

Журнал неорганической химии, 2022, T. 67, № 1, стр. 33-53
Физиологически активные соединения на основе мембранотропных каркасных носителей – производных адамантана и полиэдрических кластеров бора (Обзор)
В. В. Авдеева a, *, Т. М. Гараев b, Е. А. Малинина a, К. Ю. Жижин a, Н. Т. Кузнецов a
a Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Россия
b Национальный исследовательский центр эпидемиологии и микробиологии им. почетного академика Н.Ф. Гамалеи Минздрава России
123098 Москва, Россия
* E-mail: avdeeva.varvara@mail.ru
Поступила в редакцию 03.06.2021
После доработки 20.07.2021
Принята к публикации 29.07.2021
- EDN: KUWWGR
- DOI: 10.31857/S0044457X22010020
Аннотация
Обобщены сведения о соединениях на основе каркасных структур – кластеров бора (бороводороды, карбораны, металлокарбораны) и соединений адамантанового ряда, которые обладают физиологической активностью. Основной упор сделан на противовирусную активность соединений. Рассмотрен механизм возможного действия ингибиторов репликации штаммов вируса гриппа А, обсуждена молекулярная модель ингибитора виропоринов. Предлагаемая модель представляет собой каркасное гидрофобное ядро, выполняющее функцию мембранотропного носителя (кластер бора или фрагмент адамантана), в которое введены физиологически активные функциональные группы. Проанализирована связь структуры каркасного соединения с введенным заместителем с биологически активными свойствами такой молекулярной конструкции.
ВВЕДЕНИЕ
Одной из важнейших задач, стоящих перед современной наукой в XXI в., является борьба с социально значимыми вирусными инфекциями, которые оказывают негативное влияние на качество жизни населения. К таким заболеваниям можно отнести вирусные гепатиты (прежде всего В и С), ВИЧ-инфекцию, грипп А, новую коронавирусную инфекцию COVID-19 и др. Вакцинация как метод борьбы с социально значимыми инфекциями не всегда эффективна, а зачастую вовсе не может быть проведена, поэтому в отсутствие вакцины эффективными остаются противовирусные препараты.
Поиск и создание химических соединений, способных эффективно взаимодействовать непосредственно с вирусной частицей и тем самым ингибировать процесс ее репликации, представляются наиболее перспективным методом лечения и профилактики социально значимых вирусных инфекций. Несмотря на то, что для профилактики заболеваний, вызванных вирусом гриппа, ВОЗ готовит ежегодный прогноз для создания сезонных вакцин, заболеваемость и смертность от этой болезни и осложнений после нeе остаются высокими во всем мире. В некоторых странах от сезонного гриппа ежегодно страдает до 40% населения, а более 500 тысяч человек во всем мире ежегодно от него умирает. Несмотря на всемирные усилия по созданию средств химиотерапии и вакцин, пандемия 2009/2010 гг., вызванная вирусом гриппа A(H1N1)pdm2009, показала их крайнюю ограниченность и недостаточную эффективность. Новые штаммы высоковирулентного вируса гриппа могут появиться неожиданно и вызвать всемирные пандемии с высоким уровнем заболеваемости и смертности.
К настоящему времени практически все штаммы вируса гриппа А, вызывающие эпидемии, оказались полностью резистентными к римантадину. Ввиду этих причин поиск новых ингибиторов репликации вирусов гриппа является актуальной задачей современной науки. Для ее решения необходим анализ строения известных соединений, обладающих физиологическими свойствами, и проведение корреляций структура–свойство, что позволит определить наиболее перспективные группы соединений на основе каркасных структур для решения поставленной задачи.
В настоящем обзоре обсуждаются фармакологические свойства соединений на основе трехмерных каркасных неорганических и органических систем, состоящих из атомов бора (полиэдрические кластеры бора, рис. 1) или атомов углерода (соединения адамантанового ряда, рис. 2) соответственно.
Рис. 1.
Строение кластерных анионов бора [B10H10]2– (а), [B20H18]2– (б) и o-карборана [C2B10H12] (в).

Ван-дер-ваальсовы объемы карборанов (148, 143 и 141 Å3 для орто-, мета- и пара-карборана соответственно) сравнимы с объемом адамантана (136 Å3). Наличие в молекуле десяти BH-групп, не способных образовывать классические водородные связи, делает карборановые кластеры чрезвычайно гидрофобными. При этом гидрофобность карборанильного фрагмента сравнима с гидрофобностью адамантильной группы и может заметно варьироваться в зависимости от изомера карборана и места присоединения заместителя. Кроме того, электронный эффект карборанильного фрагмента зависит как от изомера карборана, так и от положения заместителя и варьируется от сильного электроноакцепторного для C-замещенных производных до умеренного электронодонорного для B-замещенных производных, что дает возможность в значительной степени изменять свойства соединений на их основе. Все это позволяет рассматривать карбораны как перспективные фармакофорные группы и использовать их в качестве аналогов адамантана при создании новых лекарственных препаратов [1]. В случае полиэдрических бороводородных анионов, несмотря на хорошую растворимость в воде их солей с катионами щелочных и щелочноземельных металлов, гидрофобный характер B–H-групп, препятствующий образованию устойчивой гидратной оболочки, придает им скрытый амфифильный характер [2], что также открывает хорошие перспективы для создания препаратов на их основе.
Кластерные анионы бора [BnHn]2– (n = 6–12) и их аналоги (карбораны, металлокарбораны) [3–11] являются уникальными неорганическими системами, которые предоставляют широкие возможности для создания производных, содержащих различные функциональные группы. Для кластерных анионов бора существует возможность изменения геометрического и электронного строения борного кластера: в дизайне новых соединений могут быть использованы дианионы [B10H10]2– (двухшапочная архимедова антипризма), [B12H12]2– (икосаэдр), [B20H18]2– (димерный макрополиэдр), однозаряженные монокарбораны [CB11H12]– или нейтральные дикарбораны [C2B10H12]. Строение некоторых из указанных борных кластеров представлено на рис. 1. Введение в кластер функциональных групп может приводить к понижению заряда системы и образованию моноанионов или нейтральных соединений. Возможность образования анионных соединений очень важна с точки зрения физиологии, так как позволяет синтезировать натриевые соли целевых соединений, которые обладают высокой растворимостью в воде и низкой токсичностью. Отметим, что широкие возможности варьирования структурой кластерных анионов бора в первую очередь связаны с трехмерной ароматичностью этих объектов [12–14], что позволяет им образовывать большое количество устойчивых замещенных производных с различными функциональными группами.
В свою очередь, органическая химия предоставляет неограниченные возможности изменения структуры каркасных углеродных соединений (в нашем случае соединений адамантанового ряда) для настраиваемых физиологически значимых характеристик конечных соединений. В настоящей работе рассмотрены производные адамантанов, обладающие противовирусной активностью.
Данные, приведенные в настоящем обзоре, позволяют проследить изменение физиологически активных свойств соединений в зависимости от изменения их структуры, определить влияние природы введенного в органический или неорганический каркас заместителя на свойства соединения, а также предположить наиболее перспективную область изменения структуры каркасных производных для поиска новых физиологически активных препаратов, в первую очередь перспективных в ингибировании вирусов гриппа.
ПРОИЗВОДНЫЕ СОЕДИНЕНИЙ АДАМАНТАНОВОГО РЯДА, ОБЛАДАЮЩИЕ ПРОТИВОВИРУСНОЙ АКТИВНОСТЬЮ
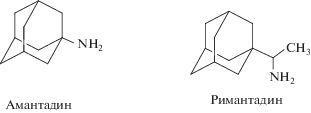
Адамантановые производные являются структурной основой многочисленных соединений, и их открытие положило начало новой области химии, изучающей подходы к синтезу, а также физико-химические и биологические свойства органических полиэдрических соединений, имеющих практическое применение в фармацевтической промышленности [15, 16]. Производные адамантана востребованы во многих областях медицины, в том числе в системной и местной терапии. Липофильность адамантанового ядра обеспечивает взаимодействие как с биологическими мембранами, содержащими липидный слой, так и с гидрофобными участками белковых молекул, входящими в структуру рецепторов. При введении в структуру фармакофоров фрагментов адамантана улучшаются фармакокинетические профили модифицированных препаратов [17, 18]. Простые аминоадамантаны (амантадин, римантадин) заняли надежное место на фармацевтическом рынке, показав свою эффективность для лечения таких вирусных заболеваний, как грипп А, герпес, гепатит С и ВИЧ [19]. Ведутся также разработки пиразоловых производных адамантана против инфекции ящура [20]. Исследователи установили, что высвобождение вирионов из клеток, инфицированных вирусом ящура, подавлялось амантадином – ингибитором функции виропорина М2 вируса гриппа А [21].
Помимо вируса гриппа римантадин ингибирует репродукцию вируса Синдбис, так как, являясь слабым липофильным основанием, способен повышать рН эндосомального содержимого и препятствовать депротонизации вируса [22]. Препарат адапромин (рис. 3а) [23] активен в отношении вирусов гриппа типа А и В, но обладает большей токсичностью, чем римантадин. Противовирусный препарат тромантадин (рис. 3б) применяется в виде мазей и активен в отношении вирусов простого герпеса 1-го и 2-го типов и вируса герпес Зостер (вирус герпеса 3-го типа) [24]. Соединения адамантан-α-аминокислот, в которых адамантан связан с углеродом боковой цепи аминокислоты связью С–С, а карбоксильная группа аминокислоты представлена эфирной или амидной группировкой (рис. 3в и 3г), активны в отношении вируса Синдбис (индекс селективности (Si) равен 2) и вируса гриппа А на уровне римантадина. В отношении вируса гриппа А S-15 активны также алкильные производные аминоадамантана (рис. 3д, 3е, 3ж) [25].

Авторы [26] показали, что триазольные и тетразольные производные адамантанов обладают высоким уровнем противовирусной активности in vitro в отношении лабораторных и циркулирующих сезонных вирусов гриппа А и умеренной активностью в отношении пандемического штамма A(H1N1)pdm2009. Причем тетразольные производные адамантанов более активны, нежели их триазольные гомологи [27]. Соединения класса азоло-адамантанов обладают высоким уровнем противовирусной активности in vitro в отношении римантадин-устойчивого штамма A/PR78/34 (H1N1) (SI > 8) [26, 27]. Однако уровень активности сильно зависит от химической структуры. Так, среди производных 1,2,4-триазолов соединение 1-(3-хлоро-1,2,4-триазол-1-ил)-3-(1-аминоэтил-1)адамантан (рис. 4а) проявляет значительную активность в отношении вируса гриппа А (SI = 10), в то время как адамантильные производные тетразола (рис. 4б, 4в, 4г) демонстрируют высокий уровень активности против вируса гриппа. Как было установлено, положение адамантильной части в тетразольном цикле может играть важную роль в усилении активности соединений этого класса.
Рис. 4.
Триазольные и тетразольные производные адамантанов, обладающие противовирусной активностью in vitro в отношении гриппа А.

Был предложен способ преодоления резистентности вирусов гриппа А к препаратам адамантанового ряда путем введения новых функциональных групп (карбоксильной, гидроксильной, имидазольной, индольной и др.) в аминоадамантановый карбоцикл с использованием для этого аминокислот, пептидов или других физиологически важных соединений [28]. Полученный ряд карбоциклических производных аминокислот и пептидов способен ингибировать высокопатогенные штаммы вирусов гриппа А, включая и такие как A/H1N1pdm09, A/H5N1, A/H3N2 и др. (рис. 5). При этом токсичность соединений была не выше римантадина, а для ряда соединений даже ниже [29]. Соединения обладают химико-терапевтическим индексом (Si) от 8 до 120 и проявляют вирулицидные свойства в отношении пандемичного вируса гриппа А/H5N1. В культурах клеток Vero-E6 (перевиваемые культуры клеток почки зеленой мартышки, клон 6) и СПЭВ (линия клеток почки эмбриона свиньи) снижение инфекционного титра вируса составило от 3 до 5 логарифмов (lg) по отношению к контролю.
Рис. 5.
N-ацилпроизводные аминоадамантанов, обладающие противогриппозной активностью в отношении штаммов вирусов, резистентных к римантадину.

Соединение римантадина с остатком аминокислоты гистидина (рис. 5a) высокоэффективно in vitro в отношении вируса гриппа птиц A/duck/Novosibirsk/56/05 (H5N1), обладающего пандемическим потенциалом. Соединение эффективно защищает монослой клеток Vero-E6 в различных схемах внесения препарата, а 50%-ная ингибирующая доза составляет в среднем 0.5 мМ. Противовирусная активность этого соединения превосходит известный отечественный препарат Арбидол [30]. Для соединения (рис. 5a) был определен механизм действия in silico и in vitro посредством сравнения результатов молекулярного докинга и противовирусных свойств искусственно созданных мутантов вируса A/PuertoRico/8/34(H1N1) с точечными аминокислотными заменами в трансмембранной области белка М2 [31].
Производное 1,3-адамантандиуксусной кислоты с двумя остатками этилового эфира треонина N,N-1,3-диациладамантил-диэтилтреонат (рис. 5б) высокоселективно подавляет репродукцию вируса гриппа А и эффективно против штаммов, резистентных к гидрохлориду римантадина [32]. Соединения, содержащие тиенилкарбоновые кислоты (рис. 5в и 5г), подавляют репродукцию вируса гриппа A/IIV-Orenburg/83/ 2012(H1N1)pdm09. Наименьшей ингибирующей дозой (ИД50 1 мМ) обладает соединение с 1-аминоадамантаном (рис. 5в). Соединения римантадина с остатком метионинсульфона эффективно ингибировали репродукцию эталонного штамма вируса гриппа A/California/07/2009 (рис. 5д и 5е). Наибольшим противовирусным эффектом обладает соединение на рис. 5е с Boc-блокированной аминогруппой (ИД50 0.65 μМ). Молекула соединения N-ациладамантил-пептидов с римантадином имеет бóльшие размеры, чем молекула адамантил-аминокислоты, но не превышающие внутренний диаметр поры канала М2 вируса гриппа. Соединение римантадина с хинальдил-Ala-Pro-OH (рис. 5з) обладает устойчивым процентом подавления цитопатического действия эталонного штамма вируса гриппа A/California/07/2009 (ИД50 0.74 мМ) [33].
Авторы [34] синтезировали производные адамантана (римантадин и амантадин), модифицированные глицил-тиазольными (рис. 6a, 6б, 6д и 6е) и глицил-тиазол-тиазольными молекулами (рис. 6в и 6г) и исследовали их противовирусную и противомикробную активность. Соединения с блокированной аминогруппой глицина Boc-Gly-Thz-амантадин, Boc-Gly-Thz-римантадин (рис. 6a и 6б) растворяли в 20 мл TFA для удаления защитной Boc-группы и также использовали в испытаниях противовирусной и фунгицидной активности. Соединения с дитиазольным мотивом содержали защитную Fmoc-группу (рис. 6c). Испытания показали, что наличие пространственно громоздкой Fmoc-группы не приводит к улучшению противовирусного эффекта аминоадамантанов: защищенные по α-аминогруппе соединения не проявляют никакой противовирусной активности. Аналог римантадина со свободной α-аминогруппой, включающий тиазольное звено (рис. 6е), продемонстрировал умеренную активность против вируса гриппа A/Hongkong/68 (H3N2). Напротив, аналог амантадина со свободной α-аминогруппой, включающий тиазольный мотив (рис. 6e), не проявляет противовирусного эффекта. Результаты показывают, что ни тиазольный цикл, ни свободная аминогруппа не являются решающими для противовирусной активности. Соединение Gly-Thz-римантадин (рис. 6е) в двух испытанных концентрациях (10 и 60 мМ) показало очень хорошую противогрибковую активность против модельного штамма грибка Yarrowia lipolytica.
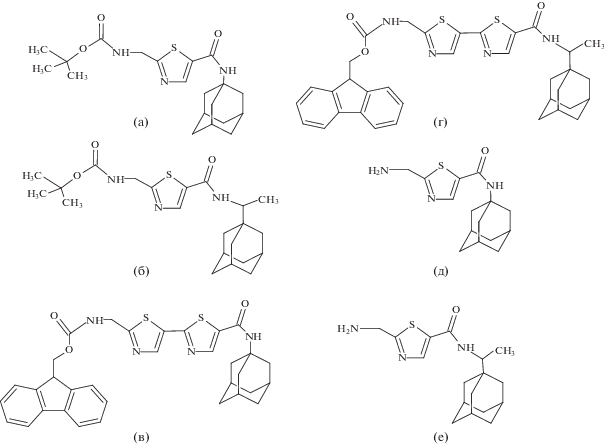
Другие соединения не обладают противогрибковой активностью. Все соединения не проявляют активности против модельных штаммов грамположительных и грамотрицательных микроорганизмов.
Спиропроизводные адамантана способны ингибировать штаммы вируса гриппа А(H1N1), А(H2N2) и А(H3N2) в концентрациях ~55 мкг/мл. Соединения (рис. 7) также проявляли активность на уровне 115 мкг/мл в отношении ВИЧ-1 in vitro, однако эти соединения не были эффективны против ВИЧ-2. Более того, активность анти-ВИЧ-1 проявлялась только для спирошестичленных аналогов, но не обнаруживалась для спиропятичленных [35].
Рис. 7.
Спиропроизводные адамантана, обладающие противовирусной активностью в отношении гриппа А и ВИЧ-1.

В последние годы интерес ученых вызывает синтез адамантилсодержащих нуклеиновых оснований и родственных им соединений и изучение их способности ингибировать репликацию ВИЧ-1. В частности, такие производные способны облегчить транспорт лекарственного средства через биологические мембраны. Производное 3'-азидо-3'-дезокситимидина (азидотимидин, AZT), содержащее фрагмент адамантана в положении 5'-нуклеозида (рис. 8а), легче, чем нативный AZT, проникает в ткани головного мозга, где вирус ВИЧ непосредственно повреждает оболочки и ткани головного мозга [36]. Известны и другие производные AZT, модифицированные молекулой адамантана [37], такие как 3'-(1-адамантил)тиоуреидопроизводное тимидина (рис. 8б) и производное по фосфатной группе, полученное взаимодействием (1-адамантилфосфонил)фосфата с монофосфатом азидотимидина (рис. 8в).

Подобно протонному каналу М2 вируса гриппа А важную роль в воспроизводстве вирусных частиц вируса гепатита С (HCV) играет ионный канал p7. Неструктурный белок p7 HCV состоит из 63 а. о. и имеет два трансмембранных домена (ТМ1 и ТМ2) [38]. Шесть субъединиц p7 образуют гексамерный агрегат, локализующийся преимущественно во внутриклеточных мембранах, который в опытах in vitro проявляет функцию ионного канала, необходимую для сборки вируса и оптимального выхода из инфицированных клеток путем изменения кислотно-щелочного равновесия внутриклеточных везикул [39]. Ранее было определено, что функционирование ионного канала p7 может быть блокировано небольшими молекулами-ингибиторами, в частности производными адамантана, что приводит к значительному спаду воспроизводства вирусных частиц [40].
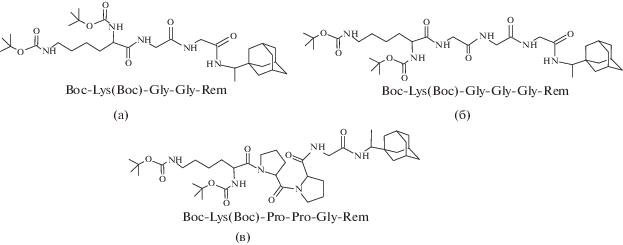
Более объемные структуры карбоциклических производных пептидов активны в отношении ингибирования репликации вируса гепатита С [4]. Молекула карбоцикла аминоадамантана, обеспеченная дополнительными функционально активными группами, в процессе взаимодействия с трансмембранным доменом белка p7 HCV способна нарушить процесс транспорта ионов через мембрану. Источником таких функционально активных групп могут быть пептидные остатки, присоединенные к римантадину методами пептидного синтеза [41].
Синтетические соединения, представленные на рис. 9, проявляют значительно меньший токсический эффект по сравнению с римантадином на культуре клеток СПЭВ. Причем соединение (рис. 9б) обладает наименьшей токсичностью, а также проявляет вирулицидную активность в отношении HCV. Снижение инфекционного титра происходит более чем на пять десятичных логарифмов (10 000 раз) по отношению к вирусному контролю in vitro.
Рис. 9.
Структурные формулы 1-(1-адамантил)этиламид-N,N-ди-трет-бутилоксикарбонилпептидов, способных ингибировать репликацию HCV in vitro.
Основной предполагаемый механизм противовирусного действия, по-видимому, сходен с действием амантадина на ионный канал p7 HCV [42]. По крайней мере, ионный канал p7 HCV представляет собой наиболее вероятную мишень для предлагаемых соединений. Остаток карбоцикла адамантана в этом случае, очевидно, исполняет роль носителя, к которому “прицеплена” функционально активная группа соответствующего пептида. Это согласуется с предложенным дизайном универсальной молекулярной модели ингибитора виропоринов.
ПРОИЗВОДНЫЕ КЛАСТЕРНЫХ АНИОНОВ БОРА, ОБЛАДАЮЩИЕ ФИЗИОЛОГИЧЕСКОЙ АКТИВНОСТЬЮ
Интерес к химии кластерных анионов бора обусловлен в первую очередь возможностью использования соединений на основе кластеров бора в медицине. Это направление исследований включает поиск путей синтеза замещенных производных кластерных анионов бора для получения новых препаратов для диагностики и терапии онкозаболеваний (в частности, для борнейтронозахватной терапии и бинарной терапии и т.д. [43–45]), контрастных агентов для МРТ-диагностики [46], получения соединений с противомикробной и противовирусной активностью [47] и др.
Одним из важнейших требований к получению препаратов с фармакологической активностью является селективность доставки препарата бора к биологической мишени, в связи с чем стратегия создания новых борсодержащих веществ для терапевтических целей основана на введении в кластер бора эффективных транспортных групп. Для реализации этого подхода необходима разработка методов, позволяющих проводить модификацию соединений кластерных анионов бора в мягких условиях и получать устойчивые в биологических средах продукты.
Введение кластерных анионов бора в биомолекулы приводит к резкому изменению гидрофильных и липофильных свойств, что дает возможность регулировать поведение полученных соединений в биологических средах. Клозо-декаборатные и клозо-додекаборатные анионы [B10H10]2– и [B12H12]2– проявляют гидрофильные свойства, тогда как дикарбододекаборан [C2B10H12] – ярко выраженный гидрофобный характер, что позволяет карборансодержащим биомолекулам эффективно связываться с гидрофобными областями протеинов, в частности рецепторов [48]. Создание биомолекул, содержащих кластерные анионы бора, позволяет добиться повышенной in vitro стабильности полученных соединений.
Наиболее изученной областью применения кластерных анионов бора и карборанов является их использование для целей борнейтронозахватной терапии (БНЗТ). В основе БНЗТ лежит ядерная реакция взаимодействия стабильного изотопа бора-10 с тепловыми нейтронами (Еn = 0.025 эВ, сечение захвата 10B 3890 барн). Образующиеся в результате реакции 10B(nth|α,γ)7Li частицы – ядра гелия (альфа-частицы) и ядра отдачи лития-7 – обладают в тканях высокой линейной потерей энергии (соответственно 200 и 350 кэВ/мкм) и небольшим суммарным пробегом (~14 мкм), соизмеримым с диаметром одной клетки. В случае же селективного накопления бора-10 в опухолевых клетках может быть достигнут избирательный радиационный эффект на клеточном уровне. В идеальном случае разрушаются только опухолевые клетки, включая сколь угодно мелкие метастазы, без повреждения нормальных тканей в облучаемом объеме. Ключевой остается задача создания борсодержащих препаратов, способных избирательно доставлять в клетки злокачественных опухолей терапевтическое количество бора-10, обеспечивать его оптимальное микрораспределение и оставаться в клетках в течение необходимого для облучения периода времени. Работы в этой области ведутся интенсивно (например, [49–54]) и требуют отдельного рассмотрения, что выходит за рамки настоящего обзора. Кластерные анионы бора пригодны для достижения этих целей по причине своей высокой химической и биологической стабильности, большого содержания бора в молекуле, низкой токсичности и возможности получения водорастворимых соединений (например, натриевых солей производных кластерных анионов бора). Следует отметить, что димерный кластерный анион бора [транс-B20H18]2– [55–57] содержит в своем составе 20 атомов бора (рис. 1) на один анион и имеет существенное преимущество по сравнению со многими другими бороводородными анионами и гетероборанами при получении соединений, пригодных для целей БНЗТ. В связи с этим изучение реакционной способности димерного аниона [транс-B20H18]2– и его производных представляет особенный интерес.
Cинтезировано большое количество карборановых аналогов разнообразных стероидных соединений. Стероиды часто выступают в качестве гормонов, взаимодействующих со специфическими рецепторами. От гидрофобного характера стероидов зависит эффективность их связывания с рецепторами. Введение в биомолекулу карборанового фрагмента способствует сильному увеличению ее гидрофобности, что приводит к усилению биологической активности данных соединений. В частности, синтезированы карборансодержащие аналоги 17-эстрадиола (рис. 10a) [58], холестерина (рис. 10б) [59], 4,5-2H-дигидротестостерона (рис. 10в) [60].


Ряд производных карборанов были протестированы в качестве потенциальных антагонистов [58, 61] и агонистов [58, 62] рецептора эстрогена (рис. 11a, 11б). Основной акцент был сделан на синтезе аналогов эстрадиола, содержащих кластерное ядро, что позволяло полученным соединениям проявлять сильные гидрофобные взаимодействия с рецептором эстрогена. Было изучено влияние типа изомера карборана и природы заместителя у карборанового кластера на биологическую активность.
Карборановые аналоги ретиноидов [63, 64] представляют интерес в области дерматологии и онкологии. Введение в ретинобензойные кислоты объемной гидрофобной карборановой группы способствует появлению антагонистического эффекта. Синтезированы и изучены биологические свойства различных дифениламинов, связанных с карборанами (рис. 12). Ряд соединений проявлял сильную агонистическую активность при концентрациях 10–8–10–9 моль. Результаты показывают, что карбораны применимы в качестве гидрофобных фрагментов биологически активных молекул.

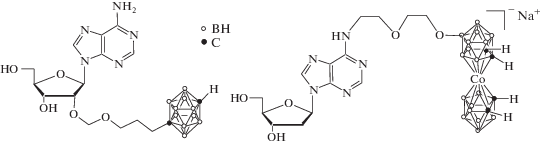
Синтезированы новые типы производных аденозина и 2'-дезоксиаденозина, содержащие кластеры бора в положениях C2', N6 или C8 [65]. Некоторые из них представлены на рис. 13. Изучено влияние этих модифицированных соединений на функцию тромбоцитов. Модификация аденозина в положении C2 пара-карбораном приводит к эффективному ингибированию функции тромбоцитов, включая агрегацию, секрецию белка и экспрессию P-селектина, индуцированную тромбином или ADP. Полученные результаты способствуют созданию нового класса аналогов аденозина, модулирующих активность тромбоцитов крови человека.
Еще одной областью применения соединений на основе кластерных анионов бора является создание биоконъюгатов на основе карбоборанильных фосфонатов [66, 67]. Существует два подхода к синтезу подобных биомолекул: создание соединений, в которых фосфонатная группа напрямую связана с карборановым кластером (рис. 14a, 14б) или отделена от кластера спейсерной группой (рис. 14в). Карбоборанильные фосфонаты проявляют высокую антихолинэстеразную активность. Соединения, в которых фосфонатная группа напрямую связана с карбораном, проявляют гаметоцидную активность, тогда как молекулы, в которых фосфонатная группа соединена с кластером через атомы серы или селена, проявляют бактерицидную активность. Карбоборанильные моно- и дифосфонаты, связанные в олигонуклеотидные последовательности, могут служить в качестве агентов для олигонуклеотидной терапии из-за повышенной сопротивляемости к усвоению нуклеазами.

Изучено противомикробное действие некоторых производных кластерных анионов бора и карборанов. В работе [68] синтезирован ряд производных о-карборанов и анионные дикарболлиды нидо-типа, изучена их антимикробная активность (рис. 15). Почти все исследованные моноанионы дикарболлида активны in vitro против грибов, таких как Candida albicans, Aspergillus fumigatus и Tricophyton asteroides, а также против грамположительных бактерий. Из соотношений структура–активность авторы пришли к выводу, что введение липофильных алкильных групп (C5–C17) или о-карборанильных групп к гидрофильным дикарболлидным анионам приводит к антимикробной активности, в то время как большинство производных карборана демонстрирует низкую активность, за исключением амино- и хлорпроизводных, которые, как оказалось, активны по отношению к Tricophyton asteroides и грамположительным бактериям.

В работе [69] изучена активность о-карборанилаланина (рис. 16) – высоколипофильного аналога аминокислоты фенилаланина – против различных патогенов растений. о-Карборанилаланин показал высокую активность (значения MIC в диапазоне от 0.00015 до 0.32 мкМ, что более чем в 1000 раз выше, чем у тридеморфа – фунгицида селективного ингибитора зооспор) против всех бесполых спор Plasrnopara halstedii.
Рис. 16.
Строение о-карборанилаланина (а) и металлокарборановых производных, активных в отношении Y. enterocolitica и P. aeruginosa (б).

Синтезированы металлокарборановые производные (рис. 16б), обладающие антибактериальной активностью против methicillin-resistant Staphylococcus aureus, Y. enterocolitica и P. aeruginosa [70, 71].
В работе [72] синтезирован ряд изониазидных производных карборана, содержащих 1,2-дикарба-клозо-додекаборан, 1,7-дикарба-клозо-додекаборан, 1,12-дикарба-клозо-додекаборан или 7,8-дикарба-нидо-ундекаборатный анион. Соединения были протестированы in vitro против штамма Mycobacterium tuberculosis (Mtb) H37Rv и его мутанта (ΔkatG), дефектного по синтезу каталазы-пероксидазы (KatG). N'-((7,8-дикарба-нидо-ундекаборанил)метилиден)изоникотиногидразид (рис. 17а) проявил наивысшую активность в отношении штамма Mtb дикого типа (MIC99 = 0.33 мкМ (Mtb), MIC99 ≥ 660 мкМ (ΔkatG)).
Рис. 17.
Строение изониазидного производного 7,8-дикарба-нидо-ундекаборана (а) и производных бис-дикарболлида кобальта [3,3'-Co(8-R(CH2CH2O)2-1,2-C2B9H10)(1',2'-C2B9H11)]− (б).
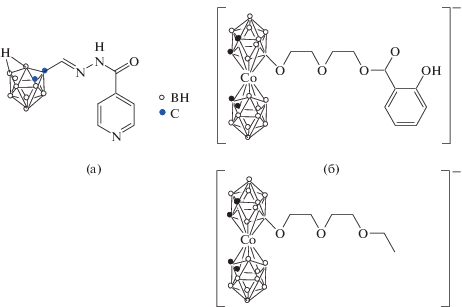
В работе [73] изучены противомикробные свойства in vitro бис-дикарболлида кобальта [3,3'-Co(1,2-C2B9H11)2]− и натриевых солей его производных [3,3'-Co(8-R(CH2CH2O)2-1,2-C2B9H10)(1',2'-C2B9H11)]− (R = −OOCCH3, –OCH3, –OCH2CH3). Полученные результаты показали, что среди исследованных соединений соединения с R = = OCH2CH3 (рис. 17б) проявляют наивысшую антимикробную активность, которая равна или даже выше активности коммерчески доступного антибиотика широкого спектра действия тиамфеникола. Установлено, что бис-дикарболлид кобальта проявляет сравнительно более низкие антибактериальные и противогрибковые свойства по сравнению с его производными. С практической точки зрения авторы подчеркивают, что метициллинрезистентный штамм Staphylococcus aureus (TSA MRSA), полирезистентные штаммы Pseudomonas aeruginosa, а также Candida spp. чувствительны к указанным на рис. 17б соединениям.
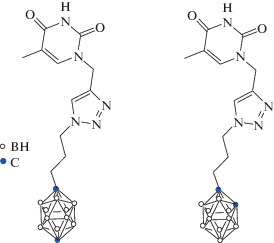
Авторами [74] синтезированы производные тимина, содержащие орто-карборан, пара-карборан и нидо-карборановые кластеры (рис. 18). Обнаружено, что полученные соединения являются ингибиторами бактериальных ферментов и могут быть использованы в качестве новых противотуберкулезных препаратов.
Ряд амидов и производных дибораоксазолов, содержащих клозо-додекаборатный анион (рис. 19), которые можно рассматривать как 3D-аналоги бензоксазолов, были синтезированы и исследованы на предмет противомикробной активности [75] против грамотрицательных (Neisseria gonorrhoeae) и грамположительных (S. aureus и E. faecalis) бактерий. Диборагетероциклы показали высокую специфическую активность против N. gonorrhoeae, но низкую активность против грамположительных бактерий.

Синтезирована серия новых аналогов пенициллина G, содержащих липофильные орто-, мета- или пара-карборан вместо фенильного кольца [76]. Аналоги пенициллина G получены путем амидирования 6-аминопеницилановой кислоты (6-APA) активными эфирами N-сукцинимидила, содержащими орто-, мета- или пара-карборан. Альтернативно были синтезированы аналоги, содержащие орто- или пара-карборан с использованием хлорангидридов орто- или пара-карборановых кислот. Полученные таким образом соединения тестировали in vitro против грамположительных бактерий Staphylococcus aureus и грамотрицательных бактерий Klebsiella pneumoniae, Enterobacter cloacae, Acinetobacter baumanii и Pseudomonas aeruginosa. Самым мощным ингибитором роста грамположительных бактерий было соединение, содержащее пара-карборановый кластер (рис. 20) (S. aureus, MIC 64 мкг/мл). Соединения, содержащие орто- или мета-карборан, соответственно, были менее активны в отношении S. aureus.

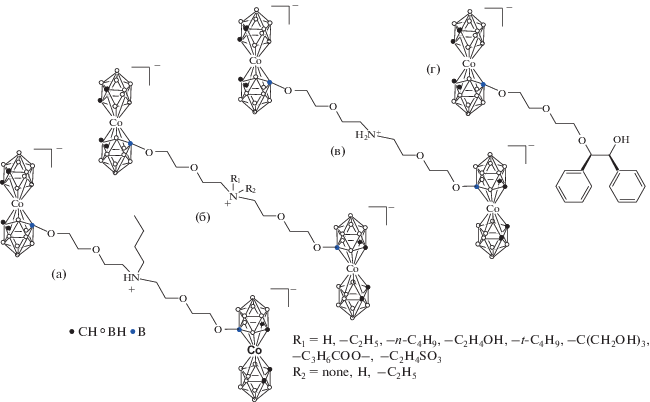
Cерия производных N-алкиламмония 8-диэтиленгликоль-бис-дикарболлида кобальта (рис. 21) была получена прямым методом [77]. Антибактериальная и противогрибковая активность семи производных обнаружена на основании определения минимальных ингибирующих концентраций (MIC80). Рост S. aureus подавлялся с высокой селективностью в присутствии соединения с Nu = NH3 в концентрации 3.8 мг/л. Авторы отмечают абсолютную избирательную активность соединения c Nu = NH2CH3 в ингибировании роста нитчатого гриба Trichosporon cutaneum.
Рис. 21.
Производные N-алкиламмония 8-диэтиленгликоль-бис(дикарболлид)кобальта. Nu = NH3, NH2CH3, NH(CH3)2, N(CH3)3, NH2C2H5, NH(C2H5)2, N(C2H5)3.

Ряд соединений, содержащих карборановые кластеры, был протестирован на наличие противовирусной активности. В частности, ряд конъюгатов пара-карборана [78], орто-карборана [79] и бис(1,2-дикарболлид)кобальта [80] с 5-этинил-уридином были получены по реакции кросс-сочетания Соногашира соответствующего борсодержащего терминального алкина и 5-йод-нуклеозида. Строение некоторых соединений представлено на рис. 22. Изучена активность полученных соединений против вирусов HCMV, EMCV, HPIV-3, HSV-1; соединения продемонстрировали цитотоксичность от низкой до умеренной в нескольких клеточных линиях. Наиболее активным соединением является 5-[(1,12-дикарба-клозо-додекаборан-2-ил]-20-дезоксиуридин (рис. 22в); для этого соединения было установлено IC50 5.5 мМ с индексом селективности выше 180. Обнаружено, что соединение проявляет противовирусную активность против HCMV и не активен против HSV-1, HPIV-3 или EMCV [78].
Рис. 22.
Строение нуклеозидных коньюгатов, содержащих орто-карборан (a), бис(1,2-дикарболлид)кобальт (б), пара-карборан (в).

Кроме того, получены конъюгаты на основе клозо-додекаборатоаминов в качестве универсальных синтонов, включая бис(клозо-додекабораты), конъюгаты клозо-додекабората с липидами и с ненатуральным нуклеозидом 8-аза-7-деаза-2'-деоксиаденозином (рис. 23) [81]. Соединения показали низкую или умеренную токсичность, однако оказались неэффективными против вирусов – не обнаружена активность против HSV-1, HPIV-3, EMCV, VSV, HMCV.

Недавно обнаружено [82–84], что новый класс производных бис(1,2-дикарболлид)кобальта является действенным специфическим и селективным конкурентным ингибитором протеазы ВИЧ (рис. 24). Наиболее активное соединение (рис. 24a) проявляет высокую активность против ВИЧ и не оказывает токсического действия на культуру клеток (IC50 = 100 нМ) [82].
Родственные производные бис(1,2-дикарболлид)кобальта изучены в работах [83, 84] (рис. 24б–24г). Замещенные металлокарбораны являются мощными специфическими и селективными конкурентными ингибиторами протеазы (PR) дикого типа и мутированных PR ВИЧ. Авторы объясняют их способность ингибировать множество устойчивых к ингибиторам протеазы видов PR благодаря их новому способу связывания на сайтах связывания за счет участия в диводородных связях и их способности регулировать положение металлокарборанового остова в субстрате PR ВИЧ. Авторы подчеркивают, что кластеры бора являются многообещающими фармакофорами для мощного специфического ингибирования устойчивых к лекарствам мутантов протеазы ВИЧ.
Описан синтез нового ингибитора нейраминидазы – карборансодержащего эфира карбоновой кислоты осельтамивир (Oseltamivir), описаны его физико-химические и спектральные характеристики [85] (рис. 25). Карборановый аналог осельтамивира оказался на порядок менее активным, чем его предшественник – соответствующий этиловый эфир, который является активным ингредиентом фармацевтических препаратов, используемых в профилактике и терапии гриппа.

В завершение обзора отметим несколько примеров, которые оправдывают сравнение фармакологических свойств неорганических и органических трехмерных систем, к которым относятся кластеры бора и адамантаны. Проведение аналогий в структурах и свойствах соединений является инструментом, необходимым для дизайна новых соединений [86]. В работе [87] проведено сравнение активности фенильных производных мета- и пара-карборана с аналогичным производным адамантана (рис. 26) по отношению к связыванию с эстрогеном. С помощью метода биолюминесценции показано, что производное 4-фенил-пара-карборана проявляет наибольшую активность среди других карборановых аналогов и в 10 раз более активно, чем 17-эстрадиол. Производное 4-фенил-пара-карборана проявляет транскрипционную активность в диапазоне концентраций от 1 × 10–10 до 1 × 10–8 моль, и его действие сравнимо с действием аналогичного производного адамантана (рис. 26a). Антагонисты эстрогенного рецептора широко используются при лечении рака молочной железы.

Авторы [88] синтезировали 48 аналогов ингибитора роста клеток Mycobacterium tuberculosis SQ109, в которой этилендиаминовый линкер заменен на окса-, тиа- или гетероциклические группы, содержащие адамантан или 1,2-карборан. Некоторые представители соединений представлены на рис. 27. Соединения были протестированы против M. tuberculosis (H37Rv и/или Erdman), Mycobacterium smegmatis, Bacillus subtilis, Escherichia coli, Saccharomyces cerevisiae, Trypanosoma brucei и двух линий клеток человека (эмбриональной почки человека HEK293T и гепатоцеллюлярной карциномы). Наиболее сильная активность была обнаружена против T. Brucei – возбудителя африканского трипаносомоза человека, это было обнаружено у 15 аналогов SQ109, которые были более активны, чем SQ109, в ингибировании роста клеток, имея значение IC50 всего 12 нМ (5.5 нг/мл) и индекс селективности ∼300. Карбораны были менее активны в отношении M. tuberculosis, но, что удивительно, обладали активностью (IC50 ~ ~ 2 мкг/мл) против грамотрицательных бактерий E. coli.
В работе [89] проведено сравнение биоизостеров на основе однотипных производных адамантана и орто-, мета- и пара-карборана и изучено их антималярийное действие (рис. 28). Показано, что введение адамантана снижает эффективность in vitro против Plasmodium falciparum по сравнению с исходным фенильным соединением, а введение карборанов приводит к ее улучшению; однако эти изменения способствуют снижению метаболической стабильности. Переход клозо-карборанов в нидо-структуру приводит к снижению токсичности соединений, а также их активности по сравнению с клозо-аналогами.
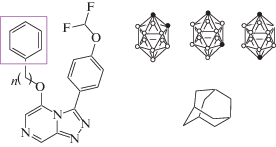
В работах [90, 91] синтезировано производное клозо-декаборатного аниона [В10Н9–O(CH2)4C(O)–His–OMe]2– с функциональной группой His-OMe, связанной с кластерным анионом бора спейсером O(CH2)4 (рис. 29б), и изучена in vitro противовирусная активность его натриевой соли в отношении вируса гриппа А/Moscow/01/2009(H1N1)pdm09. Показано, что соединение обладает противовирусной активностью при 10 и 5 мкг/мл и демонстрирует отсутствие цитотоксичности до 160 мкг/мл. Активность целевого соединения сопоставлена с адамантансодержащим аналогом – производным римантадина (рис. 29а) и показана его более высокая активность по сравнению с HCl · H–His–Rim (процент ингибирования равен 91.0 и 88.0% при концентрации 5 и 10 мкг/мл по сравнению с 47 и 62% для производного римантадина соответственно).
Рис. 29.
Аминокислотные производные адамантана (а) и клозо-декаборатного аниона (б), обладающие противовирусной активностью.

Создана квантово-механическая модель аниона [В10Н9–O(CH2)4C(O)–His–OMe]2–, проведен молекулярный докинг с моделью трансмембранной области белка M2 (PDB 2KIH) и на основании полученных данных предложен механизм действия целевого соединения [91].
Активное изучение новых методов введения функциональных групп в кластерные анионы бора расширяет возможности дизайна новых соединений, в том числе с физиологически активными свойствами [92, 93].
МОЛЕКУЛЯРНАЯ МОДЕЛЬ ИНГИБИТОРА ВИРОПОРИНОВ
В завершение обзора нам хотелось бы обсудить возможный механизм действия каркасных соединений, обладающих ингибирующим действием в отношении вирусов гриппа А. Исследования биологической активности производных адамантанового ряда возникли почти одновременно с быстрым развитием химии адамантана. Как уже было отмечено, первым соединением адамантана, нашедшим применение в медицинской химии, был амантадин (1-аминоадамантан), обладающий высокой противовирусной активностью в отношении вируса гриппа А. Возникшая впоследствии резистентность вирусов к препаратам адамантана побудила исследователей к изучению механизма действия, который связан с угнетением функции ион-селективного канала (протонного насоса) в оболочке вируса. Такие трансмембранные белки, способные к образованию проводящих каналов в оболочке самих вирусов или посттрасляционно в органеллах клетки хозяина, получили название виропорины (viroporins).
Виропорины – это группа белков, которые вовлечены во многие этапы репликационного цикла вируса, включая содействие высвобождению вирусных частиц из клеток. Эти белки также влияют на клеточные функции, включая систему клеточных визикул, транспорт гликопротеинов и проницаемость мембран. Присутствие виропоринов не является необходимым условием для репликации вирусов, но значительно увеличивает рост числа вирионов. Состоящие из 60–120 аминокислот виропорины имеют гидрофобный трансмембранный домен, который взаимодействует с липидным бислоем и нарушает его гексагональную упаковку. Некоторые виропорины содержат также другие мотивы, такие как основные аминокислотные остатки или домен, богатый ароматическими аминокислотами, которые придают белку способность взаимодействовать с межфазным липидным бислоем. Олигомеризация виропорина приводит к образованию гидрофильных пор на мембранах инфицированных вирусом клеток. Поскольку список известных виропоринов неуклонно растет, последние исследования сосредоточены на расшифровке действия виропоринов полиовируса 2B, альфавируса 6K, ВИЧ-1 Vpu, гепатита С p7 и вируса гриппа M2. Все эти белки могут усиливать проникновение ионов и малых молекул через мембраны в зависимости от градиента концентрации [41].
Типичным представителем тетрамерных ион-селективных виропоринов является протон-проводящий канал М2 вируса гриппа А. Препарат амантадин (Symmetrel, 1-адамантиламин гидрохлорид) был одним из первых изученных ингибиторов протонного канала М2 вируса гриппа А. Он использовался в клинической практике против вируса гриппа А с 1966 г. [94]. В 1985 г. было показано, что некоторые мутации в гидрофобной последовательности белка М2 могут приводить к резистентности вируса к амантадину и его аналогу римантадину (Flumantadine, 1-(1-адамантил)этиламин гидрохлорид). Эти данные позволили предположить, что белок М2 может быть мишенью для препаратов адамантанового ряда, а в 1992 г. эти предположения были подтверждены [95]. В 2020 г. авторы работы [96] выдвинули гипотезу в расчетах in silico, что амантадин блокирует ионный канал вируса SARS-CoV-2. Предполагается, что амантадин может взаимодействовать посредством водородных связей с аминокислотными остатками Phe26 и Ala22 белка виропорина Е.
Виропорины очень консервативны по своему аминокислотному составу. Мутации в этих белках могут привести к нежизнеспособности вирусов, поэтому они представляют интерес в качестве терапевтической мишени для создания новых ингибиторов функции виропоринов [97].
Известно, что виропорины ингибируются соединениями-блокаторами ионных каналов, такими как амантадин, римантадин, гексаметилен амилорид, и длинноцепочечными производными ацил-иминосахаров. На данный момент австралийские и китайские ученые проводят вторую стадию клинических испытаний синтетического препарата BIT225 ((N-[5-(1-Methyl-1H-pyrazol-4-yl)-napthalene-2-carbonyl]-guanidine)), который показал способность ингибировать функции виропоринов вируса гепатита С (HCV) и ВИЧ-1 [42] (рис. 30).
Рис. 30.
Строение некоторых известных ингибиторов виропоринов (амантадин, римантадин, гексаметилен амилорид (HMA) и длинно-цепочечные производные ацил-иминосахаров).

Ингибиторы (блокаторы) функции белка М2 вируса гриппа А, как правило, состоят из гидрофобной части молекулы (в препаратах амантадин и римантадин – адамантан), соединенной с полярной функциональной группой. В амантадине или римантадине заместитель представлен амино- или этиламиногруппой. Адамантильный остаток может быть заменен на другие гидрофобные группы, в том числе сопряженные и спиросопряженные мультициклические алканы, разветвленные ациклические алканы, производные терпенов и силаны [38, 39].
Более того, предложен новый класс положительно заряженных молекул – производные диазабициклооктана с постоянным зарядом +2, блокирующие диффузию протонов через ионный канал M2. Методами молекулярной динамики установлены состояния ионизации остатков гистидина в положении 37 при физиологических значениях рН. Заряженная молекула обладает преимуществом перед амантадином и римантадином, поскольку имеет заряд +2, что создает положительный электростатический потенциальный барьер для транспорта ионов водорода через ионный канал М2 в дополнение к стерическому барьеру [40].
Таким образом, не только нейтральные каркасные углеводороды и гетероциклы могут быть использованы для блокировки ион-проводящих каналов, но и заряженные полиэдры, такие как клозо-бороводородные анионы.
Обобщая данные противовирусной активности различных структур каркасных производных, можно сформировать молекулярную модель ингибитора виропоринов. Она представляет собой объемное каркасное ядро (нейтральное или несущее общий заряд) (рис. 31), которое выполняет функцию мембранотопного носителя функциональной группы. Эту роль могут выполнять карбоциклические алканы, такие как адамантан, норборнен или конденсированные и замещенные ароматические системы, такие как кластерные анионы бора. С другой стороны, каркасный фрагмент соединен с функциональной группой, которая способна к образованию нековалентного взаимодействия с поверхностью поры канала виропорина. В качестве источника таких групп могут выступать аминокислоты, пептиды и ряд других физиологически активных соединений.

ЗАКЛЮЧЕНИЕ
Рассмотрены производные каркасных структур – кластерных анионов бора и соединений адамантанового ряда. Обнаружено, что введение различных функциональных групп позволяет получать соединения-ингибиторы репликации вирусов, а также придает другие фармацевтические свойства целевым молекулам. Анализируя представленные данные, можно ожидать появления противовирусных свойств у производных декагидро-клозо-декаборатного аниона с боковыми аминокислотами, пептидами и некоторыми другими физиологически важными соединениями, так как в этом случае клозо-декаборатный анион действует как мембранотропный носитель, что согласуется с предложенной моделью ингибитора. Таким образом, обсуждаемые производные кластерных анионов бора можно рассматривать как новый класс объектов с многообещающей противовирусной активностью.
Список литературы
Scholz M., Hey-Hawkins E. // Chem. Rev. 2011. № 111. P. 7035.
Ďorďovič V., Tošner Z., Uchman M. et al. // Langmuir. 2016. V. 32. P. 6713. https://doi.org/10.1021/acs.langmuir.6b01995
Greenwood N.N., Earnshaw A. Chemistry of the Elements, 2nd ed., Butterworth-Heinemann, 1997.
Grimes R.N. Carboranes. London: Academic Press, 2016. 1058 p. https://doi.org/10.1016/B978-0-12-801894-1.09989-3
Boron Science: New Technologies and Applications / Ed. Hosmane N.S. CRC Press, 2012.
Boron-Based Compounds: Potential and Emerging Applications in Medicine / Eds. Hey-Hawkins E., Viñas Teixidor C. John Wiley & Sons Ltd., 2018. 470 p. https://doi.org/10.1002/9781119275602
Zhizhin K.Yu., Zhdanov A.P., Kuznetsov N.T. // Russ. J. Inorg. Chem. 2010. V. 55. № 14. P. 2089. https://doi.org/10.1134/S0036023610140019
Sivaev I.B., Prikaznov A.V., Naoufal D. // Coll. Czech. Chem. Commun. 2010. V. 75. № 11. P. 1149. https://doi.org/10.1135/cccc2010054
Sivaev I.B. // Russ. J. Inorg. Chem. 2019. V. 64. P. 955. https://doi.org/10.1134/S003602361908014X
Sivaev I.B., Bregadze V.I. Polyhedral Boron Hydrides in Use: Current Status and Perspectives. Hauppauge: Nova Science Publishers, 2009. 85 p.
Shmal’ko A.V., Sivaev I.B. // Russ. J. Inorg. Chem. 2019. V. 64. P. 1726. https://doi.org/10.1134/S0036023619140067
Сиваев И.Б. // Журн. неорган. химии. 2021. Т. 66. № 9. С. 1192. https://doi.org/10.31857/S0044457X21090154
Bruce King R. // Chem. Rev. 2001. V. 101. № 5. P. 1119. https://doi.org/10.1021/cr000442t
Chen Z., King R.B. // Chem. Rev. 2005. V. 105. P. 3613. https://doi.org/10.1021/cr0300892
Kazimierczuk Z., Gorska A., Switaj T., Lasek W. // Bioorg. Med. Chem. Lett. 2001. V. 11. P. 1197.
Spilovska K., Zemek F., Korabecny J. et al. // Curr. Med. Chem. 2016. V. 23. P. 3245. https://doi.org/10.2174/0929867323666160525114026
Igumnova N.D., Lemina E., Bitiukova I.I. et al. // Farmakol. Toksikol. V. 51. P. 38.
Su-Mi Kim, Jong-Hyeon Park, Kwang-Nyeong Lee et al. // Antivir. Res. V. 96. P. 213. https://doi.org/10.1016/j.antiviral.2012.09.009
Agnew-Francisa K.A., Williams C.M. // Synth. Catal. Adv. 2016. V. 358. P. 675.
Wassel M.S., Wael M., Gamal E. et al. // J. Appl. Vet. Sci. 2020. V. 5. № 4. P. 37.
Ao D., Guo H-C., Sun S-Q. et al. // Plos ONE. 2015. V. 10. № 5. P. e0125828.
Андронова В.Л. // Антибиотики и химиотерапия. 1996. Т. 41. № 718. С. 26.
Лаврова Л.H. // Журн. орган. химии. 1974. Bып. 4. С. 761.
Davies W.L., Grunert R.R., Haff R.F. et al. // Science. 1964. V. 144. P. 862.
Spilovska K., Zemek F., Korabecny J. et al. // Curr. Med. Chem. 2016. V. 23. № 29. P. 3245. https://doi.org/10.2174/0929867323666160525114026
Zarubaev V.V., Golod E.L., Anfimov P.M. et al. // Bioorg. Med. Chem. 2010. V. 18. № 2. P. 839.
Анфимов П.М., Киселев О.И., Зарубаев В.В. Молекулярная медицина. Москва, 2010. 124 с.
Shibnev V.A., Garaev T.M., Finogenova M.P. et al. // Bull. Exp. Biol. Med. 2012. V. 153. № 2. P. 233.
Shibnev V.A., Deryabin P.G., Garaev T.M. et al. // Russ. J. Bioorg. Chem. 2017. V. 43. № 5. P. 517.
Дерябин П.Г., Гараев Т.М., Финогенова М.П., Одноворов А.И. // Вопросы вирусологии. 2019. Т. 64. № 6. С. 268. https://doi.org/10.36233/0507-4088-2019-64-6-268-273
Garaev T.M., Odnovorov A.I., Lashkov A.A. et al. // Adv. Pharm. Bull. 2021. https://doi.org/10.34172/apb.2021.079
Щелканов М.Ю., Шибнев В.А., Финогенова М.П. и др. // Вопр. вирусол. 2014. Т. 59. № 2. С. 37.
Гараев Т.М., Одноворов А.И., Кириллова Е.С. и др. // Вопр. вирусол. 2020. Т. 65. № 1. P. 16. https://doi.org/10.36233/0507-4088-2020-65-1-16-20
Stankova I., Chuchkov K., Chayrov R. et al. // Int. J. Pept. Res. Ther. 2020. V. 26. P. 1781. https://doi.org/10.1007/s10989-019-09983-4
Aldrich P.E., Hermann E.C., Walter E. // J. Med. Chem. 1971. V. 14. № 6. P. 535.
Tsuzuki N., Hama T., Kawada M. // J. Pharm. Sci. 1994. V. 83. № 4. P. 481.
Литвинов В.П. // Химия гетероцикл. соединений. 2002. Т. 50. № 1. С. 12.
Wang J., Ma C., Jo H. et al. // J. Med. Chem. 2013. V. 56. № 7. P. 2804.
Wang J., Ma C., Fiorin G. et al. // J. Am. Chem. Soc. 2011. V. 133. P. 12834.
Vorobjev Y.N. // Molecular Biology. 2020. V. 54. № 2. P. 281. https://doi.org/10.1134/S0026893320020168
Gonzalez M.E., Carrasco L. // FEBS Lett. 2003. V. 552. P. 28.
Khoury G., Ewart G., Luscombe C. et al. // AIDS Res. Ther. 2016. V. 13. P. 7. https://doi.org/10.1186/s12981-016-0093-z
Corrias A., Ennas G., Musinu A. et al. // Chem. Mater. 1993. V. 5. P. 1722.
Fayaz Ali, Narayan S. Hosmane, Yinghuai Zhu et al. // Molecules. 2020. V. 25. № 4. P. 828.
Sivaev I.B., Bregadze V.I. // Eur. J. Inorg. Chem. 2009. P. 1433.
Goswami L.N., Ma L., Chakravarty Sh. et al. // Inorg. Chem. 2013. V. 52. P. 1694. https://doi.org/10.1021/ic3017613
Fink K., Uchman M. // Coord. Chem. Rev. 2021. V. 431. P. 213684. https://doi.org/10.1016/j.ccr.2020.213684
Fanfrlík J., Lepšík M., Horinek D. et al. // ChemPhysChem. 2006. V. 7. P. 1100. https://doi.org/10.1002/cphc.200500648
Buzharevski A., Paskaš S., Sárosi M.B. et al. // Sci. Rep. 2020. V. 10. P. 4827. https://doi.org/10.1038/s41598-020-59059-3
Hawthorne M. F. // Angew. Chem. Int. Ed. Engl. 1993. V. 32. P. 950.
Kugler M., Nekvinda J., Holub J. et al. // ChemBioChem. 2021. https://doi.org/10.1002/cbic.202100121
Grüner B., Brynda J., Das V. et al. // ChemPlusChem. 2021. V. 86. P. 352. https://doi.org/10.1002/cplu.202000723
Alberti D., Michelotti A., Lanfranco A. et al. // Sci. Rep. 2020. V. 10. Art. 19274. https://doi.org/10.1038/s41598-020-76370-1
Nelyubin A.V., Klyukin I.N., Zhdanov A.P. et al. // Russ. J. Inorg. Chem. 2019. V. 64. P. 1750. https://doi.org/10.1134/S0036023619140043
Авдеева В.В., Малинина Е.А., Жижин К.Ю. и др. // Журн. структур. химии. 2019. Т. 60. № 5. С. 726.
Hawthorne M.F., Shelly K., Li F. // Chem. Commun. 2002. P. 547.
Авдеева В.В., Малинина Е.А., Кузнецов Н.Т. // Журн. неорган. химии. 2020. Т. 65. № 3. С. 334. [Malinina E.A., Avdeeva V.V., Korolenko S.E. et al. // Russ. J. Inorg. Chem. 2020. V. 65. P. 335. https://doi.org/10.1134/S003602362003002X]
Yasuyuki Endo, Tomohiro Yoshimi, Chisato Miyaura // Pure Appl. Chem. 2003. V. 75. № 9. P. 1197.
Thiruinamagal B.T.S., Zhao Xiaobin B., Bandyopadhyaya Achintya K. et al. // Bioconjugate Chem. 2006. V. 17. № 5. P. 1141. https://doi.org/10.1021/bc060075d
Goto T., Ohta K., Suzuki T. et al. // Bioorg. Med. Chem. 2005. V. 13. № 23. P. 6414. https://doi.org/10.1016/j.bmc.2005.06.061
Endo Yasuyuki, Iijima Toru, Yamakoshi Yuko et al. // Bioorg. Med. Chem. Lett. 1999. V. 9. № 23. P. 3313. https://doi.org/10.1016/S0960-894X(99)00596-X
Zargham Emilia O., Mason Christian A., Lee Mark W.Jr. // Int. J. Cancer Clin. Res. 2019. V. 6. P. 110. https://doi.org/10.23937/2378-3419/1410110
Endo Yasuyuki, Iijima Toru, Kagechika Hiroyuki et al. // Chem. Pharm. Bull. 1999. V. 47. № 4. P. 585. https://doi.org/10.1248/cpb.47.585
Iijima T., Endo Y., Tsuji M. et al. // Chem. Pharm. Bull. (Tokyo). 1999. V. 47. № 3. P. 398. https://doi.org/10.1248/cpb.47.398
Bednarska K., Olejniczak A.B., Wojtczak B.A. et al. // ChemMedChem. V. 5. № 5. P. 749. https://doi.org/10.1002/cmdc.201000075
Leśnikowski Z.J., Schinazi R.F. // J. Org. Chem. 1993. V. 58. № 24. P. 6531. https://doi.org/Leśnikowski.1021/jo00076a001
Lesnikowski Z.J., Shi J., Schinazi R.F. // J. Organomet. Chem. 1999. V. 581. P. 156. https://doi.org/10.1016/S0022-328X(99)00129-1
Tetsushi Totani, Katsutoshi Aono, Kiyoe Yamamoto, Katsuya Tawara // J. Med. Chem. 1981. V. 24. № 12. P. 1492. https://doi.org/10.1021/jm00144a024
Oros G., Ujvary I., Nachman R.J. // Amino Acids. 1999. V. 17. P. 357.
Swietnicki W., Goldeman W., Psurski M. et al. // Int. J. Mol. Sci. 2021. V. 22. № 13. P. 6762. https://doi.org/10.3390/ijms22136762
Zheng Y., Liu W., Chen Y. et al. // Organometallics. 2017. V. 36. P. 3484. https://doi.org/10.1021/acs.organomet.7b00426
Różycka D., Korycka-Machała M., Żaczek A. et al. // Pharmaceuticals. 2020. V. 12. № 12. P. 465. https://doi.org/10.3390/ph13120465
Popova T., Zaulet A., Teixidor F. et al. // J. Organomet. Chem. 2013. V. 747. P. 229. https://doi.org/10.1016/j.jorganchem.2013.07.006
Adamska A., Rumijowska-Galewicz A., Ruszczynska A. et al. // Eur. J. Med. Chem. 2016. V. 121. P. 71.
Sun Y.J., Zhang J.L., Zhang Y.B. et al. // Chemistry. 2018. V. 24. P. 10364.
Różycka D., Leśnikowski Z., Olejniczaka A. // J. Organomet. Chem. 2019. V. 881. P. 19. https://doi.org/10.1016/j.jorganchem.2018.11.037
Kvasničková E., Masák J., Čejka J. et al. // J. Organomet. Chem. 2017. V. 827. P. 23. https://doi.org/10.1016/j.jorganchem.2016.10.037
Białek-Pietrasa M., Olejniczak A.B., Paradowska E. et al. // J. Organomet. Chem. 2015. V. 798. P. 99.
Białek-Pietrasa M., Olejniczak A.B., Paradowska E. et al. // J. Organomet. Chem. 2018. V. 865. P. 166.
Kosenko I., Ananyev I., Druzina A. et al. // J. Organomet. Chem. 2017. V. 849–850. P. 142.
Laskova J., Kozlova A., Białek-Pietras M. et al. // J. Organomet. Chem. 2016. V. 807. P. 29.
Cígler P., Kozísek M., Rezácová P. et al. // Proc. Natl. Acad. Sci. USA. 2005. V. 102. № 43. P. 15394. https://doi.org/10.1073/pnas.0507577102
Kožíšek M., Cígler P., Lepšík M. et al. // J. Med. Chem. 2008. V. 51. № 15. P. 4839. https://doi.org/10.1021/jm8002334
Rezácová P., Pokorná J., Brynda J. et al. // J. Med. Chem. 2009. V. 52. № 22. P. 7132. https://doi.org/10.1021/jm9011388
Adamska A., Olejniczak A.B., Zwoliński K., et al. // Acta Pol. Pharm. 2012. V. 69. № 6. P. 1218.
Сиваев И.Б. // Журн. неорган. химии. 2020. Т. 65. № 12. С. 1643. https://doi.org/10.31857/S0044457X20120168
Endo Yasuyuki, Iijima Toru, Yamakoshi Yuko et al. // J. Med. Chem. 1999. V. 42. № 9. P. 1501. https://doi.org/10.1021/jm9900725
Kai Li, Yang Wang, Gyongseon Yang et al. // ACS Infect. Dis. 2015. V. 1. № 5. https://doi.org/10.1021/acsinfecdis.5b00026
Tse E.G., Houston S.D., Williams C.M. et al. // J. Med. Chem. 2020. V. 63. P. 11585. https://doi.org/10.1021/acs.jmedchem.0c00746
Гараев Т.М., Гребенникова Т.В., Авдеева В.В. и др. Аминокислотное производное декагидро-клозо-декаборатного аниона и его противовирусная активность в отношении вируса гриппа А. Пат. РФ RU 2 749 006 С1 приоритет от 27.11.2020 (Опубликовано 02.06.2021).
Гараев Т.М., Авдеева В.В., Жданов А.П., и др. // XXVIII Междунар. Чугаевская конф. коорд. хим. Туапсе. 2021. С. 364.
Нелюбин А.В., Селиванов Н.А., Быков А.Ю. и др. // Журн. неорган. химии. 2020. Т. 65. № 6. С. 719. https://doi.org/10.31857/S0044457X20060136
Нелюбин А.В., Клюкин И.Н., Жданов А.П. и др. // Журн. неорган. химии. 2021. Т. 66. № 2. С. 134. https://doi.org/10.31857/S0044457X21020136
Thomaston J.L., Polizzi N.F., Konstantinidi A. et al. // J. Am. Chem. Soc. 2018. V. 140. № 45. P. 15219.
Wang C., Takeuchi K., Pinto L.H. et al. // J. Virol. 1993. V. 67. № 9. P. 5585.
Abreu G.E.A., Aguilar M.E.H., Covarrubias D.H., Durán F.R. // Med. Hypotheses. 2020. V. 140. P. 109755. https://doi.org/10.1016/j.mehy.2020.109755
Griffin S.D., Beales L.P., Clarke D.S. et al. // FEBS Lett. 2003. V. 535. P. 34.
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии