

Журнал неорганической химии, 2022, T. 67, № 8, стр. 1095-1107
Комплексообразование в процессе сорбции ионов палладия(II) химически модифицированными кремнеземами
Т. М. Буслаева a, *, Г. В. Эрлих b, Е. В. Волчкова a, П. Г. Мингалев b, Н. С. Панина c
a МИРЭА – Российский технологический университет (Институт тонких химических технологий
им. М.В. Ломоносова)
119571 Москва, пр-т Вернадского, 86, Россия
b Московский государственный университет им. М.В. Ломоносова, Химический факультет
119991 Москва, Ленинские горы, 1, Россия
c Санкт-Петербургский государственный технологический институт (Технический университет)
190013 Санкт-Петербург, Московский пр-т, 26, Россия
* E-mail: buslaevatm@mail.ru
Поступила в редакцию 19.01.2022
После доработки 31.01.2022
Принята к публикации 03.02.2022
- EDN: QVWRCK
- DOI: 10.31857/S0044457X22080050
Аннотация
Исследовано комплексообразование в процессе сорбции ионов палладия(II) из хлоридных сред химически модифицированными кремнеземами. Указанные сорбенты, благодаря использованию для их синтеза мезопористых кремнеземов со средним диаметром пор >10 нм, не изменяют своего объема в водных и органических средах и обладают высокими массообменными характеристиками. Получены зависимости сорбции ионов Pd(II) из хлоридных растворов в статических условиях в зависимости от времени, концентрации HCl, иона Cl–; построены изотермы сорбции. На основании экспериментальных данных сделан вывод о координационном механизме сорбции, который доказан совокупностью спектральных методов (ИК-, УФ-Vis- и рентгеновская фотоэлектронная спектроскопия). Выполнено моделирование структуры серосодержащих сорбентов и их комплексов с палладием квантово-химическим методом DFT M06 в базисе def2tzvp в газовой фазе и с учетом растворителя H2O. Проведенные расчеты позволили обосновать характер зависимости сорбции от концентрации кислоты и подтвердить возможность образования в фазе кремнезема, модифицированного тиосалициловой и меркаптоуксусной кислотами, комплексов палладия(II) с бидентатной координацией лигандов.
ВВЕДЕНИЕ
Сорбция в настоящее время представляет собой один из наиболее перспективных и востребованных методов выделения и разделения ионов палладия и других платиновых металлов из хлоридных растворов. К сожалению, заранее не удается учесть и предусмотреть все факторы, влияющие на сорбцию ионов различных металлов (состав раствора, солевой фон, размер пор сорбента и т.п.). Поэтому возникает необходимость в проведении многочисленных экспериментов, что, в свою очередь, порождает поток публикаций, число которых в последние 5–10 лет неизмеримо выросло, особенно в области аналитической химии с целью подготовки проб сложного состава: руды, горные породы, продукты аффинажного производства. Данному обстоятельству немало способствуют неоспоримые преимущества сорбции перед прочими методами, в частности, высокая скорость достижения равновесия и селективность [1–4]. Среди работ по сорбции ионов благородных металлов подавляющее большинство относится к сорбции ионов Pd(II) из солянокислых либо водных хлоридных растворов. В качестве сорбентов, обеспечивающих относительно селективное извлечение указанного иона, превалируют азот- и серосодержащие сорбенты различных типов [5–8]. Стоит подчеркнуть, что матрицей сорбента, как правило, служат синтетические органические полимеры, хотя встречаются и природные полимерные сорбенты, например, хитозан – линейный полисахарид, содержащий аминогруппы. Наличие функциональных гидроксильных и аминогрупп предоставляет широкие возможности для модифицирования хитозана и получения сорбентов с требуемыми механическими и структурными характеристиками [9, 10]. В [11] обсуждаются результаты сорбции ионов платиновых металлов на полученных из водорослей биомассах, модифицированных различными полиэтилениминами. Указано, что десорбция Pd(II) возможна растворами тиомочевины. Никаких принципиальных отличий от сорбции на аналогичных сорбентах на основе синтетических полимеров не обнаружено [9–11]. Рассмотрены возможности полимерных тиоэфиров в селективном сорбционном концентрировании для аналитических целей, а также ускорение сорбции за счет введения в тиоэфиры азотсодержащих групп [12]. Выявлена зависимость селективности указанных сорбентов от природы заместителя при атоме азота. Полимерные волокнистые пористые сорбенты на основе металлорганических каркасов MOFs, функционализированные четвертичными аммониевыми основаниями, эффективны для извлечения ионов палладия(II) из сильнокислых растворов и/или цианидных растворов [13, 14]. Заметим, что во многих проанализированных нами работах акцент смещен в сторону синтеза различных полимеров и сополимеров, включая кремнийорганические, которые обладают сорбционной способностью по отношению к ионам цветных, тяжелых и благородных металлов, причем преимущественно к ионам Pd(II) [8, 15, 16]. Однако доступность этих сорбентов, их селективность и возможность реализации процессов десорбции не обсуждаются.
Среди сорбентов для выделения ионов палладия(II) из хлоридных растворов особое место занимают химически модифицированные кремнеземы (ХМК), которые, несмотря на относительно невысокую сорбционную емкость по сравнению с органополимерными сорбентами, выгодно отличаются от них высокой скоростью установления сорбционно-десорбционного равновесия. Еще в 1980-е годы кремнезем с привитыми моноаминными группами SiO2–O–Si(CH2)3NH2, получаемый с использованием выпускаемого промышленностью модификатора γ-аминопропилтриэтоксисилана (C2H5O)3Si(CH2)3NH2, с успехом применяли для извлечения ионов платиновых металлов, в первую очередь Pd(II) [1]. Однако гораздо больший интерес, особенно в последние годы, вызывают кремнеземы, модифицированные серосодержащими молекулами. Силикагель, покрытый оксидом графена и модифицированный меркаптогруппами SiO2@GO–SH, успешно используется при переработке вторичного сырья. В процессе хемосорбции ионов палладия(II) идет их восстановление до металла [17].
Синтезированы кремнеземы, модифицированные меркаптопропилом, меркаптофенилом, дипропилдисульфидом, тиадиазолтиолом, дитиокарбаматом, производными тиомочевины, которые количественно извлекают ионы Pd(II) из 0.5–4 М растворов соляной кислоты [18]. При этом происходит их отделение от ионов цветных, щелочноземельных и щелочных металлов, а также от других платиновых металлов. Показано [18], что селективность выделения целевого иона повышается при переходе от тионовой серы к тиоловой. Установлено, что преобладающим механизмом сорбции палладия(II) является комплексообразование с функциональными группами сорбентов платины(IV) – ионный обмен [11, 19, 20]. Возрастание степени сульфоэтилирования аминополимерной матрицы способствует уменьшению сорбции платины(IV) и, как следствие, увеличению селективности сорбции палладия(II) относительно данного иона. Наиболее значительным этот эффект является для сорбентов на основе полиаллиламина [20]. Вывод о механизме сорбции сделан в перечисленных выше работах без привлечения каких-либо методов исследования и, скорее, по привычке, без ожидания иного результата.
В настоящей работе обобщены данные по сорбции Pd(II) из солянокислых хлоридных растворов селективными кремнеземами с привитыми серосодержащими молекулами, и ее целью является выявление особенностей комплексообразования палладия(II) в процессе сорбции.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реагентами служили PdCl2, синтезированный по методике [21], HCl, NaCl, NH3 · H2O (все марки “х. ч.”) производства ООО “ТД “Химмед”, 3-глицидилоксипропилтриметоксисилан, диизопропилэтиламин, меркаптоэтанол, меркаптоуксусная и тиосалициловая кислоты производства компании Sigma-Aldrich. Для синтеза сорбента применяли силикагель Davisil Grade 62 (W. R. Grace&Co., США), фракция 0.07–0.2 мм (60–200 меш), удельная поверхность 327 м2/г, размер пор 13.7 нм. Органические растворители – толуол, этанол, ацетонитрил (ООО “ТД “Химмед”) – использовали без предварительной перегонки.
Синтез серосодержащих сорбентов. Раствор эквимолярных количеств сероорганического соединения (меркаптоэтанола, меркаптоуксусной кислоты, тиосалициловой кислоты), 3-глицидилоксипропил-триметоксисилана и диизопропилэтиламина в 100 мл толуола перемешивали при комнатной температуре в течение 2 ч. Затем к смеси добавляли 20 г кремнезема и полученную суспензию перемешивали механической мешалкой при нагревании на кипящей водяной бане в течение 8 ч. Далее на кипящей водяной бане в токе воздуха медленно удаляли толуол до сыпучего состояния сорбента. Сорбент переносили на фильтр, промывали этанолом до исчезновения характерного запаха сероорганического соединения и высушивали на фильтре. Синтез сорбента с привитым меркаптоэтанолом описан также в [22], а синтез кремнезема, модифицированного тиосалициловой кислотой, запатентован [23]. Характеристики синтезированных сорбентов приведены в табл. 1.
Таблица 1.
Характеристики синтезированных сорбентов
| Функциональная группа | Условное обозначение | Результаты химического анализа, % | Количество привитых молекул | |||
|---|---|---|---|---|---|---|
| S | H | C | ммоль/г | групп/нм2 | ||
 |
МЭ (L1) | 3.7 | 3.67 | 18.7 | 1.16 | 2.71 |
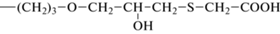 |
МУК (L2) | 1.46 | 1.55 | 7.60 | 0.46 | 1.04 |
 |
ТСК (L3) | 1.79 | 1.96 | 11.8 | 0.56 | 1.28 |
Методика эксперимента. Сорбцию Pd(II) проводили в статических условиях из солянокислых растворов (0.1–6 М HCl; $C_{{{\text{Cl}}}}^{--}$ = 10–100 г/л) при комнатной температуре. Объем растворов в каждом опыте составлял 10–15 мл, масса сорбента – 30–50 мг, СPd = 5 × 10–3 моль/л. Изотермы сорбции Pd(II) строили для растворов с исходной концентрацией металла от 1 × 10–4 до 1 × 10–2 моль/л в растворе, масса сорбента 50 ± 0.3 мг, объем раствора 10 мл. Растворы после сорбции отделяли от сорбента фильтрацией и центрифугированием (с использованием медицинской лабораторной центрифуги ЦЛн-16 с ротором, Россия) и определяли содержание палладия в растворе. Количество сорбированного металла находили по разности содержания его в растворе до и после сорбции. Образцы для исследования спектральными методами (ЭСП, ИК, РФЭС) готовили обработкой сорбентов растворами палладия(II) в 1 М HCl, концентрация металла в растворе $С_{{{\text{Pd}}}}^{{{\text{исх}}}}$ = 1 × × 10–2 моль/л, что, согласно изотермам сорбции, соответствует максимальному насыщению сорбентов ионами металла.
Методы. Структурные характеристики сорбентов определяли методом низкотемпературной адсорбции азота на автоматическом анализаторе ASAP 2020 (Micromeritics, США). Элементный анализ сорбентов на содержание углерода, водорода и азота проводили на анализаторе CHN-2400 Perkin Elmer. Содержание ионов металла в растворе определяли методом масс-спектрометрии с индуктивно связанной плазмой на масс-спектрометре Agilent 7900 (Agilent Technologies, США). Обработку результатов анализа проводили с помощью программного обеспечения Agilent Mass Hunter (Agilent Technologies, США). ИК-спектры образцов получали на ИК-фурье-спектрометре Thermo Scientific Nicolet IR200 методом нарушенного полного внутреннего отражения на приставке с кристаллом ZnSe. Спектры регистрировали в диапазоне 4000–650 см–1 (область прозрачности кристаллического ZnSe). Электронные спектры поглощения растворов записывали на спектрофотометре Specord UV-Visible “Heλios α” (США) в диапазоне длин волн 200–800 нм в кварцевых кюветах с толщиной поглощающего слоя 1 см или на кварцевой подложке – в случае сорбентов, насыщенных ионами металлов, приготовленных в виде суспензий в вазелиновом масле. Спектры диффузного отражения твердых образцов регистрировали на спектрометре Specord М-40. Спектры диффузного отражения приводили к виду, соответствующему спектрам поглощения, используя функцию Гуревича–Кубелки–Мунка, в координатах F(R) = (1 – R2)/(2R) [24]. Состав поверхности сорбентов исследовали методом рентгеновской фотоэлектронной спектроскопии на спектрометре Theta Probe фирмы Thermo Fisher Scientific (Великобритания) c использованием монохроматического AlKα-излучения. Фотоэлектронные спектры регистрировали в режиме постоянного абсолютного разрешения по энергии с шагом 0.1 эВ. Измерения выполняли при давлении в камере анализа ~5 × 10–7 Па. Анализируемые спектры аппроксимировали гауссовым профилем или их суммой, а фон, обусловленный вторичными электронами и фотоэлектронами, испытавшими потери энергии, – прямой линией. Энергетическую шкалу спектрометра калибровали по стандартной методике с использованием следующих значений энергии связи: Cu 2p3/2 – 932.7 эВ, Ag 3d5/2 – 368.3 эВ и Au 4f7/2 – 84.0 эВ. Количественный анализ проводили на основе коэффициентов элементной чувствительности, значения которых входят в программное обеспечение к прибору. Поверхностную зарядку учитывали по форме пика в спектре C1s (284.8 эВ). Заряд компенсировали с использованием низкоэнергетической электронной пушки. Энергия электронов составляла ~0.5 эВ. Электронную и геометрическую структуру ряда серосодержащих сорбентов и их комплексов с металлами платиновой группы исследовали ориентированным на расчеты энтальпий реакций с участием переходных металлов квантово-химическим методом DFT M06 [25] c использованием базисов def2szvp и def2tzvp [26] по программному комплексу GAUSSIAN'16 [27]. Отметим, что в программном комплексе [27] они вычисляются с помощью следующих соотношений:
где Eполн – полные энергии систем; ZPE – энергии нулевого колебательного уровня;
(3)
$\Delta H' = {{Е}_{{{\text{колеб}}}}} + {{Е}_{{{\text{вращ}}}}} + {{Е}_{{{\text{транс}}}}} + RT.$Колебательный вклад в термодинамические функции (включая ZPE) получают из расчетов частот нормальных колебаний соединений в гармоническом приближении. Энергии сольватации соединений рассчитывали в рамках модели поляризуемого континуума [28, 29]. Визуализацию молекулярных структур и нормальных колебаний обеспечивала графическая программа ChemСraft [30].
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
В качестве исходных сероорганических соединений нами выбраны три: меркаптоэтанол (МЭ) HSCH2CH2OH, меркаптоуксусная кислота (МУК) HSCH2COOH, известная также под названием тиогликолевая кислота, и представитель класса серосодержащих реагентов – производных тиофенола с электроноакцепторными заместителями в ароматическом ядре общей формулы HSC6H4X – тиосалициловая кислота (ТСК) о‑HSC6H4СООН. Меркаптоэтанол – доступный и дешевый реагент, интерес к которому обусловлен тем, что при его прививке к поверхности кремнезема по описанной выше методике на поверхности образуются структуры, подобные тиакраун-эфирам [22]. Меркаптоуксусная кислота – доступный и дешевый продукт, который получают в промышленности реакцией хлоруксусной кислоты с гидросульфидом натрия. От меркаптоэтанола ее отличает наличие электроноакцепторной карбоксильной группы, что должно способствовать повышению селективности связывания ионов палладия(II).
Для синтеза серосодержащих сорбентов использовали метод иммобилизации с предварительным синтезом серосодержащего кремнийорганического модификатора, аналогичный использованному в некоторых патентах Р. Айзатта [31, 32], схема которого приведена ниже:

На первом этапе исследований были получены зависимости сорбции ионов Pd(II) на синтезированных химически модифицированных кремнеземах от времени, концентрации кислоты, хлорид-иона, протона, а также построены изотермы сорбции. Установлено, что время достижения постоянных значений сорбции (сорбционного равновесия) не превышает 2 мин, что типично для сорбентов на основе модифицированных мезопористых кремнеземов. Время сорбции практически не зависит от природы функциональной группы. Во всех дальнейших экспериментах время контакта фаз составляло 5 мин.
На рис. 1 приведены зависимости сорбции ионов Pd(II) на синтезированных сорбентах, на основании которых можно сделать вывод, что рост концентрации кислоты в растворах приводит к снижению сорбции ионов палладия(II) для всех сорбентов независимо от характера привитой группы. Данные рис. 1 отражают брутто-эффект от одновременного увеличения концентрации ионов H+ и Cl–. Если же разделить эффекты и рассматривать зависимость сорбции от концентрации хлорид-ионов при постоянной кислотности раствора и зависимость сорбции от концентрации Н+ при постоянном солевом фоне, то окажется, что она зависит только от концентрации протона, а влияние солевого фона незначительно. Сорбция палладия(II) на сорбентах МЭ и ТСК практически не зависит от изменения концентрации ионов Н+ в интервале 0.01–2 моль/л и концентрации ионов Cl– в интервале 10–100 г/л. Это означает, что степень извлечения палладия на указанных сорбентах устойчива к возможным изменениям состава раствора, что имеет важное практическое значение.
Рис. 1.
Зависимость сорбции Pd(II) на серосодержащих сорбентах от концентрации HCl в растворах: СPd = 5 × 10–3 моль/л; mсорб = 0.05 г; Vр-р = 10 мл; время контакта фаз 5 мин.

На рис. 2 приведены изотермы сорбции хлоридного комплекса Pd(II) на серосодержащих ХМК. Очевидно, что в случае сорбента МЭ значение сорбционной емкости по палладию (0.61 ммоль/г) приблизительно в два раза меньше количества привитых лигандов (1.16 ммоль/г). Следовательно, один ион палладия(II) реагирует с двумя привитыми серосодержащими лигандами. Аналогичный результат получен и для сорбента МУК, хотя значение его сорбционной емкости примерно в три раза ниже (табл. 1).

Безусловно, нельзя делать вывод о механизме процесса образования комплексов в фазе сорбента только на основании стехиометрии взаимодействия иона палладия(II) с привитыми группами сорбентов. Поэтому для выяснения вопроса о механизме комплексообразования мы привлекли спектральные методы исследования – инфракрасную (ИК), электронную (UV-Vis) и рентгеновскую фотоэлектронную (РФЭС) спектроскопию.
В ИК-спектрах всех образцов присутствуют интенсивные полосы, принадлежащие матрице, а именно: 1106 см–1 (асимметричные валентные колебания Si–O–Si), 802 см–1 (симметричные валентные колебания Si–O–Si), 959 см–1 (валентные колебания силанольных групп Si–O–H и Si–O–). Модифицирование поверхности SiO2 приводит к появлению в спектрах образцов новых полос, свидетельствующих об изменении химического состава поверхности. Но их небольшая интенсивность вызвана, скорее всего, тем, что содержание привитого модификатора относительно невысоко по сравнению с содержанием SiO2. Интенсивное поглощение силанольных групп кремнезема практически экранирует поглощение, обусловленное колебаниями связей C–S–C, которое характеризуется слабой интенсивностью. Наиболее информативными оказались ИК-спектры сорбента МУК до и после сорбции иона Pd(II) (табл. 2).
Таблица 2.
Отнесение полос в ИК-спектрах сорбента МУК-D до и после сорбции палладия(II)
| Полосы поглощения, см–1 | Отнесение | |
|---|---|---|
| до сорбции | после сорбции | |
| 2941 | 2941 | Валентные колебания С–Н |
| 2876 | 2876 | Валентные колебания С–Н |
| 1729 | 1726 | Валентные колебания С=О |
| 1580 | 1632 | Асимметричные валентные колебания СОО– |
| 1371 | 1407 | Симметричные валентные колебания СОО– |
| 693–697 | Валентные колебания C–S–C | |
О связывании модификатора с поверхностью SiO2 можно судить на основании присутствия в ИК-спектрах исследованных образцов валентных колебаний связей С–Н, С=О и СОО–. Смещение полосы асимметричных валентных колебаний νas(COO–) в образце после сорбции указывает на образование связи между карбоксильной группой и ионом металла, а разница между частотами асимметричных и симметричных валентных колебаний координированной карбоксильной группы Δ = νas(COO–) – νs(COO–) = 225 см–1 соответствует монодентатному связыванию карбоксильной группы с ионом металла [33]. При таком характере комплексообразования становится понятным заметное увеличение интенсивности полосы колебаний ν(С=О) при 1726 см–1. Возможное участие в связывании палладия карбоксильной группы привитой меркаптоуксусной кислоты (наряду с атомом серы) объясняет более резкое уменьшение сорбции на сорбенте МУК с ростом кислотности раствора по сравнению с сорбентом МЭ.
Рассмотрим ЭСП сорбентов, насыщенных ионами палладия(II) и окрашенных в оранжевый цвет разных оттенков (рис. 3). Так, в ЭСП фазы сорбента МЭ + Pd(II) наблюдаются полосы поглощения при 244 (D = 2.5), 303 (D = 2.4), 353 (D = 2.3) и 399 нм (D = 2.1) (для сравнения: в ЭСП исходного сорбента присутствуют полосы поглощения при 208 (D = 1.8) и 224 нм (D = 1.7), изгиб при 247 нм (D = 1.6), а начиная с 290 нм поглощение отсутствует. В ЭСП фазы сорбента МУК + + Pd(II), т.е. насыщенного ионами Pd(II), наблюдаются полосы поглощения при 206 (D = 2.4), 244 (D = 2.1) и 306 нм (D = 2.0), 327 пл. (D = 1.9), 360–370 пл. (D = 1.6–1.5), 430–440 пл. нм (D = 1.2–1.1) (для сравнения: в ЭСП исходного сорбента можно выделить лишь слабовыраженную полосу поглощения при 204 нм (D = 0.972). В случае фазы ТСК + Pd(II) в спектре присутствуют полосы при 209 (D = 2.3), 226 (D = 2.2), 260 нм (D = 1.9), 328 пл. (D = 1.7–1.6) и 443 пл. нм (D = 0.9) (для сравнения: в ЭСП сорбента ТСК наблюдаются полосы поглощения при 264 (D = 2.1) и 314 нм (D = 1.8), а начиная с 370 нм, поглощение отсутствует). Таким образом, в ЭСП сорбентов после сорбции появляются новые полосы поглощения по сравнению с ЭСП исходных сорбентов. Во всех трех спектрах сорбентов после сорбции не наблюдаются полосы поглощения при 279 и 473 нм, характерные для тетрахлоропалладат(II)-иона [34], что связано с образованием комплексов палладия(II) с функциональными группами сорбентов. Тот факт, что спектры, полученные для образцов МУК + Pd(II) и ТСК + Pd(II), схожи, свидетельствует о близости строения сорбируемых комплексов палладия(II).
Рис. 3.
ЭСП образцов сорбентов, насыщенных ионами Pd(II), в вазелиновом масле: 1 – МЭ; 2 – МУК; 3 – ТСК.

Сделанный вывод подтверждают и спектры диффузного отражения (СДО), полученные для тех же образцов. В качестве примера на рис. 4 приведен СДО сорбента МЭ, насыщенного ионами палладия(II), в координатах F(R) = (1 – R2)/(2R). Присутствующая в этом спектре полоса при 425 нм указывает на изменение лигандного окружения по сравнению с исходным комплексом [PdCl4]2– и комплексообразование с функциональной группой привитой молекулы сорбента. Отметим также, что СДО для других серосодержащих сорбентов, насыщенных палладием(II), практически не отличаются от приведенных на рис. 4.

Еще бóльшую информацию дают рентгеновские фотоэлектронные спектры. Они позволяют судить не только о степени окисления палладия, но и природе его лигандного окружения. Величины энергии связи для палладия в целом соответствуют его степени окисления +2 [35, 36], а наличие двух компонент указывает на присутствие двух форм комплексов палладия с различным лигандным окружением (рис. 5, табл. 3) [37, 38].
Рис. 5.
Рентгеновские фотоэлектронные спектры Pd3d образцов, полученных сорбцией комплекса [PdCl4]2– на сорбентах МЭ (а) и ТСК (б).

Таблица 3.
Значения энергии связи S2p, Pd3d для модифицированных кремнеземов, насыщенных ионами палладия(II)
| Образец | Значения энергии связи, эВ | |||
|---|---|---|---|---|
| S2p3/2 | S2p1/2 | Pd3d5/2 | Pd3d3/2 | |
| Pd-МЭ | 164.0 | 165.1 | 336.9 338.4 |
342.0 343.7 |
| Pd-ТСК | 164.3 | 165.6 | 336.9 338.4 |
342.0 343.8 |
Данные РФЭ-спектров подтверждают присутствие в образцах сульфидной серы, для которой характерен спин-орбитальный дублет S2p3/2–S2p1/2 с энергией связи 164.0–165.5 эВ. Для образца Pd + МЭ содержание палладия, хлора и серы составляет 0.64, 0.96 и 0.48 ат. % соответственно. По нашему мнению, это указывает на комплексообразование палладия(II) с атомами серы сорбента с вытеснением в среднем двух ионов хлора из внутренней координационной сферы центрального атома. В образце Pd + ТСК метод РФЭС вообще не зафиксировал присутствие хлора. Возможно, на поверхности образуется хелатный комплекс с участием атома серы и атома кислорода карбоксильной группы привитой тиосалициловой кислоты.
Представлялось целесообразным обосновать высказанные выше предположения о характере связывания сорбируемого иона Pd(II) с привитой к поверхности кремнезема серосодержащей молекулой квантово-химическими расчетами электронной и геометрической структуры исследованных лигандов и комплексов с этими лигандами, а также соответствующих термодинамических характеристик реакций комплексообразования. Если обратиться к формулам серосодержащих соединений L, которые были использованы для синтеза сорбентов (табл. 1), то станет очевидным, что их можно представить в общем виде как R'SR, где R' – группа, непосредственно присоединенная к поверхности SiO2, а R – меняющийся радикал при атоме серы (L1, L2, L3). Полученные с использованием атомного базисного набора def2tzvp величины отрицательных зарядов на донорном атоме S, по которым возможна простая оценка донорной способности свободных лигандов L (они представлены в табл. 4), показали, что и в газовой фазе (g), и в водном растворе (s) они лишь слабо понижаются в ряду q(S)L1 > q(S)L2 > q(S)L3.
Таблица 4.
Распределение электронной плотности в лигандах L и комплексах Pd(II) (базис def2tzvp, g – gas, s – solution): q – заряды на атомах (по Малликену); W(А–В) – кратности связей Вайберга [39]; ∑W(А) – суммарные значения кратностей связей атома в молекуле
| Параметр | L1 | L2 | L3 | ||
|---|---|---|---|---|---|
| q(S) | –0.12g, –0.16s | –0.09g, –0.13s | 0.00g, –0.06s | ||
| ∑W(S) | 2.20 | 2.19 | 2.40 | ||
| Комплекс | [PdCl4]2– | [Pd(L1)2Сl2] | [Pd(L2)2Сl2] | [Pd(L3)2Сl2] | [Pd(bi-L2)2]2+ |
| q(Pd) | 0.41g, 0.39s | 0.24g, 0.25s | 0.21g, 0,28s | 0.13g, 0.16s | 0.32g |
| q(S) | – | 0.11g, 0.07s, 0.08g, 0.07s | 0.15g, 0.11s, 0.13g, 0.10s | 0.10g, 0.07s, 0.08g, 0.06s | 0.19g, 0.15g |
| W(Pd–Cl) | 0.62 | 0.70, 0.63 | 0.69, 0.66 | 0.63,0.59 | – |
| W(Pd–S) | – | 0.58, 0.59 | 0.58, 0.59 | 0.48, 0.47 | 0.61, 0.61 |
| W(Pd–O) | – | – | – | – | 0.39, 0.41 |
| ∑W(Pd) | 2.47 | 2.62 | 2.64 | 2.55 | 2.24 |
| ∑W(Cl) | 0.79 | 0.95, 0.92 | 0.94, 0.95 | 0.97, 0.90 | – |
| ∑W(S) | – | 2.87, 2.90 | 2.87, 2.89 | 2.90, 2.89 | 2.92, 2.90 |
Согласно анализу NBO для координационных соединений состава [Pd(L)2Сl2], вычисленные кратности связей металл–сера W(Pd–S), определяемые индексами Вайберга [39], также оказались довольно близки и в среднем равны 0.5–0.6 ед. кратности. Такие малые отличия в рассматриваемых характеристиках лигандов R'SR при весьма разных группах R объясняются принципом выравнивания электроотрицательностей, сформулированным Сандерсеном [40]: при образовании связей M–S перенос электронной плотности идет не только с донорного атома серы, но и включает ее подкачивание всей лигандной системой, в том числе неизменной группой R'. Суммарное значение индексов Вайберга для атома в соединении ∑W ассоциируется с его валентной активностью. Величина ∑W(S) в указанных соединениях также выравнивается до значений 2.9–3.0 ед. (табл. 4).
Проведенный простой анализ перераспределения электронной плотности при комплексообразовании даже в расширенном атомном базисе def2tzvp не позволил установить различия в донорной способности лигандов L. Для соединений [Pt(L)2Сl2] такой подход дал аналогичные результаты по характеристикам W(M–S) и ∑W(S), причем они оказались очень близки к вышеописанным характеристикам и межатомным расстояниям в комплексах [Pd(L)2Сl2]. Возможно, по этой причине процесс разделения комплексов Pd(II) и Pt(II) при совместном присутствии затруднителен.
Как известно [41], реакции замещения в плоскоквадратных комплексах Pd(II) и Pt(II) протекают через образование пятикоординационных интермедиатов, причем сделан вывод, что активационные барьеры в реакциях нуклеофильного замещения для комплексов Pd(II) значительно ниже, чем для аналогичных соединений Pt(II). Однако поиск интермедиатов с оценкой относительных энергий активации в реакциях нуклеофильного замещения координированных хлорид-ионов на большие и разветвленные серосодержащие лиганды представляет весьма сложную задачу. Поэтому в настоящей работе мы оценивали только термодинамические характеристики – относительные изменения энергии Гиббса, учитывающие исходные реагенты и продукты в реакциях комплексообразования. Рассматривая возможность протекания химических реакций в растворе, использовали фундаментальное требование уменьшения в реагирующей системе термодинамического потенциала Гиббса ∆G0. Энергетические характеристики соединений для последующих расчетов величин ∆G0 реакций, полученные в настоящей работе, приведены в табл. 5. Реакции комплексообразования и соответствующие им значения изменения ∆G0 в газовой фазе (g) и с учетом раcтворителя (s) приведены в табл. 6.
Таблица 5.
Рассчитанные для реакций энергетические характеристики соединений (базисы def2tzvp и * def2svp): энергии Гиббса в газовой фазе и с учетом растворителя – $G_{{{\text{gas}}}}^{0},$ $G_{{{\text{solv}}}}^{0}$ (а.е.), энергия сольватации соединений $\Delta G_{{{\text{solv}}}}^{0}$ (кДж/моль)
| Соединение | $G_{{{\text{gas}}}}^{0}$ | $G_{{{\text{solv}}}}^{0}$ | $G_{{{\text{solv}}}}^{0}$ |
|---|---|---|---|
| [PdCl4 ]2– | –1968.934530 | –1969.228277 | –771.11 |
| [PdCl4 ]2–* | –1968.264893 | –1968.564490 | –786.55 |
| [Pd(H2O)2Cl2 ]0* | –1200.716404 | –1200.740942 | –64.42 |
| [Pd(H2O)4 ]2+* | –432.544538 | –432.865633 | –842.99 |
| H2O* | –76.320502 | –76.327434 | –18.20 |
| Cl– | –460.264316 | –460.380516 | –305.07 |
| Cl–* | –460.079169 | –460.198629 | –313.63 |
| L1 | –939.161997 | –939.171869 | –25.92 |
| L1* | –938.417502 | –938.426585 | –23.85 |
| L2 | –1013.236224 | –1013.250133 | –36.52 |
| L2* | –1012.405741 | –1012.418827 | –34.36 |
| L3 | –1204.842021 | –1204.857038 | –39.42 |
| L3* | –1203.810096 | –1203.827176 | –44.84 |
| [Pd(L1)2Cl2] | –2926.765900 | –2926.801012 | –92.18 |
| [Pd(L1)2Cl2]* | –2924.937498 | –2924.974056 | –95.98 |
| [Pd(L2)2Cl2] | –3074.908712 | –3074.946646 | –99.59 |
| [Pd(L2)2Cl2] * | –3072.905786 | –3072.940017 | –89.87 |
| [Pd(bi-L2)Cl2]* | –2060.480956 | –2060.518341 | –98.15 |
| [Pd(L2)(H2O)2]2+* | –1292.392015 | –1292.650751 | –679.27 |
| [Pd(bi-L2)2]2+* | –2152.194868 | –2152.424167 | –601.99 |
| [Pd(L3)2Cl2] | –3458.126707 | –3458.158229 | –82.76 |
| [Pd(L3)2Cl2]* | –3455.735856 | –3455.766561 | –80.61 |
| [Pd(bi-L3)2]2+* | –2535.054138 | –2535.253484 | –523.35 |
Таблица 6.
Реакции комплексообразования и соответствующие им рассчитанные значения изменения энергии Гиббса ∆G0 в газовой фазе (g) и с учетом раcтворителя (s)
| № реакции | Реакция | ∆G0 (базис def2svp) |
|---|---|---|
| L1 | ||
| 1 | [PdCl4 ]2– + 2L1 ↔ [Pd(L1)2Сl2]0 + 2Cl– | +10.66g, +121.67s |
| 2 | [Pd(H2O)2 Cl2]0 + 2L1 ↔ [Pd(L1)2Сl2]0 + 2H2O | –71.13g, –91.39s |
| 3 | [Pd(H2O)4 ]2+ + 2L1 + 2Cl– ↔ [Pd(L1)2Сl2]0 + 4H2O | –1789.51g, –440.35s |
| L2 | ||
| 4 | [PdCl4 ]2– + 2L2 ↔ [Pd(L2)2Сl2]0 + 2Cl– | + 32.16g, +170.30s |
| 5 | [PdCl4 ]2– + 1L2 ↔ [Pd(bi-L2)1Сl2]0 + 2Cl– | +82.28g, +177.78s |
| 6 | [PdCl4 ]2– + 1L2 ↔ [Pd(bi-L2)2]2+ + 4Cl– | +1482.88g, +481.65s |
| 7 | [Pd(H2O)2 Cl2]0 + 2L2 ↔ [Pd(L2)2Сl2]0 + 2H2O | –11.71g, –10.04s |
| 8 | [Pd(H2O)2 Cl2]0 + 1L2 ↔ [Pd(bi-L2)1Сl2]0 + 2H2O | +0.49g, –35.28s |
| 9 | [Pd(H2O)4 ]2+ + 1L2 ↔ [Pd(bi-L2)1(H2O)2]2+ + 2H2O | –217.22g, –55.55 s |
| 10 | [Pd(H2O)4 ]2+ + 2L2 ↔ [Pd(bi-L2)2]2+ + 4H2O | –317.29g, –80.38s |
| L3 | ||
| 11 | [PdCl4 ]2– + 2L3 ↔ [Pd(L3)2Сl2]0 + 2Cl– | –104.53g, +144.46s |
| 12 | [PdCl4 ]2– + 2L3 ↔ [Pd(bi-L3)2]2+ + 4Cl– | +1350.14g, +448.52s |
| 13 | [Pd(H2O)2Cl2]0 + 2L3 ↔ [Pd(bi-L3)2]2+ + 2H2O + 2Cl– | +1268.35g, +235.45s |
| 14 | [Pd(H2O)2Cl2]0 + 2L3↔ [Pd(L3)2Сl2]0 + 2H2O | –105.71g, –68.61s |
| 15 | [Pd(H2O)4 ]2+ + 2L3 ↔ [Pd(bi-L3)2]2+ + 4H2O | –450.03g, –113.51s |
Взаимодействие реагентов [PdCl4]2– и серосодержащих лигандов L по реакциям 1, 4–6, 11, 12 (табл. 6) могло бы привести к образованию следующих продуктов: [Pd(L1)2Сl2]0, [Pd(L2)2Сl2]0, [Pd(bi-L2)Сl2]0, [Pd(bi-L2)2]2+, [Pd(L3)2Сl2]0 и [Pd(bi-L3)2]2+. Здесь лиганды, обозначенные как bi-L2 и bi-L3, содержат карбоксильную группу и могут проявлять при комплексообразовании бидентатность за счет атомов серы и кислорода (рис. 6–8).



Однако для указанных реакций получены большие положительные значения энергии Гиббса и в газовой фазе $\left( {\Delta G_{{{\text{gas}}}}^{0}} \right),$ и с учетом раcтворителя $\left( {\Delta G_{{{\text{solv}}}}^{0}} \right),$ свидетельствующие о невозможности их протекания (кроме процесса 11, где $\Delta G_{{{\text{gas}}}}^{0}$ < 0, но $\Delta G_{{{\text{solv}}}}^{0}$ > 0). Эффекты, приводящие к таким результатам, могут быть объяснены следующим образом: в исходном соединении [PdCl4]2– и указанных выше продуктах предполагаемых реакций связи Pd–Cl более прочные, чем связи Pd–S и Pd–О (при бидентатной S- и O-координации) лигандов L2 или L3. Исходя из анализа NBO, в системах [Pd(L)2Сl2]0 и [Pd(bi-L2)Сl2]0 усредненные значения индексов Вайберга для лигандов L1, L2, L3 близки к следующим: W(Pd–Cl) = 0.65, W(Pd–S) = = 0.55, W(Pd–О) = 0.39 ед. кр. (табл. 4). Возрастание величины $\Delta G_{{{\text{gas}}}}^{0}$ связано с реагентом – ионом [PdCl4]2– с четырьмя более прочными связями Pd–Cl, обеспечивающим суммарный рост энтальпии $\Delta Н_{{{\text{gas}}}}^{0},$ которая дает основной вклад в величину $\Delta G_{{{\text{gas}}}}^{0}$ в указанных реакциях.
Растворитель, особенно полярный, понижает энергию Гиббса соединений $G_{{{\text{solv}}}}^{0}$ относительно этой величины в газовой фазе $G_{{{\text{gas}}}}^{0}.$ Вычисленные относительные изменения значений $G_{{{\text{solv}}}}^{0}$ уже можно качественно сопоставлять с экспериментальными. При этом следует иметь в виду, что сольватация двухзарядного аниона [PdCl4]2– ($G_{{{\text{solv}}}}^{0}$ = = –786.55 кДж/моль) оказывается более значительной по сравнению с сольватацией двух однозарядных анионов Cl– ($2G_{{{\text{solv}}}}^{0}$ = –627.26 кДж/моль). Сольватация всех нейтральных лигандов L и хлоридных комплексов относительно мала: величина $G_{{{\text{solv}}}}^{0}$ составляет всего 24–45 и 82–100 кДж/моль для L и комплексов типа [Pd(L1)2Сl2]0 соответственно (табл. 5).
В растворах с низкой концентрацией соляной кислоты возможно существование комплексов [Pd(H2O)2Cl2]0. Для процессов 2, 7, 8, 14 с исходными [Pd(H2O)2Cl2]0 и лигандами L также были рассмотрены различные возможные продукты. В соответствии с критерием протекания реакций в растворе $\Delta G_{{{\text{solv}}}}^{0}$ < 0 в указанных реакциях могут образовываться следующие соединения: [Pd(L1)2Сl2]0, [Pd(L2)2Сl2]0, [Pd(bi-L2)Сl2]0 и [Pd(L3)2Сl2]0, причем для первого и последнего в случае реагента [Pd(H2O)2Cl2]0 рассчитанные критерии ∆G0 наиболее оптимальны. Однако для реакции 14 с лигандом L3 образование продукта [Pd(L3)2Сl2]0 не подтверждается экспериментальными данными РФЭС, которые свидетельствуют о том, что в этой реакционной системе отсутствуют продукты с координированными анионами Cl–. Отсутствие хлоридных лигандов, в принципе, могло бы указывать на образование устойчивого соединения [Pd(L3)4]2+ с монодентатной S-координацией лигандов L3 или комплекса [Pd(bi-L3)2]2+, в котором серосодержащий лиганд проявляет бидентатные свойства. Однако в первом случае формированию комплекса Pd(II) с четырьмя S-координированными лигандами L3 будут препятствовать стерические особенности объемных лигандных групп. Для продукта [Pd(bi-L3)2]2+ с двумя связями Pd–O, еще более слабыми, чем Pd–Cl или Pd–S (см. W(A–B) в табл. 4), в реакции 13 термодинамическая устойчивость продуктов должна быть меньше, чем реагентов. При наличии в растворе полностью акватированных комплексов [Pd(H2O)4]2+ из-за слабых связей металл–лиганд и, как следствие, малых величин $G_{{{\text{gas}}}}^{0}$ и $G_{{{\text{solv}}}}^{0}$ суммарные значения $\Delta G_{{{\text{gas}}}}^{0}$ и $\Delta G_{{{\text{solv}}}}^{0}$ в реакциях 3, 9–10 и 15 имеют большие отрицательные значения, что свидетельствует о возможности протекания реакций с исходным аквакомплексом. Возможно, термодинамически выгодное образование комплекса иона металла c лигандом L3, не содержащего координированных хлорид-ионов ([Pd(bi-L3)2]2+), связано именно с наличием в системе аквакомплексов Pd(II).
Таким образом, реакции комплексообразования Pd(II) со всеми рассматриваемыми лигандами L термодинамически невыгодны при большой концентрации HCl, когда доля ионов [PdCl4]2– наибольшая. При понижении концентрации HCl и наличии в растворах комплексов [Pd(H2O)2Cl2]0 допустимо замещение в них лигандов H2O на серосодержащие лиганды L1 и L2 с образованием соединений типа [Pd(L1)2Сl2]0, [Pd(L2)2Сl2]0 и [Pd(bi-L2)Сl2]0. В слабокислых растворах, которые могут содержать исходные аквакомплексы [Pd(H2O)4]2+, процессы замещения координированных молекул H2O на серосодержащие лиганды с образованием комплексов [Pd(L1)2Сl2]0, [Pd(bi‑L2)1(H2O)2]2+, [Pd(bi-L2)2]2+ и [Pd(bi-L3)2]2+ термодинамически выгодны.
ЗАКЛЮЧЕНИЕ
Изучена сорбция ионов Pd(II) из солянокислых растворов кремнеземами, химически модифицированными серосодержащими органическими соединениями. Получены зависимости сорбции из хлоридных растворов в статических условиях в зависимости от времени, концентрации HCl, ионов Cl–; построены изотермы сорбции. Совокупностью спектральных методов (ИК-, УФ-Vis-, рентгеновская фотоэлектронная спектроскопия) доказано, что сорбция протекает по координационному механизму. Квантово-химическим методом DFT M06 в базисе def2tzvp в газовой фазе и с учетом растворителя H2O выполнено моделирование структуры серосодержащих сорбентов и их комплексов с палладием, что позволило обосновать характер зависимости сорбции от концентрации кислоты и подтвердить возможность образования комплексов палладия(II) с бидентатной координацией лигандов в фазе кремнезема, модифицированного тиосалициловой и меркаптоуксусной кислотами.
Список литературы
Ehrlich G.V., Buslaeva T.M., Marutina T.A. // Russ. J. Inorg. Chem. 2017. V. 62. № 14. P. 1797. https://doi.org/10.1134/S0036023617140030
Bodnar N.M., Buslaeva T.M., Ehrlich G.V. et al. // Russ. J. Inorg. Chem. 2021. V. 66. № 4. P. 586. https://doi.org./10.1134s0036023621040045 [Боднарь Н.М., Буслаева Т.М., Эрлих Г.В. и др. // Журн. неорган. химии. 2021. Т. 66. № 4. С. 549.]
Giovanna N., Goosey E., Yildiz D.S. et al. // Johnson Mattey Technol. Rev. 2021. V. 65. № 1. P. 127. https://doi.org/10.1595/205651321X16057842276133
Didukh-Shadrina S.L., Losev V.N., Maznjak N.V., Trofimchuk A.K. // J. Anal. Chem. 2019. V. 74. № 8. P. 738. [Дидух-Шадрина С.Л., Лосев В.Н., Мазняк Н.В., Трофимчук А.К. // Журн. аналит. химии. 2019. Т. 74. № 8. С. 574.]https://doi.org/10.1134/S1061934819080069
Dubenskiy A.S., Yakurnova E.D., Krasilnikova Yu.A. et al. // Analyt. Lett. 2020. V. 53. № 8. P. 1266. https://doi.org/10.1080/00032719.2019.1700994
Kimuro T., Gandhi M.R., Kunda U.M.R. et al. // Hydrometallurgy. 2017. V. 171. P. 254. https://doi.org/10.1016/j.hydromet.2017.05.022
Rao K.U.M., Yamada M. // Sep. Sci. Technol. 2017. V. 52. № 7. P. 1153. https://doi.org/10.1080/01496395.2017.1281953
Anpilogova G.R., Murinov Yu.I. // Russ. J. Appl. Chem. 2021. V. 94. № 3. P. 316. [Анпилогова Г.Р., Муринов Ю.И. // Журн. прикл. химии. 2021. Т. 94. № 3. С. 324.]https://doi.org/10.1134/S107042722103006X
Chassary P., Vincent T., Marcano J.S., Macaskie L.E. // Hydrometallurgy. 2005. V. 76. № 1. P. 131. https://doi.org/10.1016/j.hydromet.2004.10.004
Petrova Y.S., Pestov A.V., Usoltseva M.K. et al. // Sep. Sci. Technol. 2019. V. 56. № 1. P. 42. https://doi.org/10.1080/01496395.2018.1505912
Wang S., Vincent T., Roux J.-C et al. // Chem. Eng. J. 2017. V. 325. P. 521. https://doi.org/10.1016/j.cej.2017.05.103
Dalnova O.A., Dalnova Y.S., Baranovskaya V.B., Karpov Y.A. // J. Analyt. Chem. V. 73. № 3. P. 221. [Дальнова О.А., Барановская В.Б., Дальнова Ю.С., Карпов Ю.А. // Журн. аналит. химии. 2018. Т. 73. № 3. С. 181.]https://doi.org/10.1134/S1061934818030036
Liu Y., Lin S., Liu Y. et al. // Small (Nano micro). 2019. V. 15. № 10. P. 1805242. https://doi.org/10.1002/sml.201805242
Qin Zhang, Muhar Chen, Lijiang Zhang et al. // Molecules. 2018. V. 23. № 8. P. 2086. https://doi.org/10.3390/molecules23082086
Voronkov M.G., Vlasova N.N., Oborina E.N., Belousova L.I. // Russ. Chem. Bull. 2016. V. 65. № 1. P. 245. [Воронков М.Г., Власова Н.Н., Оборина Е.Н., Белоусова Л.И. // Изв. АН. Сер. хим. 2016. № 1. С. 245.]https://doi.org/10.1007/s11172-016-1292-4
Ermakova T.G., Kusnetsova N.P., Prozorova G.F. et al. // Russ. Chem. Bull. 2017. V. 66. № 6. P. 1104. https://doi.org/10.1007/s11172-017-1861-1
Li M., Tang Si, Feng Jian et al. // Metall. Mater. Trans. B.V. 50. P. 2747. https://doi.org/10.1007/s11663-019-01697-8
Losev V., Elsuf’ev E., Borodina E. et al. // Minerals. 2021. V. 11. P. 481. https://doi.org/10.3390/min11050481
Kapitanova E.I., Sinelshchikova A.R., Petrova Y.S. et al. // Russ. Chem. Bull. 2021. V. 70. № 6. P. 1161. [Капитанова Е.И., Синельщикова А.Р., Петрова Ю.С. и др. // Изв. АН. Сер. хим. 2021. № 6. С. 1161.]https://doi.org/10.1007/s11172-021-3199y
Alifkhanova L.M., Lopunova K.Y., Marchuk A.A. et al. // Russ. J. Inorg. Chem. 2021. V. 66. № 6. P. 909. [Алифханова Л.М.К., Лопунова К.Я., Марчук А.А. и др. // Журн. неорган. химии. 2021. Т. 66. № 6. С. 814.]https://doi.org/10.1134/S0036023621060024
Brauer G. // Manual of inorganic synthesis in 6 volumes. V. 5. M.: Mir, 1985. 360 p.
Buslaeva T.M., Bodnar N.M., Gromov S.P. et al. // Russ. Chem. Bull. 2018. V. 67. P. 1190. [Буслаева Т.М., Боднарь Н.М., Громов С.П. и др. // Изв. АН. Сер. хим. 2018. № 7. С. 1190.]https://doi.org/10.1007/s11172-018-2200-x
Пат. РФ RU 2698656 с приоритетом от 25.02.2019. Опубл. 28.08.2019.
Khabibullina G.R., Buslaeva T.M., Fedotova E.S. et al. // Russ. J. Gen. Chem. 2017. V. 87. P. 963. https://doi.org/10.1134/S1070363217050127
Zhao Y., Truhlar D.G. // Theor. Chem. Account. 2008. V. 120. № 1. P. 215. https://doi.org/10.1007/s00214-007-0310-x
Weigend F., Ahlrichs R. // Phys. Chem. Chem. Phys. 2005. V. 7. № 18. P. 3297. https://doi.org/10.1039/B508541A
Frisch M.J., Trucks G.W., Schlegel H.B. et al. Gaussian, Inc., Wallingford CT, 2016.
Miertus S., Scrocco E., Tomasi J. // Chem. Phys. 1981. V. 55. № 1. P. 117. https://doi.org/10.1016/0301-0104(81)85090-2
Tomasi J., Persico M. // Chem. Rev. 1994. V. 94. № 7. P. 2027. https://doi.org/10.1021/cr00031a013]
https://www.chemcraftprog.com/ru
US patent 4,959,153. 1990.
US patent 5,039,419. 1991.
Nakamoto K. Infrared and Raman Spectra of Inorganic and Coordination compounds. A Wiley Interscience Publication, 2009. 432 p.
Буслаева Т.М., Симанова С.А. Аналитическая химия металлов платиновой группы. М.: Комкнига, 2005. 592 с. ISBN 5-484-00161-7
Diaz-Aunon J.A., Garcia-Martinez J., Roman-Martinez M.C., Salinas-Martinez de Lecea C. // Catalysis Lett. 2001. V. 76. № 1–2. P. 41. https://doi.org/10.1023/A:1016732208513
Нефедов В.И. Рентгеноэлектронная спектроскопия химических соединений. Справочник. М.: Химия, 1984. 55 с.
Ivanova A.S., Slavinskaja E.M., Stonkus O.A. et al. // Kinet. Catal. 2013. V. 54. № 1. P. 81. [Иванова А.С., Славинская Е.М., Стоункус О.А. и др. // Кинетика и катализ. 2013. Т. 54. № 1. С. 85.]https://doi.org/10.1134/S0023158413010084
Boronin A.I., Slavinskaya E.M., Danilova I.G. et al. // Catalysis Today. 2009. № 144. C. 201. https://doi.org/10.1016/j.cattod.2009.01.035
Wiberg K.B. // Tetrahedron. 1968. V. 24. № 3. P. 1083. https://doi.org/10.1016/0040-4020(68)88057-3
Sanderson R.T. Chemical Bonds and Bond Energy. N.Y.: Acad. Press, 1976. 225 p.
Panina N.S., Belyaev A.N., Eremin A.V. et al. // Russ. Chem. Bull. 2012. V. 61. № 4. P. 796. [Панина Н.С., Беляев А.Н., Еремин А.В. и др. // Изв. АН. Сер. хим. 2012. № 4. С. 794.]https://doi.org/10.1007/s1172-012-0110-x
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии