП в реакционную среду цитрат-ионов (Cit3–) приводит к изменению структурно-морфологических характеристик синтезируемых материалов. Показано [23–26], что цитрат-ионы, участвующие в биологических циклах, играют важную роль в процессах резорбции костной ткани и формирования биоапатита, например, при наноразмерной стабилизации кристаллитов ГАП в кости.
Таким образом, биоапатит по химическому составу представляет собой наноструктурный кальций-дефицитный гидроксиапатит, содержащий в своем составе силикат- и карбонат-анионы. Нами уже был проведен цикл исследований, посвященных синтезу биомиметического кальций-дефицитного гидроксиапатита (БМГАП), допированного силикат- и карбонат-анионами, в присутствии цитрат-ионов [22, 27].
В данной статье представлены результаты исследований влияния мольных отношений ${{{{C}_{{{\text{C}}{{{\text{a}}}^{{{\text{2 + }}}}}}}}} \mathord{\left/ {\vphantom {{{{C}_{{{\text{C}}{{{\text{a}}}^{{{\text{2 + }}}}}}}}} {\left( {{{C}_{{{\text{PO}}_{4}^{{3 - }}}}} + {{C}_{{{\text{CO}}_{3}^{{2 - }}}}} + {{C}_{{{\text{SiO}}_{4}^{{4 - }}}}}} \right)}}} \right. \kern-0em} {\left( {{{C}_{{{\text{PO}}_{4}^{{3 - }}}}} + {{C}_{{{\text{CO}}_{3}^{{2 - }}}}} + {{C}_{{{\text{SiO}}_{4}^{{4 - }}}}}} \right)}}$ в кристаллохимической структуре БМГАП на физико-химические и биологические свойства полученного продукта.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве исходных реагентов использовали Ca(NO3)2 · 4H2O (ч. д. а.), (NH4)2HPO4 (ч. д. а.), (C2H5O)4Si (TEOS, ос. ч.), (NH4)2CO3 (ч. д. а.) и NH4OH (х. ч. а.). Источником цитрат-ионов служил полученный ранее раствор лимонной кислоты. Синтез образцов БМГАП осуществляли методом химического осаждения из водных растворов по известной методике при температуре 22 ± 2°С и рН 10 ± 0.5 [27]. Для регулирования процессов нуклеации и роста кристаллов ГАП вводили цитрат-ионы (Cit3–) в количестве 0.46 мас. %. Реакция осаждения БМГАП протекала в соответствии с уравнением:
Полученную суспензию отстаивали при комнатной температуре для завершения процесса фазообразования в течение 24 ч, затем твердую фазу отфильтровывали от маточного раствора, тщательно промывали дистиллированной водой и высушивали при температуре 110 ± 5°С до постоянной массы.
Для проведения исследования в качестве объектов были выбраны образцы БМГАП с мольными отношениями ${{{{C}_{{{\text{C}}{{{\text{a}}}^{{2 + }}}}}}} \mathord{\left/ {\vphantom {{{{C}_{{{\text{C}}{{{\text{a}}}^{{2 + }}}}}}} {\left( {{{C}_{{{\text{PO}}_{4}^{{3 - }}}}} + {{C}_{{{\text{CO}}_{3}^{{2 - }}}}} + {{C}_{{{\text{SiO}}_{4}^{{4 - }}}}}} \right)}}} \right. \kern-0em} {\left( {{{C}_{{{\text{PO}}_{4}^{{3 - }}}}} + {{C}_{{{\text{CO}}_{3}^{{2 - }}}}} + {{C}_{{{\text{SiO}}_{4}^{{4 - }}}}}} \right)}}$ = = 1.50, 1.55, 1.60, где степени замещения фосфат-ионов карбонат- и силикат-анионами во всех случаях были постоянными и составляли х = y = 1. Таким образом, по химическому составу все образцы были кальций-дефицитными. Образцам были присвоены индексы БМГАП1.50, БМГАП1.55 и БМГАП1.60 соответственно. Объектом для сравнения в работе являлся стехиометрический немодифицированный ГАП с мольным отношением Ca/P = 1.67, синтезированный в отсутствие силикат- и карбонат-анионов, но в присутствии цитрат-ионов по уравнению:
Физико-химические и биологические исследования синтезированных образцов проводили комплексными методами анализа. Фазовый состав изучали на дифрактометре Rigaku-SmartLab. Съемку вели при ускоряющем напряжении 40 кВ, токе рентгеновской трубки 250 мА в интервале углов 2θ = 20°–70°, размер шага составлял 0.02°, скорость регистрации – 3 град/мин. Идентификацию соединений, входящих в состав исследуемых образцов, выполняли с помощью программы PDXL Qualitative Analysis по базам данных PDF 2008. Размер кристаллитов D и степень кристалличности Xs синтезированных образцов определяли по стандартной методике [28, 29]. ИК-спектры поглощения измеряли на ИК-Фурье-спектрометре IRPrestige-21 (Shimadzu, Япония) в интервале волновых чисел от 400 до 4000 см–1. Морфологию и размеры частиц в форме гидродисперсий и в виде высушенных порошков БМГАП определяли по микрофотографиям, полученным с помощью просвечивающей (ПЭМ) и сканирующей (СЭМ) электронной микроскопии на приборах JEM-2100 (JEOL, Япония) и Quanta-200 3D (FEI, США) соответственно. Обработку полученных изображений осуществляли при помощи программного обеспечения ImageJ. Измерения удельной поверхности (Sуд) проводили методом БЭТ на автоматизированной сорбционной установке TriStar II 3020 (Micromeritics, США). Величину и знак поверхностного заряда синтезированных образцов оценивали посредством измерения электрокинетического потенциала. Измерения ζ-потенциала частиц выполняли с применением анализатора серии Zetasizer Nano ZS (Malvern Instruments, Великобритания). Биорезорбируемость синтезированных образцов БМГАП в физиологическом (ω(NaCl) = 0.9%) и модельном SBF-растворе (рН 7.4) оценивали по суммарной концентрации ионов Ca2+ в растворе через фиксированное время экспозиции методом комплексонометрического титрования [30].
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Исследование фазового состава синтезированных образцов методом РФА показало, что все образцы БМГАП, полученные при варьировании мольных отношений в интервале 1.50–1.60, являются однофазными нанокристаллическими (рис. 1а) и могут быть отнесены к гексагональной сингонии (пр. гр. P63/m) и идентифицированы как модификация ГАП (по ICDD карта № 01-072-1243).
Рис. 1.
Дифрактограммы синтезированных образцов: а – общий вид; б – дифракционный пик (211) и его аппроксимация тремя функциями Лоренца.
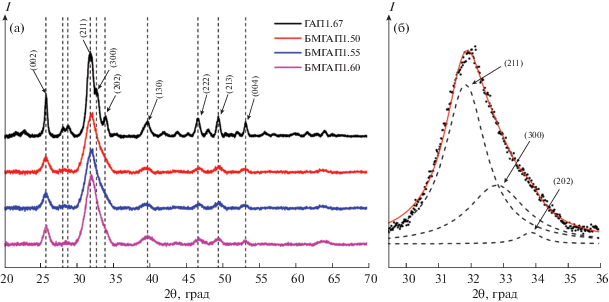
Следует отметить, что дифрактограммы порошков БМГАП отличаются более широкими линиями и меньшей интенсивностью по сравнению со стехиометрическим немодифицированным ГАП1.67 (рис. 1б). Наблюдаемое уменьшение интенсивности характеризующих пиков синтезированных образцов БМГАП, по-видимому, связано с внедрением силикат- и карбонат-анионов в кристаллохимическую структуру ГАП. Таким образом, полученные рентгенограммы полностью идентичны таковым для типичного ГАП.
Кристаллохимические и текстурные характеристики порошкообразных ГАП и БМГАП приведены в табл. 1.
Таблица 1.
Параметры кристаллической решетки, размер кристаллитов и удельная поверхность образцов ГАП1.67 и БМГАП
| Образец | a = b, нм | с, нм | Vгекс, нм3 | D, нм | Xs, % | Sуд, м2/г |
|---|---|---|---|---|---|---|
| БМГАП1.50 | 0.942(8) | 0.685(4) | 0.527(6) | 7.52(0) | 88.18 | 192.51 |
| БМГАП1.55 | 0.952(3) | 0.685(1) | 0.538(0) | 8.22(7) | 90.28 | 193.53 |
| БМГАП1.60 | 0.948(8) | 0.687(4) | 0.535(9) | 11.52(3) | 89.15 | 184.45 |
| ГАП1.67 | 0.942(2) | 0.687(6) | 0.528(6) | 70.30(5) | 93.93 | 74.41 |
| Ca10(PO4)6(OH)2 (01-072-1243) | 0.943(2) | 0.688(1) | 0.530(1) | – | – |
Как видно из табл. 1, повышение мольного отношения в интервале 1.50–1.67 в кристаллической структуре ГАП обусловливает монотонное увеличение параметра с. Для параметра а данная зависимость не наблюдается. Наибольшее значение параметра а (0.9523 нм) фиксируется у образца БМГАП1.55. При дальнейшем увеличении мольного отношения параметр а начинает уменьшаться и достигает минимума (0.9422 нм) у ГАП1.67. Объем элементарной ячейки меняется аналогично изменению параметров а и с. Вместе с тем определенных закономерностей в изменении параметров кристаллической решетки БМГАП от величины мольных отношений нами не выявлено. По-видимому, этот вопрос требует проведения отдельных исследований.
Обнаружено также, что при снижении степени дефицитности ионов Ca2+ в диапазоне мольных отношений 1.50–1.60 происходит увеличение размеров кристаллитов от 7.52 до 11.52 нм. По мере приближения к стехиометрическому значению их размер изменяется скачкообразно и достигает максимума (70.30 нм) у образца ГАП1.67.
Результаты измерений удельной поверхности синтезированных порошков методом БЭТ показали, что все исследуемые образцы БМГАП обладают высокоразвитой поверхностью по сравнению со стехиометрическим немодифицированным ГАП1.67. Максимальная величина удельной поверхности (193.53 м2/г) зафиксирована у образца БМГАП1.55. Установлено, что с возрастанием мольного отношения до стехиометрического удельная поверхность синтезированных образцов понижается и достигает минимума (74.41 м2/г) у ГАП1.67.
ИК-Фурье-спектры всех порошков БМГАП, допированных силикат- и карбонат-анионами, в интервале заданных мольных отношений содержат полосы поглощения групп ${\text{PO}}_{{\text{4}}}^{{{\text{3}}--}},$ ${\text{CO}}_{{\text{3}}}^{{2--}}$ и OH–, характерные для апатитовой фазы.
По данным ИК-Фурье-спектроскопии (рис. 2), у всех образцов ГАП1.67 и БМГАП присутствуют полосы поглощения с волновыми числами в интервале 607–565 см–1, соответствующие деформационным колебаниям фосфатных групп (мода ν4). Полосы в области 1092–1034 см–1 могут быть отнесены к асимметричным валентным колебаниям связей Р–О в составе ${\text{PO}}_{{\text{4}}}^{{{\text{3}}--}}$-тетраэдров (мода ν3), а полосы в интервале 962–959 см–1 вызваны симметричными валентными модами ν1. Полоса поглощения при 470 см–1 соответствует деформационным колебаниям О–Р–О (мода ν2). Широкая полоса в интервале от 3700 до 2500 см–1 может быть отнесена к модам валентных колебаний адсорбционной воды и гидроксильных групп. Кроме того, обнаруживаются полосы деформационных колебаний связей О–Н в молекулах структурно связанной воды и в гидроксид-ионах в интервале 1603–1635 см–1. Предположительно, смещение полосы деформационных колебаний воды на 1600 см–1, а также значительное увеличение ее интенсивности могут быть связаны с увеличением содержания карбонат-ионов в структуре ГАП. Полоса при 630 см–1 в спектре ГАП может быть отнесена к либрационным колебаниям ОН-группы. Из рис. 2 видно, что введение как силикатных, так и карбонатных анионов в структуру ГАП приводит к исчезновению этой полосы. Согласно [31, 32], при замещении группы ${\text{PO}}_{{\text{4}}}^{{{\text{3}}--}}$ на ${\text{SiO}}_{{\text{4}}}^{{4--}}$ может происходить потеря одной OH-группы для компенсации заряда в элементарной ячейке ГАП. В случае допирования гидроксиапатита ${\text{CO}}_{{\text{3}}}^{{2--}}$-анионами возможна потеря одной ОН-группы за счет карбонатного замещения B-типа.
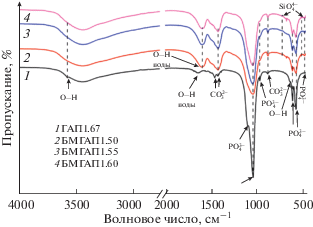
У стехиометрического немодифицированного ГАП1.67 присутствуют пики, соответствующие максимальной интенсивности колебаний группы ${\text{PO}}_{{\text{4}}}^{{{\text{3}}--}}$ при 1092 и 602 см–1, но отсутствуют полосы поглощения, характерные для силикатных групп. У образцов БМГАП происходит уменьшение интенсивности колебаний фосфатных групп в результате замещения части фосфат-анионов на силикат- и карбонат-анионы. В спектрах всех образцов фиксируются также полосы в интервалах 873–871 см–1 (мода ν2) и 1456–1417 см–1 (мода ν3), относящиеся соответственно к деформационным и валентным колебаниям связей С=О в составе карбонат-аниона. В случае стехиометрического немодифицированного ГАП1.67 это может быть связано с адсорбцией углекислого газа из атмосферы воздуха в процессе синтеза. Для образцов БМГАП характерно уширение и увеличение интенсивности полосы с максимумом при 1417 см–1, которая соответствует колебаниям ${\text{CO}}_{{\text{3}}}^{{2--}}$ (мода ν3). Это, на наш взгляд, связано с наложением полосы поглощения при 1417 см–1 на полосу при 1458 см–1, которое происходит за счет вхождения в структуру ГАП карбонатных анионов. В отличие от ИК-Фурье-спектра стехиометрического ГАП1.67, в спектрах порошков БМГАП, допированных силикат- и карбонат-анионами, наблюдаются характерные малоинтенсивные полосы поглощения деформационных колебаний связей Si–O и симметричных валентных колебаний мостиковых связей Si–O–Si в силикатных тетраэдрах при 504 (мода ν2) и ~720 см–1 (мода ν1) соответственно. Наличие данных сигналов [31, 33] свидетельствует о внедрении силикат-анионов в решетку ГАП.
Морфологическое исследование синтезированных образцов ГАП1.67 и БМГАП в форме гидродисперсий после синтеза было проведено методом ПЭМ. На рис. 3 представлены ПЭМ-изображения образцов ГАП1.67 и БМГАП1.50.

По данным ПЭМ (рис. 3), внедрение силикат- и карбонат-анионов в структуру ГАП приводит к изменению формы и среднего размера частиц. Видно, что частицы немодифицированного ГАП1.67 имеют игольчатую форму длиной 45–100 нм и шириной 12–25 нм (рис. 3а). В то же время для образцов БМГАП1.50 (рис. 3б) характерно наличие сферической формы с размером частиц в интервале от 13 до 32 нм. Согласно данным [34], сферическая форма частиц ГАП является важным фактором для инициации провоспалительной реакции, а игольчатая форма кристаллов ГАП является наименее биосовместимой. В [35] сообщалось, что игольчатая форма кристаллов ГАП с размером частиц микрометрового порядка (от 0.1 до 20 мкм) инициирует активацию NLRP3 инфламасомы и выраженную секрецию IL-1β макрофагами и дендритными клетками костного мозга мышей in vitro. В то же время при внутрибрюшинной инъекции микроразмерного и субмикронного ГАП воспалительный ответ не развивался при введении частиц ГАП сравнимого размера, но гладкой сферической формы.
Дополнительное структурно-морфологическое исследование ГАП и БМГАП в порошкообразном состоянии после высушивания гидродисперсий было проведено методом сканирующей электронной микроскопии. Конечные продукты синтеза после высушивания гидрогеля до постоянной массы и измельчения в фарфоровой ступке представляли собой тонкодисперсные порошки белого цвета. На рис. 4 представлены СЭМ-микрофотографии исследуемых материалов после сушки.

Анализ СЭМ-изображений полученных образцов ГАП и БМГАП подтвердил, что данные порошкообразные субстанции представляют собой полидисперсные кристаллические системы, которые имеют схожую морфологию и состоят из агломератов наночастиц с размерами в диапазоне от 2 до 23.5 мкм для немодифицированного ГАП1.67 и от 1 до 10 мкм для БМГАП. На микрофотографиях (рис. 4) присутствуют уже не наноразмерные частицы БМГАП, диспергированные в жидкой фазе, как на рис. 3, а агломерированные микрочастицы продукта, высушенного и измельченного до состояния порошка. В процессе сушки кристаллы ГАП теряют свою гидратную оболочку и происходит их агрегация.
Для кальций-фосфатных материалов, в том числе БМГАП, применяемых в качестве субстратов для изготовления костных имплантатов, помимо биосовместимости, биоактивности и остеоиндуктивности крайне важными являются электроповерхностные свойства синтезированных продуктов. Известно, что в случае помещения биоматериала внутрь организма сразу возникает биологическая ответная реакция на глубине в несколько нанометров от поверхности. В то же время наличие поверхностного заряда на границе между биоматериалом и биологической средой оказывает существенное влияние на процессы адсорбции белка и клеточной адгезии. На отрицательно заряженных кальций-фосфатных материалах происходит преимущественная адсорбция катионов кальция с образованием апатитового слоя, усиливая фиксацию белков и клеток к поверхности. С этой точки зрения частицы ГАП, несущие регулируемый отрицательный поверхностный заряд, имеют заметное преимущество. Значения ζ-потенциала синтезированных образцов ГАП и БМГАП приведены на рис. 5.
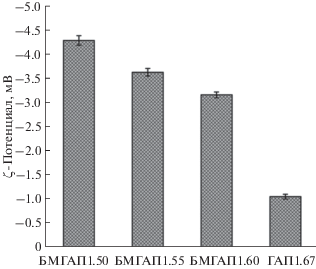
Согласно результатам измерения ζ-потенциала синтезированных образцов (рис. 5) методом динамического рассеяния света, в водной суспензии при рН 7 частицы всех образцов несут отрицательный заряд. Установлено, что с увеличением мольного отношения в кристаллохимической структуре ГАП в интервале 1.50–1.67 наблюдается закономерное уменьшение ζ-потенциала по абсолютной величине (от 4.29 до 1.05 мВ). Так, у образца у БМГАП1.50, что отвечает наибольшей степени дефицитности ионов Ca2+, зафиксировано максимальное отрицательное значение ζ-потенциала (4.29 мВ), далее по мере приближения мольного отношения к стехиометрическому значению ζ-потенциал уменьшается и достигает минимума у ГАП1.67. Таким образом, для синтезированных образцов БМГАП имеет место широкий диапазон варьирования величины отрицательного поверхностного заряда, что позволяет управлять их адгезионными свойствами по отношению к клеткам и протеинам.
Важной характеристикой кальций-фосфатных материалов медицинского назначения является их резорбируемость. Согласно [8], в соответствии с делением процесса резорбции на две фазы (медленную и быструю), различающиеся по уровню рН в зоне резорбции, а также по участию клеток в этом процессе, оценка биорезорбируемости материалов может быть основана на изучении растворимости биоматериала в воде или модельных средах. Биорезорбируемость гидроксиапатитовых биоматериалов зависит от нескольких факторов, таких как фазовый состав, размер кристаллов, удельная поверхность, в том числе и от мольного отношения в кристаллохимической структуре ГАП. В настоящей работе биорезорбируемость синтезированных образцов оценивали по способности к биорезорбции, т.е. интенсивности растворения в физиологическом растворе NaCl (ω(NaCl) = 0.9%), изотоничном плазме крови человека, и в SBF-растворе, имитирующем межтканевую жидкость организма.
В табл. 2 приведены результаты определения растворимости образцов ГАП1.67 и БМГАП в интервале мольных отношений 1.50–1.67 в физиологическом растворе при комнатной температуре 22°С и температуре человеческого тела 37°С (время экспозиции 7 сут).
Из данных табл. 2 видно, что образцы БМГАП обладают улучшенной степенью биорезорбции по сравнению со стехиометрическим немодифицированным ГАП1.67. Такое поведение хорошо согласуется с данными [10], согласно которым замещения на анионы с меньшим зарядом и/или бóльшим радиусом с большей энергией гидратации ведут к повышению растворимости. Для исследуемых образцов БМГАП замещения фосфат-ионов на силикат- и карбонат-анионы приводят к искажению кристаллической решетки, создают напряжения и нестабильность в структуре ГАП, что и обусловливает возрастание растворимости материала. Следует также отметить, что по мере увеличения мольного отношения в образцах БМГАП наблюдается тенденция к уменьшению растворимости. Так, у образца БМГАП1.50 зафиксирована максимальная концентрация ионов Ca2+ (4.10 ммоль/л), переходящих в жидкую фазу при контакте твердого вещества с физиологическим раствором. Минимальная концентрация ионов Ca2+ (1.10 ммоль/л) при принятом времени экспозиции наблюдалась у стехиометрического ГАП1.67. Таким образом, после выдерживания образцов в течение 7 сут в физиологическом растворе 0.9% NaCl концентрация ионов Ca2+ для образцов БМГАП была в 1.9–2.4 раза выше таковой для стехиометрического немодифицированного ГАП1.67. Кроме того, из данных табл. 2 видно, что с возрастанием температуры от 22 до 37°С для всех синтезированных образцов ГАП и БМГАП также отмечается повышение растворимости.
Для оценки биологической активности синтезированных образцов ГАП и БМГАП в условиях in vitro их выдерживали в модельном SBF-растворе (рН 7.4) при постоянной температуре 37 ± 0.5°С в течение 14 сут. По окончании времени выдержки образцов в насыщенном SBF-растворе их биорезорбируемость оценивали по количественному выходу Са2+. Результаты представлены на рис. 6.
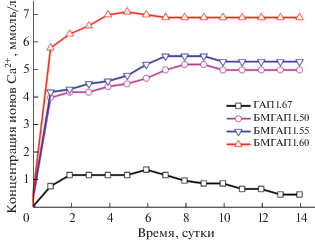
Из рис. 6 видно, что для всех изучаемых образцов наблюдается схожий характер кривых растворения в SBF-растворе. Так, в течение первых суток выход ионов Ca2+ из ГАП1.67 и БМГАП в SBF-раствор быстро растет и через 5–7 сут достигает максимального равновесного значения C(Ca2+). После этого периода времени количество ионов Ca2+, переходящих в жидкую фазу, незначительно уменьшается и далее находится на постоянном уровне. Данный факт, по-видимому, связан с образованием малорастворимого апатитового слоя, формирующегося на поверхности БМГАП, который затрудняет его дальнейшее растворение; как следствие, происходит незначительное уменьшение концентрации ионов Ca2+ в растворе. Известно, что высвобождаемые из кальций-фосфатных материалов подобных БМГАП ионы Ca2+, ${\text{PO}}_{{\text{4}}}^{{{\text{3}}--}},$ ${\text{SiO}}_{{\text{4}}}^{{4--}}$ и OH– играют важную роль в формировании новой апатитовой фазы в биологических средах.
Следует отметить, что все образцы БМГАП имеют более высокую биорезорбируемость в сравнении с немодифицированным ГАП1.67. Из анализа кинетических кривых следует, что по мере увеличения мольного отношения в диапазоне 1.50–1.67 их биорезорбируемость последовательно уменьшается. Максимальный выход ионов Ca2+ в SBF-раствор наблюдается у образца БМГАП1.50, а минимальный – у стехиометрического образца ГАП1.67.
ЗАКЛЮЧЕНИЕ
Методом химического осаждения из водных растворов синтезированы образцы наноструктурного биомиметического гидроксиапатита. Показано, что при увеличении мольного отношения в интервале 1.50–1.67 происходит изменение параметров решетки а, с, а также возрастание среднего размера кристаллитов от 7.52 до 70.30 нм и уменьшение величины удельной поверхности. Установлено, что в водной суспензии (рН 7) частицы суспензии и частицы всех образцов несут отрицательный заряд. Выявлена закономерность изменения величины ζ-потенциала синтезированных порошков в исследованном диапазоне мольных отношений.
Исследования биоактивности образцов методом динамического растворения в физиологическом 0.9%-ном растворе NaCl и в SBF-растворе показали, что скорость резорбции ГАП1.67 и БМГАП уменьшается с возрастанием мольного отношения в кристаллохимической структуре исследуемых образцов.
Список литературы
Liangzhi G., Weibin Z., Yuhui S. // RSC Adv. 2016. V. 6. № 115. P. 114707. https://doi.org/10.1039/C6RA24469F
Trunec M., Chlup Z. // Ceram. Int. 2017. V. 43. № 14. P. 11 265. https://doi.org/10.1016/j.ceramint.2017.05.177
Ларионов Д.С., Кузина М.А., Евдокимов П.В. и др. // Журн. неорган. химии. 2020. V. 65. № 3. P. 309. https://doi.org/10.31857/S0044457X20030071
Голованова О.А. // Журн. неорган. химии. 2020. V. 65. № 3. P. 302. https://doi.org/10.31857/S0044457X20030046
Kolmas J., Krukowski S., Laskus A. et al. // Ceram. Int. 2016. V. 42. № 2. P. 2472. https://doi.org/10.1016/j.ceramint.2015.10.048
Szcześ A., Hołysz L., Chibowski E. // Adv. Coll. Interf. Sci. 2017. V. 249. P. 321. https://doi.org/10.1016/j.cis.2017.04.007
Eliaz N., Metoki N. // Materials. 2017. V. 10. № 4. P. 334. https://doi.org/10.3390/ma10040334
Hendi A.A. // J. Alloys Compd. 2017. V. 712. P. 147. https://doi.org/10.1016/j.jallcom.2017.04.021
Vallet-Regí M., Arcos D. // J. Mater. Chem. 2005. V. 15. № 15. P. 1509. https://doi.org/10.1039/B414143A
Сафронова Т.В., Путляев В.И. // Наносистемы: физика, химия, математика. 2013. V. 4. P. 24.
Chapter 1. Biological Apatites in Bone and Teeth, in: Nanoscience & Nanotechnology Series, Royal Society of Chemistry, Cambridge, 2008. P. 1. https://doi.org/10.1039/9781847558923-00001
Chapter 3. Biomimetic Nanoapatites on Bioceramics, in: Nanoscience & Nanotechnology Series, Royal Society of Chemistry, Cambridge, 2008. P. 61. https://doi.org/10.1039/9781847558923-00061
Salinas A.J., Esbrit P., Vallet-Regí M. // Biomater. Sci. 2013. V. 1. № 1. P. 40. https://doi.org/10.1039/C2BM00071G
Guth K., Campion C., Buckland T. et al. // Adv. Eng. Mater. 2010. V. 12. № 4. P. B113. https://doi.org/10.1002/adem.200980026
Cameron K., Travers P., Chander C. et al. // J. Biomed. Mater. Res. 2013. V. 101A. № 1. P. 13. https://doi.org/10.1002/jbm.a.34261
Putlayev V., Veresov A., Pulkin M. et al. // Mat.-wiss. u. Werkstofftech. 2006. V. 37. № 6. P. 416. https://doi.org/10.1002/mawe.200600007
Munir G., Koller G., Di Silvio L. et al. // J. R. Soc. Interface. 2011. V. 8. № 58. P. 678. https://doi.org/10.1098/rsif.2010.0548
Landi E., Celotti G., Logroscino G. et al. // J. Eur. Ceram. Soc. 2003. V. 23. № 15. P. 2931. https://doi.org/10.1016/S0955-2219(03)00304-2
Landi E., Uggeri J., Sprio S. et al. // J. Biomed. Mater. Res. 2010. V. 94A. № 1. P. 59. https://doi.org/10.1002/jbm.a.32671
Boanini E., Gazzano M., Bigi A. // Acta Biomaterialia. 2010. V. 6. № 6. P. 1882. https://doi.org/10.1016/j.actbio.2009.12.041
Королева М.Ю., Каракатенко Е.Ю., Юртов Е.В. // Коллоид. журн. 2020. V. 82. № 3. P. 324. https://doi.org/10.31857/S0023291220030052
Трубицын М.А., Хоанг Вьет Хунг, Фурда Л.В. // Вестник технологического университета. 2020. V. 23. P. 19.
Wu Y.-J., Tseng Y.-H., Chan J.C.C. // Cryst. Growth Des. 2010. V. 10. № 10. P. 4240. https://doi.org/10.1021/cg100859m
Hu Y.-Y., Rawal A., Schmidt-Rohr K. // Proceedings of the National Academy of Sciences. 2010. V. 107. № 52. P. 22425. https://doi.org/10.1073/pnas.1009219107
Jana N.R., Gearheart L., Murphy C.J. // J. Phys. Chem. B. 2001. V. 105. № 19. P. 4065. https://doi.org/10.1021/jp0107964
Hoang V.H., Troubitsin M.A., Furda L.V. et al. // JBBBE. 2020. V. 47. P. 1. https://doi.org/10.4028/www.scientific.net/JBBBE.47.1
Troubitsin M., Hoang V.H., Furda L. // Bulletin of Belgorod State Technological University named after. V.G. Shukhov. 2020. P. 106. https://doi.org/10.34031/2071-7318-2020-5-3-106-113
Cullity B.D., Weymouth J.W. // Am. J. Phys. 1957. V. 25. № 6. P. 394. https://doi.org/10.1119/1.1934486
Singh G., Singh S., Prakash S. // Surf. Coat. Technol. 2011. V. 205. № 20. P. 4814. https://doi.org/10.1016/j.surfcoat.2011.04.064
Шарло Г. // Методы аналитической химии. Количественный анализ неорганических соединений. M., 1966.
Marchat D., Zymelka M., Coelho C. et al. // Acta Biomaterialia. 2013. V. 9. № 6. P. 6992. https://doi.org/10.1016/j.actbio.2013.03.011
Mostafa N.Y., Hassan H.M., Abd Elkader O.H. // J. Am. Ceram. Soc. 2011. V. 94. № 5. P. 1584. https://doi.org/10.1111/j.1551-2916.2010.04282.x
Palard M., Champion E., Foucaud S. // J. Solid State Chem. 2008. V. 181. № 8. P. 1950. https://doi.org/10.1016/j.jssc.2008.04.027
Laquerriere P., Grandjean-Laquerriere A., Addadi-Rebbah S. et al. // Biomaterials. 2004. V. 25. № 13. P. 2515. https://doi.org/10.1016/j.biomaterials.2003.09.034
Lebre F., Sridharan R., Sawkins M.J. et al. // Sci Rep. 2017. V. 7. № 1. P. 2922. https://doi.org/10.1038/s41598-017-03086-0
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии