

Журнал общей биологии, 2020, T. 81, № 2, стр. 83-95
Регуляторное влияние мезенхимных стромальных клеток на развитие фиброза печени: клеточно-молекулярные механизмы и перспективы клинического применения
О. В. Паюшина 1, *, Д. А. Цомартова 1, Е. В. Черешнева 1, М. Ю. Иванова 1, С. Л. Кузнецов 1
1 Первый МГМУ им. И.М. Сеченова Минздрава России (Сеченовский Университет)
119991 Москва, Трубецкая ул., 8, стр. 2, Россия
* E-mail: payushina@mail.ru
Поступила в редакцию 27.05.2019
После доработки 21.10.2019
Принята к публикации 01.11.2019
Аннотация
Фиброз печени, прогрессирование которого способно привести к печеночной недостаточности, портальной гипертензии и гепатоцеллюлярной карциноме, представляет собой актуальную медицинскую проблему. Механизмы, лежащие в основе его развития, включают избыточную продукцию внеклеточного матрикса активированными клетками Ито, подавление фибролиза, оксилительное повреждение и апоптоз гепатоцитов, воспаление, капилляризацию эндотелия синусоидов. Развитие патологического процесса обусловлено взаимным влиянием нескольких клеточных популяций посредством различных сигнальных путей. В настоящее время эффективность воздействия на фиброгенез в печени с помощью фармакологических агентов, хирургического вмешательства или воздействия физических факторов невелика. Большие надежды в этом отношении возлагаются на клеточную терапию с использованием мезенхимных стромальных клеток. Их гепатопротективный и регенеративный эффект обусловлен в первую очередь паракринной секрецией широкого спектра регуляторных молекул, которые способствуют выживанию и пролиферации гепатоцитов, предотвращают активацию и стимулируют апоптоз клеток Ито, оказывают иммуномодулирующее, проангиогенное и фибролитическое действие. Комплексное влияние мезенхимных стромальных клеток на многие клеточные популяции, участвующие в развитии фиброза печени, дает основание рассматривать эти клетки и их секреторные продукты (в частности, внеклеточные везикулы) как перспективный ресурс для клинического использования.
Хронические заболевания печени, вызываемые травмами, вирусными инфекциями, алкоголем, токсинами и метаболическими ядами, представляют собой актуальную медицинскую проблему. В настоящее время они входят в число наиболее частых причин нетрудоспособности и смертности населения многих развитых стран. Несмотря на высокую способность печени к регенерации, длительное воздействие повреждающих факторов способно привести к развитию фиброза, терминальной стадией которого является цирроз печени – необратимое замещение ее паренхимы рубцовой тканью с грубым нарушением структуры органа. Дальнейшее развитие патологического процесса приводит к печеночной недостаточности и портальной гипертензии; во многих случаях на фоне цирроза печени развивается гепатоцеллюлярная карцинома (Hernandez-Gea, Friedman, 2011; Schuppan, 2015). Несмотря на определенные успехи в поиске путей воздействия на процесс фиброгенеза в печени с помощью фармакологических агентов (Schuppan, 2015), а также хирургического вмешательства или воздействия физических факторов (Могилевец и др., 2013), в настоящее время эффективность лечения фиброза и цирроза печени невелика. Большие надежды в этом отношении возлагаются на клеточную терапию, в качестве одного из перспективных ресурсов для которой рассматриваются мезенхимные стромальные клетки (МСК) (Lysy et al., 2008; Петракова и др., 2012).
Согласно критериям, разработанным Международным обществом клеточной терапии, МСК определяются как адгезивные к пластику клетки, экспрессирующие поверхностные антигены CD73, CD90 и CD105 при отсутствии кроветворных маркеров и способные дифференцироваться в остеогенном, адипогенном и хондрогенном направлениях (Dominici et al., 2006). Они обладают широким спектром потенций и, по некоторым данным, могут быть индуцированы к дифференцировке в функционально активные гепатоцитоподобные клетки, способные улучшать состояние печени при трансплантации экспериментальным животным с ее повреждением (Ewida et al., 2017; Zhou et al., 2017; Zhang et al., 2018). Однако способность этих клеток эффективно репопулировать паренхиму печени остается дискуссионной (Lysy et al., 2008; Ewida et al., 2017).
Значительно более перспективным подходом представляется использование МСК не для замещения утраченных гепатоцитов, а для создания микроокружения, способствующего полноценной регенерации. Как было показано на моделях повреждения различных тканей и органов, благотворное влияние МСК на репаративные процессы опосредовано главным образом паракринной секрецией широкого спектра регуляторных молекул, оказывающих иммуномодулирующее, ангиогенное, антифибротическое действие, а также стимулирующих пролиферацию и дифференцировку резидентных клеток-предшественников (Паюшина, 2015). Для понимания механизмов влияния МСК на процесс фиброгенеза в печени и перспектив их применения для лечения фиброза и цирроза прежде всего необходимо рассмотреть клеточные и молекулярные события, определяющие патогенез этих заболеваний.
ПАТОГЕНЕТИЧЕСКИЕ МЕХАНИЗМЫ ФИБРОЗА ПЕЧЕНИ
Источники фиброгенных клеток
Наиболее частыми причинами фиброза печени у человека являются злоупотребление алкоголем, вирусные и аутоиммунные гепатиты, холестаз, метаболические нарушения, воздействие токсинов, а в экспериментах на животных он может быть смоделирован перевязкой общего желчного протока, введением гепатотропных ядов (четыреххлористого углерода, тиоацетамида), высокожировой или холиндефицитной диетой (Liedtke et al., 2013).
Механизмы, лежащие в основе фиброза печени различной этиологии, в целом сходны. Ключевым моментом в его развитии является активация клеток Ито с потерей ими липидных включений, содержащих витамин А, и приобретением фенотипа миофибробластов. В активированном состоянии они продуцируют большое количество внеклеточного матрикса, преобладающими компонентами которого являются коллагеновые белки (Hernandez-Gea, Friedman, 2011; Schuppan, 2015). Наряду с клетками Ито в фиброгенезе могут участвовать фибробласты, локализованные в области портальных триад, а также, возможно, клетки, окружающие центральные вены. В нормальной печени они находятся в покоящемся состоянии, однако при ряде патологических состояний трансформируются в миофибробласты (Penz-Österreicher et al., 2011). Фиброгенные клетки могут возникать также из мезотелиальных, которые при повреждении печени способны мигрировать вглубь органа и дифференцироваться в клетки Ито или миофибробласты (Lua, Asahina, 2016). Обсуждается также гипотеза об их образовании из клеток печеночной паренхимы в результате эпителио-мезенхимного перехода (Hernandez-Gea, Friedman, 2011). В ее пользу свидетельствует возможность индукции эпителио-мезенхимного перехода в первичной культуре гепатоцитов под действием профиброгенных цитокинов, однако в экспериментах по отслеживанию судьбы генетически меченых клеток в ходе развития фиброза печени у мыши было показано, что ни гепатоциты, ни холангиоциты источниками миофибробластов не являются (Seki, Brenner, 2015).
Избыточному накоплению внеклеточного матрикса способствует не только его усиленное отложение, но и подавление фибролиза вследствие снижения секреции и активности матриксных металлопротеиназ (MMP-1, -3, -13) и усиления продукции тканевых ингибиторов металопротеиназ (TIMP-1, -2) активированными клетками Ито или портальными миофибробластами (Schuppan, 2015). В свою очередь, состояние внеклеточного матрикса влияет на клетки Ито и миофибробласты, модулируя их пролиферацию и активацию через взаимодействие с рецепторами адгезии, а также определяя доступность и активность факторов роста и матриксных металлопротеиназ (Hernandez-Gea, Friedman, 2011).
Ключевые факторы активации фиброгенных клеток
Активация фиброгенных клеток – сложный процесс, в котором задействованы различные сигнальные пути. В частности, активацию и пролиферацию клеток Ито способны вызывать такие цитокины, как фактор стволовых клеток (SCF), фактор роста соединительной ткани (CTGF), кислый и основной факторы роста фибробластов (aFGF, bFGF), фактор роста сосудистого эндотелия (VEGF), инсулиноподобные факторы роста (IGF-1, IGF-2) (Hernandez-Gea, Friedman, 2011). Однако центральную роль в фиброгенном ответе на повреждение печени играют трансформирующий фактор роста β1 (TGF β1) и фактор роста тромбоцитарного происхождения (PDGF-B).
Связывание TGF β1 с его рецептором II типа приводит к димеризации последнего с рецептором TGF β I типа; полученный димер связывает факторы транскрипции SMAD2 и SMAD3, подвергается фосфорилированию и высвобождается в цитозоль, где связывается со SMAD4. Образовавшийся комплекс переносится в ядро и усиливает транскрипцию генов, в частности, кодирующих проколлаген I и III типов (Hernandez-Gea, Friedman, 2011; Seki, Brenner, 2015). TGF β1 способствует приобретению фенотипа миофибробластов как клетками Ито, так и портальными фибробластами, а также клетками мезотелия (Lua et al., 2016). Кроме того, сигналинг через TGF β1 подавляет апоптоз клеток Ито, стимулирует их пролиферацию, препятствует деградации внеклеточного матрикса, ингибируя продукцию матриксных металлопротеиназ и усиливая продукцию их ингибиторов (Fabregat et al., 2016; Xu et al., 2016). TGF β1 продуцируют различные популяции клеток печени, в том числе и сами клетки Ито, что обеспечивает аутокринную и паракринную регуляцию их активации (Seki, Brenner, 2015).
PDGF-B, источником которого в поврежденной печени служат прежде всего макрофаги, инфильтрирующие ее в ходе воспалительной реакции, и клетки Купфера, оказывает мощное митогенное действие на клетки Ито и портальные фибробласты. На ранней стадии активации этих клеток в них происходит быстрая индукция рецептора PDGFR-β. Связываясь с лигандом, рецептор димеризуется и аутофосфорилируется, после чего активирует несколько сигнальных путей, включая RAS-MAPK, PI3K-AKT/PKB, PKC, RHOA, PLCγ, JAK/STAT. Это приводит к усилению пролиферации, миграции и выживания клеток, а также к их трансформации в миофибробласты с усилением продукции коллагенов I и III типов (Berardis et al., 2015; Borkham-Kamphorst, Weiskirchen, 2016; Ying et al., 2017). Экспрессия рецептора PDGFR-β в активированных клетках Ито может усиливаться под действием TGF β1, чем, видимо, и обусловлено стимулирующее влияние последнего на пролиферацию этих клеток (Shah et al., 2013).
Роль различных популяций клеток печени в развитии фиброза
Гепатоциты составляют основную массу клеток печени, и их повреждение становится первым шагом к развитию фиброза. При длительном воздействии повреждающих факторов из них выделяются свободные радикалы, а также профиброгенные и провоспалительные цитокины. В частности, в экспериментах in vitro на линии гепатоцитов HepG2 показано, что обработка гепатотоксикантами вызывает в них оксидативный стресс и апоптоз, сопровождающиеся усилением продукции PDGF-B и TGF β1, что может служить одним из первых сигналов к приобретению клетками Ито фенотипа миофибробластов (Mekala et al., 2018). В свою очередь, сигналинг через TGF β1 способен вызывать гибель гепатоцитов, а также индуцировать экспрессию ими CTGF, что еще более усиливает фиброгенез (Seki, Brenner, 2015). К активации клеток Ито ведет также воздействие продуцируемых поврежденными гепатоцитами свободных радикалов, IGF, фактора некроза опухолей α (TNF α), эпидермального фактора роста (EGF), интерлейкина (ИЛ)-33 (Bataller, Brenner, 2005; Seki, Brenner, 2015).
В последнее время появляются данные о том, что в развитии фиброза играют роль образующиеся в гепатоцитах инфламмасомы, активация которых вызывает выброс провоспалительных цитокинов (Alegre et al., 2017), и экспрессия ряда некодирующих РНК. Так, при фиброзе в гепатоцитах повышается уровень lincRNA-p21, что приводит к усилению сигналинга TGF β1, способствует апоптозу гепатоцитов и подавляет их пролиферацию (Tu et al., 2017). Показано также, что снижение выраженности фиброза может быть достигнуто путем ингибирования в гепатоцитах малой РНК miRNA-221-3p (Tsay et al., 2019).
Важная роль в патогенезе фиброза принадлежит холангиоцитам. Так, при холестазе они начинают пролиферировать и стимулируют портальные миофибробласты к отложению коллагена вокруг желчных протоков, а также выделяют воспалительные цитокины, такие как TNF α (Penz-Österreicher et al., 2011; Hall et al., 2017). Показано стимулирующее влияние пролиферирующих холангиоцитов на активацию клеток Ито посредством продукции центральных фиброгенных медиаторов – TGF β1, PDGF, Sonic Hedgehog (Schuppan, 2015; Borkham-Kamphorst, Weiskirchen, 2016). Гиперплазия холангиоцитов с приобретением ими фиброгенных свойств отмечена не только в случае холестаза, но и при повреждениях печени другой этиологии – в частности, при ее токсическом поражении (Hall et al., 2017).
В печени присутствуют по крайней мере две популяции стволовых и родоначальных клеток: одна из них, локализованная в канальцах Геринга, дает начало гепатоцитам и холангиоцитам, другая, локализованная в области крупных внутрипеченочных и внепеченочных желчных протоков, способна дифференцироваться в тех же направлениях, а также в β-клетки поджелудочной железы. При хроническом повреждении печени они участвуют в ее регенерации, но под влиянием патологически измененного микроокружения размножение этих клеток, сопровождающееся секрецией лигандов Hedgehog, остеопонтина и TGF β1, приводит к активации клеток Ито и портальных миофибробластов и, следовательно, к прогрессированию фиброза (Overi et al., 2018). На модели перевязки желчного протока показано также, что в развитие холестатического фиброза печени вносит вклад дифференцировка родоначальных клеток в холангиоциты, опосредованная сигнальным путем NOTCH (Zhang et al., 2016).
Фиброз печени сопровождается капилляризацией эндотелия синусоидов с потерей фенестр и формированием базальной мембраны. При этом не только нарушается снабжение гепатоцитов кислородом и питательными веществами, но и прекращается продукция эндотелием оксида азота (NO), способствующего поддержанию покоящегося состояния клеток Ито, и тем самым создаются условия для их активации (Xu et al., 2017a). Кроме того, патологически измененные эндотелиоциты выделяют фиброгенные цитокины, в том числе TGF β1 и PDGF, и сами участвуют в избыточном отложении внеклеточного матрикса, усиленно продуцируя коллагены I и IV типов и фибронектин (Berardis et al., 2015; Natarajan et al., 2017). Клетки Ито и миофибробласты способны активировать эндотелиальные клетки и стимулировать их пролиферацию, секретируя VEGF и ангиопоэтин-1, что в условиях фибротического микроокружения ведет к дальнейшему прогрессированию заболевания (Berardis et al., 2015; Seki, Brenner, 2015).
Ключевую роль в фиброгенезе играют и резидентные макрофаги печени – клетки Купфера. В ответ на повреждение печеночной паренхимы они высвобождают активные формы кислорода (АФК), активирующие клетки Ито и вызывающие некроз и апоптоз гепатоцитов (Luangmonkong et al., 2018), и провоспалительные цитокины ИЛ-6, ИЛ-13 и TNF α, что ведет к привлечению в печень клеток врожденного иммунитета (Natarajan et al., 2017; Xu et al., 2017a). В печени, пораженной фиброзом, клетки Купфера являются одними из главных продуцентов TGF β1 и PDGF (Seki, Brenner, 2015; Ying et al., 2017). За счет усиления синтеза TIMP-1 и снижения продукции матриксных металлопротеиназ они также компрометируют фибролиз (Schuppan, 2015).
В большинстве случаев развитию фиброза предшествует воспаление, которое в печени, в отличие от большинства других органов, чаще всего является стерильным (неинфекционным). Под влиянием факторов, выделяемых активированными клетками Купфера, печень инфильтрируют различные популяции клеток иммунной системы, включая моноциты крови, приобретающие фенотип макрофагов. Эти инфильтрирующие клетки продуцируют различные цитокины с провоспалительным или прямым профибротическим действием, такие как TNF α, ИЛ-1b, ИЛ-14, TGF β1 (Schuppan, 2015; Koyama, Brenner, 2017). Макрофаги, как резидентные, так и инфильтрирующие, оказывают также ангиогенный эффект, выделяя факторы, стимулирующие пролиферацию и миграцию эндотелиоцитов, в частности, VEGF (You et al., 2013). В развитии фиброза участвуют различные популяции T-лимфоцитов. Так, Т-хелперы типа Th17 продуцируют ИЛ-17 – провоспалительный и профиброгенный цитокин, стимулирующий клетки Ито к повышению экспрессии коллагена I типа и TGF β1, а клетки Купфера – к экспрессии TNF α, ИЛ-1b и ИЛ-6 (Seki, Brenner, 2015; Koyama, Brenner, 2017). Т-хелперы типа Th2 также усиливают фиброз за счет секреции ИЛ-4 и ИЛ-13, стимулирующих дифференцировку фиброгенных клеток и макрофагов, тогда как регуляторные Т-клетки могут как способствовать фиброгенезу, секретируя TGF β1, так и подавлять его, секретируя антифиброгенный фактор ИЛ-10. Антифибротическое действие оказывают также естественные киллеры, способные вызывать гибель активированных клеток Ито, и Т-хелперы типа Тh1 (Berardis et al., 2015).
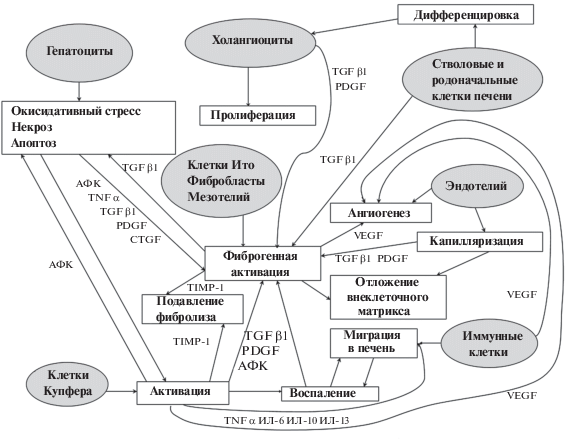
Таким образом, инициация и прогрессирование фиброза печени – сложный процесс, обусловленный взаимным влиянием нескольких клеточных популяций посредством различных сигнальных путей. Схема основных клеточных и молекулярных механизмов фиброгенеза в печени представлена на рис. 1.
Рис. 1.
Клеточные популяции, участвующие в развитии фиброза печени, и их роль в патогенетическом процессе. АФК – активные формы кислорода, ИЛ – интерлейкин, CTGF – фактор роста соединительной ткани, PDGF – фактор роста тромбоцитарного происхождения, TGF – трансформирующий фактор роста, TIMP – тканевый ингибитор металопротеиназ, TNF – фактор некроза опухолей, VEGF – фактор роста сосудистого эндотелия.
РЕГУЛЯТОРНЫЕ ФУНКЦИИ МСК В ПЕЧЕНИ
МСК, присутствующие практически во всех тканях и органах, в настоящее время рассматривают как универсальные регуляторы тканевого гомеостаза, обеспечивающие стабильное функционирование тканей и их восстановление после повреждений (Паюшина, 2015; Eom et al., 2015). Печень плода в период активного гемопоэза содержит значительное количество МСК, организующих в ней кроветворное микроокружение (Паюшина и др., 2011, 2012). Однако и в постнатальный период в этом органе также обнаруживают МСК, в целом сходные по потенциям к дифференцировке, антигенному фенотипу и профилю экспрессии генов с соответствующими клетками костного мозга (Pan et al., 2011; Malagola et al., 2016). Результаты экспериментов по сокультивированию МСК с гепатоцитами дают основания полагать, что их роль в нормальной печени половозрелого организма состоит в поддержании жизнеспособности и функциональной активности клеток печеночной паренхимы (Gu et al., 2009). При повреждении печени в ее регенерации могут участвовать не только эти резидентные МСК, но и приходящие из других источников, прежде всего из костного мозга. Они способны к миграции в область повреждения печеночной ткани в ответ на выделяемый ею фактор стромальных клеток-1 (SDF-1), взаимодействующий с рецептором CXCR4 на поверхности МСК (Xie et al., 2013; Zhang et al., 2015). Возможно, направленной миграции МСК способствуют также АФК, образующиеся в печеночной паренхиме при оксидативном стрессе, через активацию сигнальных путей ERK1/2 и JNK1/2 (Novo et al., 2011).
Хотя МСК обладают фиброгенным потенциалом и в некоторых случаях могут сами вносить вклад в развитие фиброза печени, дифференцируясь в миофибробласты (Bonzo et al., 2008; Baertschiger et al., 2009), в большинстве работ, выполненных на экспериментальных животных, отмечено благотворное влияние трансплантации этих клеток на структурно-функциональное состояние печени при ее повреждениях различной этиологии. В частности, уменьшение выраженности фибротических изменений в печени после введения МСК по сравнению с контрольными группами было показано на моделях интоксикации четыреххлористым углеродом (Шагидулин и др., 2013; Hao et al., 2017; Fu et al., 2018b; Milosavljevic et al., 2018) и тиоацетамидом (Jang et al., 2014; Quintanilha et al., 2014), перевязки общего желчного протока (Duman et al., 2012), жировой болезни печени, вызванной высокожировой диетой (Wang et al., 2018). В ряде случаев при этом наблюдали улучшение не только морфологических и цитохимических, но и функциональных показателей, таких как уровни аланинтрансаминазы, аспартаттрансаминазы, билирубина и альбумина в сыворотке крови (Шагидулин и др., 2013; Quintanilha et al., 2014; Raafat et al., 2015; Ewida et al., 2017).
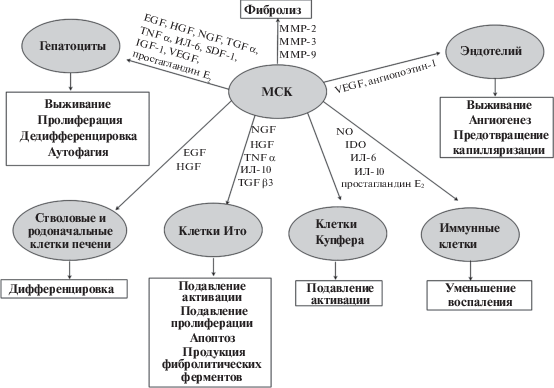
Имеются и данные клинических испытаний, свидетельствующие об улучшении гистологической картины печени и биохимических показателей крови (уровней альбумина и креатинина) и уменьшении печеночной недостаточности через 3–12 месяцев после трансплантации МСК пациентам с циррозом печени (Лукашик и др., 2013; Eom et al., 2015). Улучшение состояния печени после введения МСК обусловлено комплексным влиянием вырабатываемых ими регуляторных молекул практически на все клеточные популяции, участвующие в развитии фиброза (рис. 2), что может приводить не только к остановке патологического процесса, но в определенной степени и к его регрессии.
Рис. 2.
Основные механизмы влияния МСК на развитие фиброза печени. ИЛ – интерлейкин, EGF – эпидермальный фактор роста, HGF – фактор роста гепатоцитов, IDO – индоламин-2,3-диоксигеназа, IGF – инсулиноподобный фактор роста, MMP – матриксная металлопротеиназа, NGF – фактор роста нервов, SDF – фактор стромальных клеток, TGF – трансформирующий фактор роста, TNF – фактор некроза опухолей, VEGF – фактор роста сосудистого эндотелия.
Влияние МСК на клетки печеночной паренхимы
Как было показано в экспериментах in vitro, выделяемые МСК факторы способны защищать гепатоциты от окислительного стресса, уменьшая содержание в них АФК (Quintanilha et al., 2014). Известно, что под действием оксидантов в гепатоцитах повышается уровень микроРНК miR143, что ведет к усилению экспрессии проапоптотического гена BAX; протективный эффект секреторных продуктов МСК по крайней мере отчасти связан с тем, что под их влиянием уровень miR143 снижается (Xu et al., 2017c). Противоапоптотическое действие МСК на гепатоциты неоднократно продемонстрировано не только на клеточных культурах, обработанных токсинами (Zhang et al., 2011; Hirata et al., 2016; Xu et al., 2017b), но и на моделях повреждения печени in vivo (Poll et al., 2008; Francois et al., 2013; Cai et al., 2015). При этом в печени животных, получивших инъекцию МСК, отмечено улучшение показателей редокс-гомеостаза, в частности, снижение вызванной повреждением продукции АФК и повышение активности супероксиддисмутазы (Cho et al., 2012; Francois et al., 2013; Liu et al., 2014). По некоторым данным МСК подавляют старение гепатоцитов (Zhang et al., 2011). Кроме того, они стимулируют пролиферацию гепатоцитов (Poll et al., 2008; Cai et al., 2015) и активируют в них аутофагию (Park et al., 2015), способствуя соответственно клеточной и внутриклеточной регенерации печеночной паренхимы.
Митогенное влияние МСК на гепатоциты обусловлено продукцией фактора роста гепатоцитов (HGF), фактора роста нервов (NGF), TGF α, TNF α, ИЛ-6, а противоапоптотическое действие – продукцией SDF-1, HGF, IGF-1 и VEGF (Eom et al., 2015; Guo et al., 2016). В подавлении апоптоза гепатоцитов и стимуляции их пролиферации задействован также секретируемый МСК простагландин E2, вызывающий активацию белка YAP и усиление экспрессии связанных с YAP генов (Liu et al., 2019).
Существует также предположение, что выделяемые МСК факторы, в частности EGF и HGF, могут оказывать влияние на стволовые и родоначальные клетки печени, стимулируя их дифференцировку в гепатоциты и холангиоциты (Eom et al., 2015). Кроме того, имеются сообщения о способности кондиционированной МСК среды индуцировать дедифференцировку зрелых гепатоцитов в овальные клетки, экспрессирующие маркеры стволовых и родоначальных клеток печени (EpCAM, цитокератин-19, альфа-фетопротеин) и способные к созреванию в пролиферирующие гепатоциты, что тоже может вносить вклад в восстановление поврежденной паренхимы (Wu, Lee, 2017).
Влияние МСК на фиброгенные клетки
Способность МСК предотвращать прогрессирование фиброза печени в значительной мере обусловлена их ингибирующим влиянием на отложение компонентов внеклеточного матрикса миофибробластами, прежде всего активированными клетками Ито. При сокультивировании с этими клетками МСК подавляют их активацию, о чем свидетельствуют снижение экспрессии гладкомышечного актина (SMA), являющегося маркером миофибробластов, и уменьшение отложения коллагена (Chen et al., 2011; Sun et al., 2015; Qiao et al., 2018). Кроме того, МСК предотвращают пролиферацию активированных клеток Ито, блокируя их в фазе G0/G1 клеточного цикла (Wang et al., 2009, 2012; Chen et al., 2011; Sun et al., 2015; Qiao et al., 2018), и индуцируют их апоптотическую гибель (Lin et al., 2009; Huang et al., 2016). Результатом этого является снижение численности активированных клеток Ито, наблюдаемое в поврежденной печени экспериментальных животных после трансплантации МСК и сопровождающееся уменьшением выраженности фиброза (Zhang et al., 2011; Qiao et al., 2018).
Подобными эффектами, как in vitro, так и in vivo, обладает и кондиционированная МСК среда (Huang et al., 2016; Fu et al., 2018b; Qiao et al., 2018), что свидетельствует о преимущественно паракринном характере их влияния на клетки Ито. В то же время МСК могут подавлять пролиферацию и активацию клеток Ито через прямые межклеточные взаимодействия, опосредованные сигнальными путями NOTCH (Chen et al., 2011) и TLR4/MYD88/NF- κB (Wang et al., 2012).
Показано, что после трансплантации МСК в пораженной фиброзом печени снижается содержание главного профиброгенного цитокина, вызывающего активацию клеток Ито – TGF β1 (Jang et al., 2014; Park et al., 2015). Более того, воздействие МСК на клетки Ито ведет к снижению экспрессии ими рецепторов TGF β I и II типов, а также транскрипционных факторов SMAD2/3 и SMAD4, опосредующих фиброгенный эффект TGF β1 (Sun et al., 2015). Паракринное ингибирующее влияние МСК на пролиферацию активированных клеток Ито и отложение коллагена связано в первую очередь с секрецией ими ИЛ-10, HGF, TNF α и TGF β3, а стимуляция апоптоза – с продукцией HGF и NGF (Eom et al., 2015). В индукции апоптоза клеток Ито вырабатываемым МСК NGF участвуют факторы транскрипции NF-κB и BCL-XL (Lin et al., 2009), а угнетение пролиферации этих клеток выделяемыми МСК HGF и TGF β3 связано с усилением экспрессии p21 (Cip1) и p27 (Kip1) и снижением экспрессии циклина D1 (Wang et al., 2009). Ингибирующее влияние МСК на экспрессию клетками Ито профиброгенных генов опосредовано снижением фосфорилирования киназы ERK1/2 (Wang et al., 2009) и подавлением сигналлинга через NADPH оксидазу (Qiao et al., 2018) и Hedgehog (Hyun et al., 2015).
Иммуномодулирующие свойства МСК
В связи с тем, что неотъемлемым компонентом патологического процесса при фиброзе печени является воспаление, способность МСК регулировать его выраженность играет важную роль в их благотворном влиянии на состояние пораженной паренхимы. МСК способны подавлять пролиферацию и индуцировать апоптоз эффекторных T-клеток, тогда как пролиферация регуляторных Т-клеток под их влиянием, напротив, усиливается. Они также вызывают апоптоз нейтрофилов, ингибируют размножение B-клеток и продукцию иммуноглобулинов, подавляют созревание дендритных клеток, усиливают выработку дендритными и регуляторными Т-клетками противовоспалительных цитокинов, в частности ИЛ-10 (Паюшина, Домарацкая, 2014; Eom et al., 2015). Иммуномодулирующие эффекты МСК опосредованы экспрессией этими клетками таких факторов, как NO, простагландин Е2, индоламин-2,3-диоксигеназа (IDO), ИЛ-6, ИЛ-10 (Eom et al., 2015).
Имеются данные, что введение МСК животным после интоксикации четыреххлористым углеродом предотвращает инфильтрацию печени нейтрофилами и экспрессию в ней провоспалительных цитокинов ИЛ-6, ИЛ-12, TNF α и интерферона-γ (Pulavendran et al., 2010; Luo et al., 2019). На той же экспериментальной модели показано, что под влиянием МСК в печени снижается уровень ИЛ-17, тогда как содержание обладающих противовоспалительным действием ИЛ-10, IDO и кинуренина, напротив, повышается. При этом МСК IDO-зависимым образом подавляют пролиферацию T-хелперов типа Th17, продукция которыми ИЛ-17 способна активировать клетки Ито, и способствуют экспансии Т-регуляторных клеток с фенотипом CD4+ FoxP3+ ИЛ-10+ (Milosavljevic et al., 2018). На модели перевязки общего желчного протока показано также, что подавление фиброза после введения животным МСК сопровождается увеличением числа естественных киллеров как в паренхиме, так и в портальных областях (Duman et al., 2012).
В поврежденной печени МСК оказывают регуляторное влияние на макрофаги, снижая степень инфильтрации паренхимы этими клетками (Huang et al., 2016), а также подавляя их активацию с приобретением провоспалительного фенотипа М1 и стимулируя приобретение противовоспалительного фенотипа М2 (Luo et al., 2019). В экспериментах in vitro показана способность МСК вызывать апоптоз макрофагов (Huang et al., 2016). Помимо макрофагов, инфильтрирующих печень при фиброзе, влиянию МСК подвержены и ее резидентные макрофаги – клетки Купфера. Выделяемые МСК факторы способны предотвращать их провоспалительную активацию, что было продемонстрировано как на клеточных культурах, обработанных бактериальным липополисахаридом, так и на животных с повреждением печени (Kubo et al., 2015; Liang et al., 2019).
Влияние МСК на состояние капилляров печени
Влияние выделяемых МСК регуляторных молекул на сосудистую систему печени было исследовано на модели ее радиационного поражения, при котором некроз печеночной паренхимы, могущий впоследствии приводить к фиброзу, в значительной степени обусловлен апоптотической гибелью эндотелиальных клеток синусоидных капилляров. Введение облученному животному МСК или кондиционированной ими среды способствует выживанию эндотелиоцитов, активируя в них сигнальные пути AKT и ERK. При этом введенные МСК локализуются в периваскулярных областях и экспрессируют ангиогенные факторы VEGF и ангиопоэтин-1 (Francois et al., 2013; Chen et al., 2015).
В экспериментах in vitro было показано, что при сокультивировании с активированными клетками Ито МСК секретируют повышенное количество VEGF (Hao et al., 2017), что позволяет предполагать их проангиогенный эффект при фиброзе, вызываемом не только радиацией, но и иными причинами. Усиление ангиогенеза в печени, как было отмечено выше, способно усугублять течение фиброза, однако в комплексе с другими эффектами МСК, приводящими к нормализации состояния микроокружения, стимуляция роста сосудов может играть и положительную роль, приводя к улучшению кровоснабжения паренхимы и тем самым способствуя ее регенерации. Во всяком случае известно, что трансплантация МСК животным с фиброзом печени, индуцированным четыреххлористым углеродом, не вызывает усиления капилляризации синусоидов (Ren et al., 2010) и, более того, способна приводить к исчезновению уже развившейся капилляризации (Maksud et al., 2014).
Фибролитическая активность МСК
МСК способны не только предотвращать избыточное отложение в печени внеклеточного матрикса, но и стимулировать фибролиз за счет регуляции продукции матриксных металлопротеиназ и их ингибиторов. Показано, что под влиянием МСК в печени, пораженной фиброзом, возрастает содержание MMP-1, -2, -9, -13, -14, а содержание TIMP-1 снижается (Kim et al., 2014; Kubo et al., 2015; Mohamed et al., 2016; Rengasamy et al., 2017; Luo et al., 2019). При этом МСК как сами продуцируют фибролитические ферменты (Okura et al., 2015), так и стимулируют их продукцию активированными клетками Ито (Fu et al., 2018a).
Таким образом, МСК оказывают на патологически измененную печень многостороннее регуляторное влияние, способствующее улучшению ее структурно-функционального состояния, предотвращению прогрессирования фиброза и созданию условий для регенерации поврежденной паренхимы, что подчеркивает важность биологической роли этих клеток и определяет перспективность их использования в лечении хронических заболеваний печени.
ПРОБЛЕМЫ ИСПОЛЬЗОВАНИЯ МСК ДЛЯ РЕГЕНЕРАЦИИ ПЕЧЕНИ
Помимо всего сказанного, привлекательность МСК для клинического применения, в том числе при фиброзе и циррозе печени, связана с их доступностью для аутологичной или аллогенной трансплантации, легкостью культивирования, неиммуногенным фенотипом (Паюшина, Домарацкая, 2014; Eom et al., 2015). Однако наряду с многочисленными работами, свидетельствующими о благотворном влиянии МСК на поврежденную печень, встречаются и сообщения о неэффективности их использования для регенерации этого органа (Alves et al., 2017; Witte et al., 2017). Введенные МСК быстро элиминируются из печени за счет активности естественных киллеров (Liu et al., 2017) и макрофагов (Майбородин и др., 2019), что снижает эффективность их трансплантации. К гибели трансплантированных клеток приводит и воздействие неблагоприятного микроокружения воспаленной печени; отчасти эту проблему можно преодолеть прекондиционированием МСК либо реципиента различными физическими или химическими факторами, а также генетической модификацией клеток с целью усиления их выживаемости, миграционной способности и регенеративного потенциала (Hu et al., 2019).
До сих пор не определен оптимальный способ трансплантации МСК. Так, при внутривенном введении они преимущественно оседают в легких, чем может быть обусловлено отмечаемое в некоторых работах отсутствие влияния МСК на состояние воспаленной печени in vivo при наличии такого влияния в совместной культуре с тканью печени (Witte et al., 2017). В то же время прямое введение МСК в печень может приводить к развитию воспалительных повреждений вследствие травмирования паренхимы и внесения клеточного детрита (Майбородин и др., 2019). Нельзя сбрасывать со счетов и риск проявления фиброгенных потенций МСК (Bonzo et al., 2008; Baertschiger et al., 2009), а также их участия в развитии гепатоцеллюлярной карциномы вследствие злокачественной трансформации или создания опухолевого микроокружения (Yin et al., 2018). Кроме того, клиническое использование МСК затруднено гетерогенностью популяции и изменением характеристик клеток в ходе культивирования.
АНТИФИБРОТИЧЕСКИЙ ПОТЕНЦИАЛ ВНЕКЛЕТОЧНЫХ ВЕЗИКУЛ МСК
Эффективной и безопасной альтернативой трансплантации МСК может стать введение их секреторных продуктов, в первую очередь внеклеточных везикул, перспективы терапевтического применения которых в настоящее время активно исследуются. Выделяют два основных типа внеклеточных везикул, различающиеся по способу образования, размеру и маркерам – микровезикулы, формирующиеся путем отшнуровки выпячиваний плазмалеммы, и экзосомы, образующиеся внутри мультивезикулярных эндосомальных телец. Везикулы обоих типов содержат набор биологически активных молекул, прежде всего белков и микроРНК, и способны передавать их клеткам-мишеням. Именно продукцией внеклеточных везикул в значительной мере опосредовано регуляторное влияние МСК на регенеративные процессы (Lou et al., 2017; Fiore et al., 2018). Как показал анализ микроРНК, содержащихся в продуцируемых МСК экзосомах, их мишенями являются в первую очередь гены и сигнальные пути, регулирующие ангиогенез и ремоделирование внеклеточного матрикса, в том числе сигнальные пути TGF β и PDGF. В частности, присутствующие в этих экзосомах малая регуляторная РНК Let-7i и молекулы семейства miR-29 непосредственно влияют на экспрессию гена COL1A1; мишенью miR-29a и miR-29b является также ген FBN1, кодирующий фибронектин (Ferguson et al., 2018).
В экспериментах in vitro показано, что внеклеточные везикулы, полученные от МСК, стимулируют пролиферацию гепатоцитов (Du et al., 2017), предотвращают окислительное повреждение и апоптотическую гибель этих клеток (Yan et al., 2017) и вызывают их дедифференцировку с приобретением характеристик стволовых и родоначальных клеток печени (Wu, Lee, 2017), а также подавляют активацию клеток Ито и Купфера (Ohara et al., 2018).
При введении экспериментальным животным с хроническим повреждением печени четыреххлористым углеродом экзосомы от МСК оказывают антиоксидативный и антиапоптотический эффект, существенную роль в котором играет присутствующая в них глутатионпероксидаза 1 (Yan et al., 2017; Jiang et al., 2018). Кроме того, под влиянием экзосом в печени этих животных снижается выраженность воспаления и уменьшается отложение коллагенов I и III типов. Улучшение состояния печени сопровождается снижением содержания в ней TGF β1 и фосфорилирования SMAD2 (Li et al., 2013). Уменьшается также число клеток Купфера и активированных клеток Ито (Ohara et al., 2018). На модели фиброза, индуцированного тиоацетамидом, показано, что после введения полученных от МСК внеклеточных везикул в печени уменьшается отложение коллагена, некроз, содержание каспаз, экспрессия проапоптотического гена BAX, а также генов, кодирующих провоспалительные цитокины (TNF α, ИЛ-2) и TIMP. Экспрессия же антиапоптотического гена BCL-2, а также содержание коллагеназ (MMP-9, MMP-13) и противовоспалительных цитокинов TGF β1 и ИЛ-10 повышается. При этом противовоспалительное и противофибротическоое действие везикул сопоставимо с таковым самих МСК, от которых эти везикулы были получены (Mardpour et al., 2018).
Таким образом, имеющиеся в литературе данные свидетельствуют о возможности успешного использования внеклеточных везикул от МСК в терапии фиброза печени. По сравнению с клетками везикулы легче нарабатывать и хранить, они менее иммуногенны ввиду меньшего содержания мембранных белков, и их введение реципиенту не связано с риском образования опухоли (Lou et al., 2017). Более того, возможно направленное изменение состава и свойств внеклеточных везикул путем генетической модификации образующих их клеток, в частности, введение в них конкретных микроРНК, оказывающих гепатопротективный эффект (Fiore et al., 2018).
ЗАКЛЮЧЕНИЕ
Фиброз печени представляет собой трудноизлечимое патологическое состояние, в развитие которого вовлечены многие клеточные популяции, составляющие паренхиму и строму этого органа. Благодаря способности МСК оказывать регуляторное действие на такие биологические процессы как апоптоз, клеточная пролиферация и дифференцировка, ангиогенез, воспаление, ремоделирование внеклеточного матрикса, эти клетки рассматривают как перспективный ресурс для клеточной терапии, направленной на предотвращение прогрессирования фиброза и улучшение структурно-функционального состояния печени. Преимущественно паракринный характер влияния МСК на резидентные клетки печени делает возможным клиническое использование компонентов их секретома, прежде всего внеклеточных везикул. Однако успешное применение МСК и их секреторных продуктов для лечения фиброза печени требует дальнейшего исследования клеточных и молекулярных механизмов, лежащих в основе их терапевтического эффекта, а также подтверждения эффективности и безопасности их введения в клинических испытаниях.
Список литературы
Лукашик С.П., Алейникова О.В., Цыркунов В., Исайкина Я.И., Романова О.Н. и др., 2013. Мезенхимальные стволовые клетки как перспективный метод терапии фиброза/цирроза печени // Эксперим. и клинич. гастроэнтерология. № 12. С. 3–7.
Майбородин И.В., Фигуренко Н.Ф., Еловский А.А., Михеева Т.В., Маслов Р.В. и др., 2019. Возможность развития воспалительных повреждений интактной печени после инъекции мультипотентных стромальных клеток в эксперименте // Новости хирургии. Т. 27. № 1. С. 5–15.
Могилевец Э.В., Гарелик П.В., Батвинков Н.И., 2013. Методы стимуляции регенерации при циррозе печени // Новости хирургии. Т. 21. № 3. С. 103–109.
Паюшина О.В., 2015. Локализация и функции мезенхимных стромальных клеток in vivo // Журн. общ. биологии. Т. 76. № 2. С. 161–172.
Паюшина О.В., Домарацкая Е.И., 2014. Мезенхимные стромальные клетки как ресурс для регенерации // Жизнь без опасностей. Здоровье. Профилактика. Долголетие. Т. 9. № 4. С. 54–63.
Паюшина О.В., Домарацкая Е.И., Старостин В.И., 2012. Клеточный состав и регуляторные функции зародышевой печени // Цитология. Т. 54. № 5. С. 369–380.
Паюшина О.В., Буторина Н.Н., Никонова Т.М., Кожевникова М.Н., Шевелева О.Н., Старостин В.И., 2011. Сравнительное исследование клонального роста и дифференцировки мезенхимных стромальных клеток из печени зародышей крысы на разных сроках пренатального развития // Цитология. Т. 53. № 11. С. 859–867.
Петракова О.С., Черниогло Е.С., Терских В.В., Калистратова Е.Н., Васильев А.В., 2012. Использование клеточных технологий в лечении патологий печени // Acta Naturae. Т. 4. № 3. С. 18–33.
Шагидулин М.Ю., Горкун А.А., Онищенко Н.А., Крашенинников М.Е., Ильинский И.M. и др., 2013. Использование МСК различной онтогенетической зрелости для коррекции хронического фиброзирующего повреждения печени // Вестн. трансплантологии и искусственных органов. Т. 15. № 3. С. 73–82.
Alegre F., Pelegrin P., Feldstein A.E., 2017. Inflammasomes in liver fibrosis // Semin. Liver Dis. V. 37. № 2. P. 119–127.
Alves A.K.S., Lanzoni V., Fuziy R.A., Franco R.M.A.M.M., Maeda C.T. et al., 2017. Does mesenchymal stem cell improve the liver regeneration after the 70% hepatectomy? // Acta Cir. Bras. V. 32. № 7. P. 515–522.
Baertschiger R.M., Serre-Beinier V., Morel P., Bosco D., Peyrou M. et al., 2009. Fibrogenic potential of human multipotent mesenchymal stromal cells in injured liver // PLoS One. V. 4. № 8. P. e6657.
Bataller R., Brenner D.A., 2005. Liver fibrosis // J. Clin. Invest. V. 115. № 2. P. 209–218.
Berardis S., Dwisthi Sattwika P., Najimi M., Sokal E.M., 2015. Use of mesenchymal stem cells to treat liver fibrosis: Current situation and future prospects // World J. Gastroenterol. V. 21. № 3. P. 742–758.
Bonzo L.V., di, Ferrero I., Cravanzola C., Mareschi K., Rustichell D. et al., 2008. Human mesenchymal stem cells as a two-edged sword in hepatic regenerative medicine: Engraftment and hepatocyte differentiation versus profibrogenic potential // Gut. V. 57. № 2. P. 223–231.
Borkham-Kamphorst E., Weiskirchen R., 2016. The PDGF system and its antagonists in liver fibrosis // Cytokine Growth Factor Rev. V. 28. P. 53–61.
Cai Y., Zou Z., Liu L., Chen S., Chen Y. et al., 2015. Bone marrow-derived mesenchymal stem cells inhibits hepatocyte apoptosis after acute liver injury // Int. J. Clin. Exp. Pathol. V. 8. № 1. P. 107–116.
Chen S., Xu L., Lin N., Pan W., Hu K., Xu R., 2011. Activation of Notch1 signaling by marrow-derived mesenchymal stem cells through cell-cell contact inhibits proliferation of hepatic stellate cells // Life Sci. V. 89. № 25–26. P. 975–981.
Chen Y.X., Zeng Z.C., Sun J., Zeng H.Y., Huang Y., Zhang Z.Y., 2015. Mesenchymal stem cell-conditioned medium prevents radiation-induced liver injury by inhibiting inflammation and protecting sinusoidal endothelial cells // J. Radiat. Res. V. 56. № 4. P. 700–708.
Cho K.A., Woo S.Y., Seoh J.Y., Han H.S., Ryu K.H., 2012. Mesenchymal stem cells restore CCl4-induced liver injury by an antioxidative process // Cell Biol. Int. V. 36. № 12. P. 1267–1274.
Dominici M., Le Blanc K., Mueller I., Slaper-Cortenbach I., Marini F. et al., 2006. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement // Cytotherapy. V. 8. № 4. P. 315–317.
Du Y., Li D., Han C., Wu H., Xu L. et al., 2017. Exosomes from human-induced pluripotent stem cell-derived mesenchymal stromal cells (hiPSC-MSCs) protect liver against hepatic ischemia/ reperfusion injury via activating sphingosine kinase and sphingosine-1-phosphate signaling pathway // Cell. Physiol. Biochem. V. 43. № 2. P. 611–625.
Duman D.G., Zibandeh N., Ugurlu M.U., Celikel C., Akkoc T. et al., 2019. Mesenchymal stem cells suppress hepatic fibrosis accompanied by expanded intrahepatic natural killer cells in rat fibrosis model // Mol. Biol. Rep. V. 46. № 3. P. 2997–3008.
Eom Y.W., Shim K.Y., Baik S.K., 2015. Mesenchymal stem cell therapy for liver fibrosis // Korean J. Intern. Med. V. 30. № 5. P. 580–589.
Ewida S.F., Abdou A.G., El-Rasol Elhosary A.A., El-Ghane Metawe S.A., 2017. Hepatocyte-like versus mesenchymal stem cells in CCl4-induced liver fibrosis // Appl. Immunohistochem. Mol. Morphol. V. 25. № 10. P. 736–745.
Fabregat I., Moreno-Càceres J., Sánchez A., Dooley S., Dewidar B. et al., 2016. TGF-β signalling and liver disease // FEBS J. V. 283. № 12. P. 2219–2232.
Ferguson S.W., Wang J., Lee C.J., Liu M., Neelamegham S. et al., 2018. The microRNA regulatory landscape of MSC-derived exosomes: A systems view // Sci. Rep. V. 8. № 1. P. 1419.
Fiore E.J., Domínguez L.M., Bayo J., García M.G., Mazzolini G.D., 2018. Taking advantage of the potential of mesenchymal stromal cells in liver regeneration: Cells and extracellular vesicles as therapeutic strategies // World J. Gastroenterol. V. 24. № 23. P. 2427–2440.
Francois S., Mouiseddine M., Allenet-Lepage B., Voswinkel J., Douay L. et al., 2013. Human mesenchymal stem cells provide protection against radiation-induced liver injury by antioxidative process, vasculature protection, hepatocyte differentiation, and trophic effects // BioMed Res. Int. V. 2013. P. 151679.
Fu Q., Ohnishi S., Sakamoto N., 2018a. Conditioned medium from human amnion-derived mesenchymal stem cells regulates activation of primary hepatic stellate cells // Stem Cells Int. V. 2018. P. 4898152.
Fu X., Jiang B., Zheng B., Yan Y., Wang J. et al., 2018b. Heterogenic transplantation of bone marrow-derived rhesus macaque mesenchymal stem cells ameliorates liver fibrosis induced by carbon tetrachloride in mouse // PeerJ. V. 6. P. e4336.
Gu J., Shi X., Chu X., Zhang Y., Ding Y., 2009. Contribution of bone marrow mesenchymal stem cells to porcine hepatocyte culture in vitro // Biochem. Cell Biol. V. 87. № 4. P. 595–604.
Guo Y., Chen B., Chen L.J., Zhang C.F., Xiang C., 2016. Current status and future prospects of mesenchymal stem cell therapy for liver fibrosis // J. Zhejiang Univ. Sci. B. V. 17. № 11. P. 831–841.
Hall C., Sato K., Wu N., Zhou T., Kyritsi K. et al., 2017. Regulators of cholangiocyte proliferation // Gene Expr. V. 17. № 2. P. 155–171.
Hao T., Chen J., Zhi S., Zhang Q., Chen G., Yu F., 2017. Comparison of bone marrow-vs. adipose tissue-derived mesenchymal stem cells for attenuating liver fibrosis // Exp. Ther. Med. V. 14. № 6. P. 5956–5964.
Hernandez-Gea V., Friedman S.L., 2011. Pathogenesis of liver fibrosis // Annu. Rev. Pathol. V. 6. P. 425–456.
Hirata M., Ishigami M., Matsushita Y., Ito T., Hattori H. et al., 2016. Multifaceted therapeutic benefits of factors derived from dental pulp stem cells for mouse liver fibrosis // Stem Cells Transl. Med. V. 5. № 10. P. 1416–1424.
Hu C., Zhao L., Duan J., Li L., 2019. Strategies to improve the efficiency of mesenchymal stem cell transplantation for reversal of liver fibrosis // J. Cell. Mol. Med. V. 23. № 3. P. 1657–1670.
Huang B., Cheng X., Wang H., Huang W., la Gahu Z. et al., 2016. Mesenchymal stem cells and their secreted molecules predominantly ameliorate fulminant hepatic failure and chronic liver fibrosis in mice respectively // J. Transl. Med. V. 14. P. 45.
Hyun J., Wang S., Kim J., Kim G.J., Jung Y., 2015. MicroRNA125b‑mediated Hedgehog signaling influences liver regeneration by chorionic plate-derived mesenchymal stem cells // Sci. Rep. V. 5. P. 14135.
Jang Y.O., Kim M.Y., Cho M.Y., Baik S.K., Cho Y.Z., Kwon S.O., 2014. Effect of bone marrow-derived mesenchymal stem cells on hepatic fibrosis in a thioacetamide-induced cirrhotic rat model // BMC Gastroenterol. V. 14. P. 198.
Jiang W., Tan Y., Cai M., Zhao T., Mao F. et al., 2018. Human umbilical cord MSC-derived exosomes suppress the development of CCl4-induced liver injury through antioxidant effect // Stem Cells Int. V. 2018. P. 6079642.
Kim M.D., Kim S.S., Cha H.Y., Jang S.H., Chang D.Y. et al., 2014. Therapeutic effect of hepatocyte growth factor-secreting mesenchymal stem cells in a rat model of liver fibrosis // Exp. Mol. Med. V. 46. P. e110.
Koyama Y., Brenner D.A., 2017. Liver inflammation and fibrosis // J. Clin. Invest. V. 127. № 1. P. 55–64.
Kubo K., Ohnishi S., Hosono H., Fukai M., Kameya A. et al., 2015. Human amnion-derived mesenchymal stem cell transplantation ameliorates liver fibrosis in rats // Transplant. Direct. V. 1. № 4. P. e16.
Li T., Yan Y., Wang B., Qian H., Zhang X. et al., 2013. Exosomes derived from human umbilical cord mesenchymal stem cells alleviate liver fibrosis // Stem Cells Dev. V. 22. № 6. P. 845–854.
Liang X., Li T., Zhou Q., Pi S., Li Y. et al., 2019. Mesenchymal stem cells attenuate sepsis-induced liver injury via inhibiting M1 polarization of Kupffer cells // Mol. Cell. Biochem. V. 452. № 1–2. P. 187–197.
Liedtke C., Luedde T., Sauerbruch T., Scholten D., Streetz K. et al., 2013. Experimental liver fibrosis research: Update on animal models, legal issues and translational aspects // Fibrogenesis Tissue Repair. V. 6. № 1. P. 19.
Lin N., Hu K., Chen S., Xie S., Tang Z. et al., 2009. Nerve growth factor-mediated paracrine regulation of hepatic stellate cells by multipotent mesenchymal stromal cells // Life Sci. V. 85. № 7–8. P. 291–295.
Liu J.J., Hu X.J., Li Z.R., Yan R.H., Li D. et al., 2017. In vivo bioluminescence imaging of transplanted mesenchymal stromal cells and their rejection mediated by intrahepatic NK cells // Mol. Imaging Biol. V. 19. № 1. P. 31–40.
Liu Y., Ren H., Wang J., Yang F., Li J. et al., 2019. Prostaglandin E2 secreted by mesenchymal stem cells protects against acute liver failure via enhancing hepatocyte proliferation // FASEB J. V. 33. № 2. P. 2514–2525.
Liu Z., Meng F., Li C., Zhou X., Zeng X. et al., 2014. Human umbilical cord mesenchymal stromal cells rescue mice from acetaminophen-induced acute liver failure // Cytotherapy. V. 16. № 9. P. 1207–1219.
Lou G., Chen Z., Zheng M., Liu Y., 2017. Mesenchymal stem cell-derived exosomes as a new therapeutic strategy for liver diseases // Exp. Mol. Med. V. 49. № 6. P. e346.
Lua I., Asahina K., 2016. The role of mesothelial cells in liver development, injury, and regeneration // Gut Liver. V. 10. № 2. P. 166–176.
Lua I., Li Y., Zagory J.A., Wang K.S., French S.W. et al., 2016. Characterization of hepatic stellate cells, portal fibroblasts, and mesothelial cells in normal and fibrotic livers // J. Hepatol. V. 64. № 5. P. 1137–1146.
Luangmonkong T., Suriguga S., Mutsaers H.A.M., Groothuis G.M.M., Olinga P., Boersema M., 2018. Targeting oxidative stress for the treatment of liver fibrosis // Rev. Physiol. Biochem. Pharmacol. V. 175. P. 71–102.
Luo X.Y., Meng X.J., Cao D.C., Wang W., Zhou K. et al., 2019. Transplantation of bone marrow mesenchymal stromal cells attenuates liver fibrosis in mice by regulating macrophage subtypes // Stem Cell Res. Ther. V. 10. № 1. P. 16.
Lysy P.A., Campard D., Smets F., Najimi M., Sokal E.M., 2008. Stem cells for liver tissue repair: Current knowledge and perspectives // World J. Gastroenterol. V. 14. № 6. P. 864–875.
Malagola E., Teunissen M., Laan L.J., van der, Verstegen M.M., Schotanus B.A. et al., 2016. Characterization and comparison of canine multipotent stromal cells derived from liver and bone marrow // Stem Cells Dev. V. 25. № 2. P. 139–150.
Maksud A.R., Gennadiyevich P.A., Elkhan J.R., 2014. Influence of mesenchymal stem cells transplantation on regeneration activity of cirrhotic liver // Euroasian J. Hepatogastroenterol. V. 4. № 2. P. 83–86.
Mardpour S., Hassani S.N., Mardpour S., Sayahpour F., Vosough M. et al., 2018. Extracellular vesicles derived from human embryonic stem cell-MSCs ameliorate cirrhosis in thioacetamide-induced chronic liver injury // J. Cell. Physiol. V. 233. № 12. P. 9330–9344.
Mekala S., Tulimilli S.V., Geesala R., Manupati K., Dhoke N.R., Das A., 2018. Cellular crosstalk mediated by platelet-derived growth factor BB and transforming growth factor β during hepatic injury activates hepatic stellate cells // Can. J. Physiol. Pharmacol. V. 96. № 8. P. 728–741.
Milosavljevic N., Gazdic M., Simovic Markovic B., Arsenijevic A., Nurkovic J. et al., 2018. Mesenchymal stem cells attenuate liver fibrosis by suppressing Th17 cells – an experimental study // Transpl. Int. V. 31. № 1. P. 102–115.
Mohamed H.E., Elswefy S.E., Rashed L.A., Younis N.N., Shaheen M.A., Ghanim A.M., 2016. Bone marrow-derived mesenchymal stem cells effectively regenerate fibrotic liver in bile duct ligation rat model // Exp. Biol. Med. (Maywood). V. 241. № 6. P. 581–591.
Natarajan V., Harris E.N., Kidambi S., 2017. SECs (sinusoidal endothelial cells), liver microenvironment, and fibrosis // BioMed Res. Int. V. 2017. P. 4097205.
Novo E., Busletta C., Bonzo L.V., Povero D., Paternostro C. et al., 2011. Intracellular reactive oxygen species are required for directional migration of resident and bone marrow-derived hepatic pro-fibrogenic cells // J. Hepatol. V. 54. № 5. P. 964–974.
Ohara M., Ohnishi S., Hosono H., Yamamoto K., Yuyama K. et al., 2018. Extracellular vesicles from amnion-derived mesenchymal stem cells ameliorate hepatic inflammation and fibrosis in rats // Stem Cells Int. V. 2018. P. 3212643.
Okura H., Soeda M., Morita M., Fujita M., Naba K. et al., 2015. Therapeutic potential of human adipose tissue-derived multi-lineage progenitor cells in liver fibrosis // Biochem. Biophys. Res. Commun. V. 456. № 4. P. 860–865.
Overi D., Carpino G., Cardinale V., Franchitto A., Safarikia S. et al., 2018. Contribution of resident stem cells to liver and biliary tree regeneration in human diseases // Int. J. Mol. Sci. V. 19. № 10. P. E2917.
Pan Q., Fouraschen S.M., Kaya F.S., Verstegen M.M., Pescatori M. et al., 2011. Mobilization of hepatic mesenchymal stem cells from human liver grafts // Liver Transpl. V. 17. № 5. P. 596–609.
Park M., Kim Y.H., Woo S.Y., Lee H.J., Yu Y. et al., 2015. Tonsil-derived mesenchymal stem cells ameliorate CCl4-induced liver fibrosis in mice via autophagy activation // Sci. Rep. V. 5. P. 8616.
Penz-Österreicher M., Österreicher C.H., Trauner M., 2011. Fibrosis in autoimmune and cholestatic liver disease // Best Pract. Res. Clin. Gastroenterol. V. 25. № 2. P. 245–258.
Poll D., van, Parekkadan B., Cho C.H., Berthiaume F., Nahmias Y. et al., 2008. Mesenchymal stem cell-derived molecules directly modulate hepatocellular death and regeneration in vitro and in vivo // Hepatology. V. 47. № 5. P. 1634–1643.
Pulavendran S., Vignesh J., Rose C., 2010. Differential anti-inflammatory and anti-fibrotic activity of transplanted mesenchymal vs. hematopoietic stem cells in carbon tetrachloride-induced liver injury in mice // Int. Immunopharmacol. V. 10. № 4. P. 513–519.
Qiao H., Zhou Y., Qin X., Cheng J., He Y., Jiang Y., 2018. NADPH oxidase signaling pathway mediates mesenchymal stem cell-induced inhibition of hepatic stellate cell activation // Stem Cells Int. V. 2018. P. 1239143.
Quintanilha L.F., Takami T., Hirose Y., Fujisawa K., Murata Y. et al., 2014. Canine mesenchymal stem cells show antioxidant properties against thioacetamide-induced liver injury in vitro and in vivo // Hepatol. Res. V. 44. № 10. P. E206–E217.
Raafat N., Abdel Aal S.M., Abdo F.K., El Ghonaimy N.M., 2015. Mesenchymal stem cells: In vivo therapeutic application ameliorates carbon tetrachloride induced liver fibrosis in rats // Int. J. Biochem. Cell. Biol. V. 68. P. 109–118.
Ren H., Zhao Q., Cheng T., Lu S., Chen Z. et al., 2010. No contribution of umbilical cord mesenchymal stromal cells to capillarization and venularization of hepatic sinusoids accompanied by hepatic differentiation in carbon tetrachloride-induced mouse liver fibrosis // Cytotherapy. V. 12. № 3. P. 371–383.
Rengasamy M., Singh G., Fakharuzi N.A., Siddikuzzaman, Balasubramanian S. et al., 2017. Transplantation of human bone marrow mesenchymal stromal cells reduces liver fibrosis more effectively than Wharton’s jelly mesenchymal stromal cells // Stem Cell Res. Ther. V. 8. № 1. P. 143.
Schuppan D., 2015. Liver fibrosis: Common mechanisms and antifibrotic therapies // Clin. Res. Hepatol. Gastroenterol. V. 39. Suppl. 1. P. S51–S59.
Seki E., Brenner D.A., 2015. Recent advancement of molecular mechanisms of liver fibrosis // J. Hepatobilliary Pancreat. Sci. V. 22. № 7. P. 512–518.
Shah R., Reyes‑Gordillo K., Arellanes‑Robledo J., Lechuga C.G., Hernández‑Nazara Z. et al., 2013. TGF-β 1 up-regulates the expression of PDGF β receptor mRNA and induces a delayed PI3K-, AKT- and p70(S6K)-dependent proliferative response in activated hepatic stellate cells // Alcohol. Clin. Exp. Res. V. 37. № 11. P. 1838–1848.
Sun X.E., Zhang X.Q., Liu M.M., 2015. Effect of bone marrow mesenchymal stem cells on the TGF-β1/Smad signaling pathway of hepatic stellate // Genet. Mol. Res. V. 14. № 3. P. 8744–8754.
Tsay H.C., Yuan Q., Balakrishnan A., Kaiser M., Möbus S. et al., 2019. Hepatocyte-specific suppression of microRNA-221-3p mitigates liver fibrosis // J. Hepatol. V. 70. № 4. P. 722–734.
Tu X., Zhang Y., Zheng X., Deng J., Li H. et al., 2017. TGF-β-induced hepatocyte lincRNA-p21 contributes to liver fibrosis in mice // Sci. Rep. V. 7. № 1. P. 2957.
Wang H., Zhang H., Huang B., Miao G., Yan X. et al., 2018. Mesenchymal stem cells reverse high‑fat diet‑induced non‑alcoholic fatty liver disease through suppression of CD4+ T lymphocytes in mice // Mol. Med. Rep. V. 17. № 3. P. 3769–3774.
Wang J., Bian C., Liao L., Zhu Y., Li J. et al., 2009. Inhibition of hepatic stellate cells proliferation by mesenchymal stem cells and the possible mechanisms // Hepatol. Res. V. 39. № 12. P. 1219–1228.
Wang P.P., Xie D.Y., Liang X.J., Peng L., Zhang G.L. et al., 2012. HGF and direct mesenchymal stem cells contact synergize to inhibit hepatic stellate cells activation through TLR4/NF-kB pathway // PLoS One. V. 7. № 8. P. e43408.
Witte S.F.H., de, Merino A.M., Franquesa M., Strini T., Zoggel J.A.A., van, et al., 2017. Cytokine treatment optimises the immunotherapeutic effects of umbilical cord-derived MSC for treatment of inflammatory liver disease // Stem Cell Res. Ther. V. 8. № 1. P. 140.
Wu H.H., Lee O.K., 2017. Exosomes from mesenchymal stem cells induce the conversion of hepatocytes into progenitor oval cells // Stem Cell Res. Ther. V. 8. № 1. P. 117.
Xie J., Wang W., Si J.W., Miao X.Y., Li J.C. et al., 2013. Notch signaling regulates CXCR4 expression and the migration of mesenchymal stem cells // Cell. Immunol. V. 281. № 1. P. 68–75.
Xu F., Liu C., Zhou D., Zhang L., 2016. TGF-β/SMAD pathway and its regulation in hepatic fibrosis // J. Histochem. Cytochem. V. 64. № 3. P. 157–167.
Xu M., Wang X., Zou Y., Zhong Y., 2017a. Key role of liver sinusoidal endothelial cells in liver fibrosis // Biosci. Trends. V. 11. № 2. P. 163–168.
Xu T.B., Li L., Luo X.D., Lin H., 2017b. BMSCs protect against liver injury via suppressing hepatocyte apoptosis and activating TGF-β1/Bax singling pathway // Biomed. Pharmacother. V. 96. P. 1395–1402.
Xu X., Li D., Li X., Shi Q., Ju X., 2017c. Mesenchymal stem cell conditioned medium alleviates oxidative stress injury induced by hydrogen peroxide via regulating miR143 and its target protein in hepatocytes // BMC Immunol. V. 18. № 1. P. 51.
Yan Y., Jiang W., Tan Y., Zou S., Zhang H. et al., 2017. hucMSC exosome-derived GPX1 is required for the recovery of hepatic oxidant injury // Mol. Ther. V. 25. № 2. P. 465–479.
Yin Z., Jiang K., Li R., Dong C., Wang L., 2018. Multipotent mesenchymal stromal cells play critical roles in hepatocellular carcinoma initiation, progression and therapy // Mol. Cancer. V. 17. № 1. P. 178.
Ying H.Z., Chen Q., Zhang W.Y., Zhang H.H., Ma Y. et al., 2017. PDGF signaling pathway in hepatic fibrosis pathogenesis and therapeutics (Review) // Mol. Med. Rep. V. 16. № 6. P. 7879–7889.
You Q., Holt M., Yin H., Li G., Hu C.J., Ju C., 2013. Role of hepatic resident and infiltrating macrophages in liver repair after acute injury // Biochem. Pharmacol. V. 86. № 6. P. 836–843.
Zhang D., Jiang M., Miao D., 2011. Transplanted human amniotic membrane-derived mesenchymal stem cells ameliorate carbon tetrachloride-induced liver cirrhosis in mouse // PLoS One. V. 6. № 2. P. e16789.
Zhang S., Lv C., Yang X., Han Z., Zhang S. et al., 2015. Corticosterone mediates the inhibitory effect of restraint stress on the migration of mesenchymal stem cell to carbon tetrachloride-induced fibrotic liver by downregulating CXCR4/7 expression // Stem Cells Dev. V. 24. № 5. P. 587–596.
Zhang S., Zhu Z., Wang Y., Liu S., Zhao C. et al., 2018. Therapeutic potential of Bama miniature pig adipose stem cells induced hepatocytes in a mouse model with acute liver failure // Cytotechnology. V. 70. № 4. P. 1131–1141.
Zhang X., Du G., Xu Y., Li X., Fan W. et al., 2016. Inhibition of notch signaling pathway prevents cholestatic liver fibrosis by decreasing the differentiation of hepatic progenitor cells into cholangiocytes // Lab. Invest. V. 96. № 3. P. 350–360.
Zhou X., Cui L., Zhou X., Yang Q., Wang L. et al., 2017. Induction of hepatocyte-like cells from human umbilical cord-derived mesenchymal stem cells by defined microRNAs // J. Cell. Mol. Med. V. 21. № 5. P. 881–893.
Дополнительные материалы отсутствуют.
Инструменты
Журнал общей биологии