

Океанология, 2020, T. 60, № 2, стр. 281-292
Коромантийный цикл углерода и происхождение абиогенных углеводородов
Н. О. Сорохтин 1, *, Л. И. Лобковский 1, Н. Е. Козлов 2, **
1 Институт океанологии им. П.П. Ширшова РАН
Москва, Россия
2 Геологический институт Кольского НЦ РАН
Апатиты, Россия
* E-mail: nsorokhtin@ocean.ru
** E-mail: kozlov.n.e@yandex.ru
Поступила в редакцию 20.12.2017
После доработки 28.06.2018
Принята к публикации 02.07.2018
Аннотация
В работе исследованы процессы трансформации и переноса углерода в коре и мантии. Так, затянутые в зонах субдукции осадки дегидратируются, разрушаются и преобразуются метаморфическими процессами. Часть углеродистых соединений погружается в подлитосферную мантию и переносится конвективными течениями в рифтовые зоны срединно-океанических хребтов. Там они снова трасформируются, образуя новые химические соединения, и выносятся гидротермами на поверхность в виде растворенных во флюиде карбонатов, различных углеводородов и углекислого газа. Выпадая из растворов, они отлагаются на морском дне в виде осадков, образуя карбонатные и углеродсодержащие структурно-вещественные комплексы. В результате проявления многоступенчатого механизма физико-химических преобразований в коромантийных областях Земли углеродистые соединения приобретают черты абиогенного (мантийного) происхождения, хотя и являются изначально экзогенными образованиями. Коромантийный цикл углерода является частью глобального процесса циклического переноса углерода из атмосферы в мантию и обратно.
ВВЕДЕНИЕ
Геодинамическая эволюция океанов тесно связана с процессами конвейерного обогащения земной коры многими химическими элементами и соединениями, которые накапливаются на ее поверхности в конвергентных и дивергентных областях Земли. Исследование процессов геохимического цикла углерода между различными резервуарами традиционно описывают явления его трансформации в коре, гидросфере и атмосфере, при которой важную роль играют живые организмы [4, 12, 16]. Вопросы поведения изотопов этого элемента в геохимическом цикле подробно рассматривались в работе [5]. Интересный и неоднозначный вариант природы геохимического цикла углерода с привлечением экспериментальных данных о возможных мантийных потоках и резервуарах был предложен Добрецовым и Шацким [8], а так же [7, 21]. Они считают, что глубинный цикл углерода имеет значительную подпитку из контактовых зон ядра и нижней мантии Земли за счет его выноса восходящими плюмами в присутствии воды и кислорода. Проведенные нами исследования закономерностей коромантийного взаимодействия слагающих данные геосферы структурно-вещественных комплексов позволило обосновать наличие глубинной ветви геохимического цикла углерода без привлечения механизмов его генерации во внешнем ядре и нижней мантии, а также без существенного количества воды и кислорода в последней.
Основным поставщиком углерода, формирующего его коромантийную ветвь в глобальном цикле, являются отлагающиеся на морском дне осадочные комплексы. Они содержат большое количество соединений данного элемента и представлены карбонатными осадками биогенного и хемогенного происхождения, а также органическим веществом из пелагических и терригенных отложений и углеродистых сланцев, сносимых с окраин континентов. Затянутые в зоны поддвига плит они претерпевают ряд изменений и погружаются в подлитосферную мантию, где практически полностью разрушаются, переплавляются и, в основном, выносятся на поверхность в виде магматических тел и флюидных растворов. Часть соединений углерода и его мономинеральная фракция капсулируются, достигают уровней подлитосферной мантии и переносятся верхнемантийными конвективными течениями в области разгрузки под рифтовыми зонами литосферных плит, где в виде новых соединений снова попадают в гидросферу Земли (рис. 1 ).
В настоящей работе рассмотрен механизм многоступенчатой трансформации углеродистых соединений и переход органического углерода в неорганический и обратно. Это позволяет рассматривать коромантийную ветвь глобального цикла углерода, как движение и трансформацию экзогенного углерода в природе без добавления мантийной составляющей данного компонента. Авторы подчеркивают, что при написании соответствующих химических реакций они ориентировались на опубликованные теоретические и экспериментальные данные. Там, где они отсутствуют, рассуждения о возможном протекании реакций в метаморфически нагруженных условиях следует рассматривать как возможные.
ТЕРМОДИНАМИКА ЗОН СУБДУКЦИИ
Происходящие в зонах поддвига плит процессы дегидратации и анатексиса океанической коры развиваются по достаточно сложной многоступенчатой схеме. При этом пространственно-временнáя изменчивость метаморфических преобразований заключается в том, что породные ассоциации пододвигающейся океанической литосферной плиты испытывают прогрессивный метаморфизм в зоне контакта с надвигающимся континентом. Они последовательно проходят стадии преобразования от нижних ступеней к высшим. Формирующийся в этих условиях минерализованный и газонасыщенный флюид перемещается вверх по разломам и, остывая, вызывает ретроградные контактово-метасоматические изменения окружающих горных пород. Многочисленные протрузии ультраосновного состава и офиолиты, пройдя пик изменений, также испытывают ретроградные процессы. Наряду с этим, осадочные толщи окраины континента сносятся в океан, смешиваются с пелагическими осадками и совместно с ними затягиваются в зону поддвига плит. Содержащие органическое вещество терригенные осадки существенно увеличивают приток углерода в общий баланс и испытывают прогрессивный метаморфизм, омываясь насыщенными гидротермальными растворами. В процессе метаморфических преобразований пород океанической коры оливин, энстатит, магнетит и другие ее тугоплавкие минералы, а также гранаты, возникающие на глубинах эклогитового перехода, в большинстве своем удаляются из системы вместе с погружающейся в мантию литосферной плитой. При этом, водные флюиды, кремнезем и литофильные соединения ассимилируются формирующимися в зонах поддвига плит силикатными расплавами и преимущественно отжимаются вверх по разломам.
В этих условиях плавление осадков и осадочных пород в зонах субдукции происходит в основном за счет диссипации энергии вязкого трения внутри них и трения на контакте литосферных плит. К этому же добавляется и величина глубинного теплового потока, пронизывающего континентальные литосферные плиты, тогда как водонасыщенность затягиваемых вещественных комплексов, напротив, снижает температуру их плавления. Все это позволяет предположить, что температура в зазоре между плитами примерно соответствует геотерме континентальной плиты или чуть выше нее. Следовательно, попавшие в зону субдукции образования начинают плавиться только на глубинах, где геотерма континентальной плиты пересекается с температурой плавления осадков. При этом температура плавления большинства силикатов в присутствии воды с повышением давления до 5–10 кбар резко снижается до 600‒700°С [10]. Аналогичным образом ведут себя водонасыщенные карбонаты [32] и многие другие соединения. Отмеченные закономерности позволяют заключить, что алюмосиликатные водонасыщенные осадки начинают плавиться уже на глубинах около 50‒70 км, а карбонатные – около 80 км.
Глубже критического уровня пересечения континентальной геотермы с кривой начала плавления осадочного вещества степень плавления осадочных пород резко возрастает. Поэтому на больших глубинах должна происходить дифференциация образовавшихся расплавов и их разделение по плотности. Тяжелые железистые и сульфидные фракции уходят вниз и, в конце концов, погружаются в глубины конвектирующей мантии. Легкие же компоненты, состоящие из отделившихся флюидов, карбонатных и силикатных расплавов, не имеют возможности подняться вверх и накапливаются (консервируются) в нижних горизонтах континентальных плит, постепенно формируя там очаги щелочно-ультраосновных, карбонатитовых и лампроит-кимберлитовых магм. По-видимому, в этой зоне процессы дегидратации океанической коры и фрагментов осадочных толщ протекают не в полной мере, поэтому оставшаяся часть кристаллизационной воды, углерода, углекислого газа и некоторых других летучих компонентов может погружаться в конвектирующую мантию.
Затягивание в зону субдукции осадков приводит к процессу их многоступенчатого разложения, трансформации и выделения мономинерального углерода. На глубинах около 120–150 км наблюдается фазовый переход от графита к алмазу, ниже которого располагается область устойчивости последнего. Именно на таких глубинах кристаллизуются алмазы и возникают характерные минеральные ассоциации в эклогитах и гранатовых перидотитах алмаз-пироповой фации глубинности [19]. С другой стороны, известно [40], что на глубинах около 350 км ромбический оливин должен переходить в более плотную кубическую модификацию (шпинелевую фазу) – рингвудит. Однако этот минерал еще нигде не встречался в кимберлитах или алмазных включениях, что, по-видимому, ограничивает максимальную глубину формирования алмазоносных пород 300 км [9].
Приведенные данные позволяют предположить, что на глубинах превышающих 250–300 км углерод снова переходит в фазу графита и, попадая в область устойчивости карбидов металлов, образует с ними разнообразные соединения. В природе известно небольшое количество минералов карбидов, которые встречаются в метеоритах, кимберлитах, метаморфизованных ультраосновных породах и в шунгитах. Наиболее известными являются когенит (Fe,Ni,Co)3C), муассанит (SiC), танталкарбид (Ta,Nb)C), ниобокарбид (Nb,Ta)C), хамрабаевит (Ti,V,Fe)C) и соединения ванадия (V8C7 и V2C) и хрома (Cr2C3). Столь ограниченное количество минералов карбидов металлов объясняется их глубинным происхождением и склонностью к разложению в низких термобарических условиях и в присутствии воды. Скорее всего, в верхней мантии минералы карбидов металлов имеют более широкое распространение и, следовательно, могут существовать карбиды кальция, алюминия, марганца, железа и ряда других.
УГЛЕРОД В ЗОНАХ ПОДДВИГА ПЛИТ
Все химические реакции в зонах поддвига плит необратимы, проходят с поглощением или выделением тепла и в разных окислительно-восстановительных условиях. В реализации всех перечисленных процессов важную роль играет геологическое время, приводящее, в конечном итоге, физико-химические параметры развития складчатой системы в равновесное состояние.
Современные осадочные толщи на морском дне содержат до 20–40% воды, а в диагенетированных разностях содержание падает до 10–15%. При этом, в глинистых породах образуются гидрослюды – иллит, смектит, монтмориллонит, каолин и диаспор. Содержится в них и большое количество органического вещества (0.5–1.0%).
На ранней стадии метаморфических преобразований затягиваемые в зону субдукции осадки и осадочные породы претерпевают процессы их интенсивной дегидратации. Вначале теряется поровая (свободная) вода, затем кристаллизационная, после чего в них развивается сложный ряд эндотермических метаморфических преобразований, сопровождаемых освобождением воды, CO2, кремнезема, щелочей (особенно калия) и литофильных элементов. В зонах проявления максимальных сжатий породы уплотняются и частично запечатывают образующиеся растворы, создавая высокое давление флюида и расширяя поле устойчивости водосодержащих минералов.
Большая часть образующихся таким образом флюидных потоков перемещается снизу вверх из области высоких давлений в зоны тектонической тени. При существовании тангенциального градиента давления в проницаемой среде всегда будет наблюдаться их движение и переход из одной метаморфической фации в другую. Примечательно, что на больших глубинах, в зонах субдукции должна стираться грань контакта литосферных плит. Возникает данный эффект благодаря близости химического состава вещества третьего слоя океанической и подкоровой континентальной литосфер. В результате этого, погружающаяся в мантию деградирующая океаническая кора совместно с остатками осадочного чехла оказывается зажатой между близкими по составу комплексами мантийного происхождения. Это неизбежно приводит к разобщению и изоляции крупных и мелких объемов вещества, капсулированию коровых расплавов, метаморфизованных твердых коровых пород, рассеянного вещества, флюидных растворов и газово-жидких включений. Флюид на этих глубинах приобретает черты сверхкритической жидкости. Далее, в условиях вязкого течения среды “капсулы” с коровым веществом попадают в конвектирующую мантию и разносятся на большие расстояния, отрываясь от погружающейся к ядру литосферной плиты или совместно перемещаясь в область восходящего конвективного потока в случае ее выполаживания (рис. 1 ).
Рис. 1.
Коромантийный цикл углерода: 1 – океаническая литосфера; 2 – континентальная кора; 3 – подкоровая литосфера континента; 4 – зона перехода подкоровой литосферы континента к литосфере океанического типа; 5 – направление конвективных течений в верхней мантии; 6 – направление миграции соединений углерода.
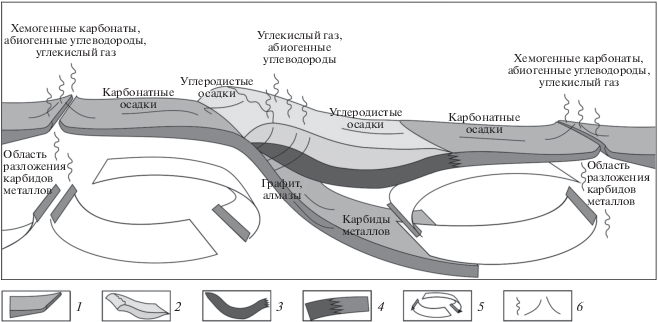
Попадающие в зону поддвига плит карбонаты трансформируются и разрушаются с выделением СО2. Это приводит к связыванию одних оснований в силикатных фазах, а других в карбонатах. Процесс разложения карбонатов может протекать по следующим эндотермическими реакциям:
(1)
$\mathop {{\text{CaC}}{{{\text{O}}}_{{\text{3}}}}}\limits_{{\text{кальцит}}} \, + {\text{Si}}{{{\text{O}}}_{2}} + {{T}^{^\circ }}{\text{C}} \to \mathop {{\text{CaSi}}{{{\text{O}}}_{3}}}\limits_{{\text{волластонит}}} + {\text{C}}{{{\text{O}}}_{2}}{\kern 1pt} \uparrow ,$(2)
$\mathop {{\text{MgC}}{{{\text{O}}}_{3}}}\limits_{{\text{магнезит}}} \, + {\text{Si}}{{{\text{O}}}_{2}} + {{T}^{^\circ }}{\text{C}} \to \mathop {{\text{MgSi}}{{{\text{O}}}_{3}}}\limits_{{\text{энстатит}}} \, + {\text{C}}{{{\text{O}}}_{2}}{\kern 1pt} \uparrow ,$(3)
$\mathop {2{\text{FeC}}{{{\text{O}}}_{3}}}\limits_{{\text{сидерит}}} \, + {\text{Si}}{{{\text{O}}}_{2}} + {{T}^{^\circ }}{\text{C}} \to \mathop {{\text{F}}{{{\text{e}}}_{2}}{\text{Si}}{{{\text{O}}}_{4}}}\limits_{{\text{фаялит}}} \, + 2{\text{C}}{{{\text{O}}}_{2}}{\kern 1pt} \uparrow {\kern 1pt} .$В условиях высоких давлений (40–50 кбар), характерных для нижних частей континентальных плит, распад карбонатов, по-видимому, должен сопровождаться окислением двухвалентного железа с образованием плотных кристаллических структур магнетита и восстановлением СО2 до окиси углерода:
(4)
$\begin{gathered} \mathop {3{\text{CaC}}{{{\text{O}}}_{3}}}\limits_{{\text{кальцит}}} \, + \mathop {3{\text{FeTi}}{{{\text{O}}}_{3}}}\limits_{{\text{ильменит}}} \, + {{T}^{^\circ }}{\text{C}} \to \\ \to \mathop {3{\text{CaTi}}{{{\text{O}}}_{3}}}\limits_{{\text{перовскит}}} \, + \mathop {{\text{F}}{{{\text{e}}}_{3}}{{{\text{O}}}_{4}}}\limits_{{\text{магнетит}}} \, + 2{\text{C}}{{{\text{O}}}_{2}} + {\text{CO}}{\kern 1pt} \uparrow {\kern 1pt} . \\ \end{gathered} $На еще больших глубинах происходит образование гранатов, корунда и кальцита, с выделением углекислого газа:
(5)
$\begin{gathered} 3{\text{Ca}}\mathop {{\text{[A}}{{{\text{l}}}_{{\text{2}}}}{\text{S}}{{{\text{i}}}_{{\text{2}}}}{{{\text{O}}}_{8}}{\text{]}}}\limits_{{\text{анортит}}} \,{\kern 1pt} + {\kern 1pt} \,\mathop {6{\text{MgC}}{{{\text{O}}}_{3}}}\limits_{{\text{магнезит}}} \to \\ \to \mathop {2{\text{M}}{{{\text{g}}}_{{\text{3}}}}{\text{A}}{{{\text{l}}}_{2}}[{\text{Si}}{{{\text{O}}}_{4}}]}\limits_{{\text{пироп}}} \,{\kern 1pt} + {\kern 1pt} \,\mathop {3{\text{CaC}}{{{\text{O}}}_{3}}}\limits_{{\text{кальцит}}} \, + \mathop {{\text{A}}{{{\text{l}}}_{2}}{{{\text{O}}}_{3}}}\limits_{{\text{корунд}}} \, + 3{\text{C}}{{{\text{O}}}_{2}}{\kern 1pt} \uparrow {\kern 1pt} . \\ \end{gathered} $(6)
$\begin{gathered} \mathop {{\text{CaC}}{{{\text{O}}}_{3}}}\limits_{{\text{кальцит}}} \, + \mathop {{\text{M}}{{{\text{g}}}_{2}}{\text{Si}}{{{\text{O}}}_{4}}}\limits_{{\text{форстерит}}} \, + {{T}^{^\circ }}{\text{C}} \to \\ \to \mathop {{\text{CaMgSi}}{{{\text{O}}}_{4}}}\limits_{{\text{монтичеллит}}} \, + \mathop {{\text{MgO}}}\limits_{{\text{периклаз}}} \, + {\text{C}}{{{\text{O}}}_{2}}{\kern 1pt} \uparrow {\kern 1pt} . \\ \end{gathered} $(7)
$\begin{gathered} 4{\text{F}}{{{\text{e}}}_{2}}{\text{Si}}{{{\text{O}}}_{4}} + 4{{{\text{H}}}_{2}}{\text{S}} + {\text{C}}{{{\text{O}}}_{2}} = \\ = 2{\text{Fe}}{{{\text{S}}}_{2}} + 2{\text{F}}{{{\text{e}}}_{3}}{{{\text{O}}}_{4}} + 4{\text{Si}}{{{\text{O}}}_{2}} + 2{{{\text{H}}}_{2}}{\text{O}} + {\text{C}}{{{\text{H}}}_{{\text{4}}}}{\kern 1pt} \uparrow , \\ \end{gathered} $(8)
$\begin{gathered} 3.5{\text{F}}{{{\text{e}}}_{{\text{2}}}}{\text{Si}}{{{\text{O}}}_{4}} + 14{{{\text{H}}}_{2}}{\text{S}} + 2{\text{C}}{{{\text{O}}}_{2}} = \\ = 7{\text{Fe}}{{{\text{S}}}_{2}} + 3.5{\text{Si}}{{{\text{O}}}_{2}} + 11{{{\text{Н}}}_{{\text{2}}}}{\text{O}} + {{{\text{С}}}_{2}}{{{\text{Н}}}_{6}}{\kern 1pt} \uparrow {\kern 1pt} . \\ \end{gathered} $На глубинах более 120–150 км алмазы образуются путем восстановления углерода по реакциям взаимодействия окиси углерода и углекислого газа с метаном или другими углеводородами органического и абиогенного происхождения, затянутыми по зонам субдукции вместе с осадками на большие глубины. Известно, что океанские и сносимые с окраин континента осадки и осадочные породы часто содержат повышенные концентрации органического вещества. Попадая в зоны поддвига плит, оно подвергается термолизу и гидролизу и быстро проходит все стадии преобразования в углеводороды, нитраты и соединения аммония. Часть из этих подвижных соединений вместе с поровыми водами выжимается из зон субдукции наверх, а некоторое их количество вместе с терригенными осадками продолжает путь в глубины мантии.
Известно [11], что устойчивость всех без исключения углеводородов существенно уменьшается с повышением температуры и давления. Происходит это за счет разрыва углеродных связей в длинных цепях сложных углеводородных молекул. В результате такого крекинг-процесса в системе постепенно уменьшается содержание углеводородов сложного состава и возрастает концентрация простых углеводородов. Наибольшей устойчивостью обладает метан, выдерживающий нагревы (при обычных давлениях) до 1200°С. Поэтому при достаточной длительности реакции в условиях высоких температур и давлений, в конце концов, все органическое вещество может превращаться в метан, водород и свободный углерод. Однако температурное разрушение углеводородов – эндотермический процесс и не может привести к образованию кристаллических фаз углерода. Освобождающийся углерод остается распыленным.
Для образования кристаллических форм углерода возможно, например, протекание экзотермической реакции соединения углеводородов с окисью углерода и углекислым газом [26]:
(9)
${\text{С}}{{{\text{Н}}}_{4}} + {\text{С}}{{{\text{О}}}_{2}} \to 2{\text{С}} + 2{{{\text{Н}}}_{2}}{\text{О}} + {{T}^{^\circ }}{\text{C}}.$(10)
$\begin{gathered} 4{{{\text{C}}}_{n}}{{{\text{H}}}_{{2n \pm k}}} + (2n \pm k){\text{C}}{{{\text{O}}}_{2}} \to \\ \to (6n \pm k){\text{C}} + 2(2n \pm k){{{\text{H}}}_{2}}{\text{O}}. \\ \end{gathered} $Помимо углеводородов чисто органического происхождения в образовании кристаллического углерода или алмаза может принимать участие и абиогенный метан, образующийся, например, по реакции (29). Данные реакции становятся возможными ввиду многостадийности процесса дегидратации и гидратации в зоне субдукции. Так как процессы гидратации ультраосновных пород наиболее характерны для условий рифтогенеза, то описывающие их химические реакции будут рассмотрены ниже.
Часто в алмазах можно встретить включения сульфидов, особенно, пирротина, что делает возможной следующую реакцию освобождения углерода:
(11)
$\begin{gathered} 2{\text{FeS}} + {\text{C}}{{{\text{O}}}_{2}} \to 2{\text{FeO}} + {{{\text{S}}}_{2}} + {\text{C}} \\ {\text{или}}\,\,2{\text{FeS}} + {\text{C}}{{{\text{H}}}_{4}} \to 2{{{\text{H}}}_{2}}{\text{S}} + 2{\text{Fe}} + {\text{C}}. \\ \end{gathered} $Помимо углеводородов чисто органического происхождения в кимберлитах, эклогитах и гранатовых перидотитах, сформировавшихся из пород океанской коры, могут возникать простейшие углеводороды абиогенного типа и, особенно, метан. Образуясь за счет биогенного вещества и претерпев ряд физико-химических преобразований, они по сути своего формирования и некоторым характеристикам являются уже абиогенными. Таким образом, происходит стирание грани между органической и неорганической природой углеводородов, т.к. все они формируются за счет экзогенного вещества.
Для образования метана необходимо существенное количество водорода, который можно получить благодаря окислению двухвалентного (силикатного) железа до стехиометрии магнетита:
(12)
${{{\text{H}}}_{2}}{\text{O}} + 3{\text{FeO}} \to {\text{F}}{{{\text{e}}}_{3}}{{{\text{O}}}_{4}} + {{{\text{H}}}_{2}} + {{T}^{^\circ }}{\text{C}}.$Синтез метана происходит за счет экзотермических реакций путем простого соединения СО и СО2 с водородом или водой. В присутствии катализаторов, например, никеля, карбоната никеля или самородного железа реакции эти значительно ускоряются и начинают протекать уже начиная с 250‒400°С (при нормальных давлениях). Все эти реакции сопровождаются выделением тепла, поэтому есть основания ожидать, что при более высоких РТ условиях, характерных для зон поддвига плит, они способны протекать и без катализатора:
(13)
$\begin{gathered} {\text{CO}} + 3{{{\text{H}}}_{2}} \to {\text{C}}{{{\text{H}}}_{4}} + {{{\text{H}}}_{2}}{\text{O}} + {{T}^{^\circ }}{\text{C}} \\ {\text{или}}\,\,{\text{C}}{{{\text{O}}}_{2}} + 4{{{\text{H}}}_{2}} \to {\text{C}}{{{\text{H}}}_{4}} + 2{{{\text{H}}}_{2}}{\text{O}} + {{T}^{^\circ }}{\text{C}}. \\ \end{gathered} $При сравнительно низких температурах зеленосланцевой и эпидот-амфиболитовой фаций метаморфизма (до 400–500°С) абиогенный синтез метана может происходить по реакции серпентинизации железосодержащих оливинов в присутствии углекислого газа:
(14)
$\begin{gathered} \mathop {4{\text{F}}{{{\text{e}}}_{2}}{\text{Si}}{{{\text{O}}}_{4}}}\limits_{{\text{оливин}}} \, + \mathop {12{\text{M}}{{{\text{g}}}_{2}}{\text{Si}}{{{\text{O}}}_{4}}}\limits_{{\text{форстерит}}} \, + 18{{{\text{H}}}_{2}}{\text{O}} + {\text{C}}{{{\text{O}}}_{2}} \to \\ \to \mathop {4{\text{M}}{{{\text{g}}}_{6}}[{\text{S}}{{{\text{i}}}_{4}}{{{\text{O}}}_{{10}}}]{{{({\text{OH}})}}_{8}}}\limits_{{\text{серпентин}}} \, + \mathop {4{\text{F}}{{{\text{e}}}_{2}}{{{\text{O}}}_{3}}}\limits_{{\text{гематит}}} \, + {\text{C}}{{{\text{H}}}_{4}} + {{T}^{^\circ }}{\text{C}}. \\ \end{gathered} $При более высоких температурах (более 660–700°С) эта реакция, по-видимому, протекает как побочная при формировании метасоматических кристаллов пироксенов (клиноэнстатита)
(15)
$\begin{gathered} 4{\text{F}}{{{\text{e}}}_{2}}{\text{Si}}{{{\text{O}}}_{4}} + 4{\text{M}}{{{\text{g}}}_{2}}{\text{Si}}{{{\text{O}}}_{4}} + 2{{{\text{H}}}_{2}}{\text{O}} + {\text{C}}{{{\text{O}}}_{2}} \to \\ \to 8{\text{MgSi}}{{{\text{O}}}_{3}} + 4{\text{F}}{{{\text{e}}}_{2}}{{{\text{O}}}_{3}} + {\text{C}}{{{\text{H}}}_{{\text{4}}}} + {{T}^{^\circ }}{\text{C}}. \\ \end{gathered} $Рассматривая приведенные реакции выделения метана, не следует забывать, что изотопы углерода легко фракционируют между СН4 и СО2. В абиогенном метане, как и в метане органического происхождения, всегда преимущественно концентрируется легкий изотоп углерода 12С.
В работах Кенней [36] и Кучерова [37] была экспериментально доказана возможность формирования сложных углеводородов, вплоть до C10H22 с использованием твердого оксида железа, мрамора и воды. Данная реакция становится возможной при температуре 1500°C и давлении выше 30 кбар, что соответствует глубинам более 100 км.
Отмеченные здесь и некоторые другие обменные реакции между углеродом и водородсодержащими соединениями должны приводить к формированию сложного состава флюидной фазы кимберлитов. Особо интересны в этом отношении газово-жидкие включения в алмазах, сохранивших в запечатанном состоянии составы тех флюидов, из которых они в свое время кристаллизовались. Проведенные Мелтоном и Гиардини [38] исследования составов этих включений показали, что в них содержится от 10 до 60% Н2О; от 2 до 50% Н2; от 1 до 12% СН4; от 2 до 20% СО2; от 0 до 45% СО; от 2 до 38% N2; около 0.5–1.2% Ar. Кроме того, оказалось, что в этих включениях встречается около 0.5% эти лена (С2Н4) и от 0.05 до 3% этилового спирта (С2Н5ОН). Свободный кислород в таких включениях нигде не обнаружен, что указывает на восстановительные условия образования алмазов. Весь этот специфический набор газов, по нашему мнению, свидетельствует о преимущественно экзогенном происхождении флюидной фазы, из которой кристаллизовались алмазы в кимберлитах.
Таким образом, необходимые для образования алмазов углеводороды могли поступать в кимберлиты как за счет термолиза органических веществ, затянутых вместе с карбонатными осадками и терригенными породами в зоны поддвига плит, так и благодаря восстановлению углекислого газа при окислении железа и железосодержащих силикатов.
Погружаясь еще глубже в конвектирующую мантию, углерод и некоторые закапсулированные твердые минеральные соединения и газово-жидкие включения деградировавших осадочных комплексов не образуют крупных скоплений. Скорее всего, это многочисленные, но мелкие (мм и доли мм) рассеянные частицы вещества, образующие устойчивый геохимический шлейф коровой направленности в мантии, распространяющийся в плоскости перемещения конвективных потоков.
На глубинах около 200‒300 км углерод может взаимодействовать с водородом (см. реакцию (12)) [31]:
где показатели n и m являются коэффициентами.
Возможно, благодаря именно этой реакции в алмазах встречаются жидкие включения сложных углеводородов, вплоть до спирта. Далее, полученные соединения вступают в реакцию с окислами различных металлов с получением карбидов:
(17)
$({\text{Ме}}){\text{О}} + {\text{C}}n{\text{H}}m \to ({\text{Ме}}){\text{С}} + {{{\text{Н}}}_{2}} + {{{\text{Н}}}_{2}}{\text{О}},$Восстановление окислов металлов углеродом с образованием карбидов в условиях дефицита кислорода может протекать по реакции [17]:
Например, при температурах 700–800°С образование карбида молибдена в присутствии метана и водорода идет по реакции [35]:
(19)
${\text{Mo}} + {{{\text{H}}}_{2}} + {\text{C}}{{{\text{H}}}_{4}} \to {\text{MoC}} + {\text{H}} + {\text{CH}}.$В близком температурном диапазоне может формироваться карбид лития за счет его спекания с кальцитом, который в избытке присутствует в зоне поддвига плит (реакция (5)) [13].
(20)
${\text{L}}{{{\text{i}}}_{2}}{\text{C}}{{{\text{O}}}_{3}} + 4{\text{C}} \to {\text{L}}{{{\text{i}}}_{2}}{{{\text{C}}}_{2}} + 3{\text{C}}.$При температурах выше 900°С углерод может образовывать с железом твердый раствор с образованием карбида железа (Fe3C и Fe2C).
В породах мантийного состава в присутствии углерода происходит восстановления железа и образование его теллурической фазы. В близких условиях формируется и когенит (FeNiCo)3C).
УГЛЕРОД В РИФТОВЫХ ЗОНАХ
В процессе раздвига литосферных плит в океанических рифтовых системах возникают открытые трещины, через которые из мантии на поверхность поднимаются базальтовые расплавы. Перекрытая толщей воды океаническая литосфера гидратируется, а в нижних ее горизонтах формируется серпентинитовый слой за счет перекристаллизации оливинсодержащих ультраосновных пород. При этом перегретые до 250–350○C гидротермальные системы океанической литосферы могут проникать на большую глубину и достигать верхнемантийных горизонтов [3].
Широко развитые на морском дне гидротермальные системы рифтовых зон выносят в гидросферу огромное количество эндогенного вещества [2, 14], которое генерируется в океанической литосфере и верхней мантии. В результате этого выносится кремнезем, кальций, магний, марганец, сульфиды металлов, метан, карбонаты, сульфаты и многие другие соединения.
В депрессионных структурах на севере хребта Хуан-де-Фука в Тихом океане описаны проявления метана (CH4), этана (C2H6), пропана (C3H8), бутана (C4H10), бензола (C6H6) и толуола (C7H8), которые находятся в ассоциации с H2O и CO2 [33]. В гидротермальных полях Срединно-Атлантического хребта обнаружены выходы углеводородов, представленных метаном (СН4), этаном (С2Н6), этиленом (С2Н4), пропаном (C3H8) и бутаном (C4H10) [39]. Естественно ожидать, что в мантии не может генерироваться такое разнообразие углеводородов, и все они являются продуктами распада корового (экзогенного) вещества или образуются за счет процессов приповерхностного изменения пород мантийного состава.
Происходящие в рифтовых зонах эндогенные процессы приводят к генерации углеродистых соединений двумя основными способами. Первый из них заключается в переносе конвективными течениями мантии закапсулированных и рассеянных фрагментов соединений и мономинеральных фаз корового вещества из зон субдукции, а второй реализуется благодаря гидратации мантийных пород океанической литосферы.
Перемещаясь в подрифтовые зоны над восходящими конвективными потоками мантии, карбиды металлов, твердые частицы корового вещества и газово-жидкие включения выносятся наверх и достигают уровней гидратации океанской литосферы (рис. 2 ). В области устойчивости минерализованных водных флюидов они легко разлагаются с выделением целого спектра углеводородов и гидроокислов металлов. При этом следует отметить, что температура плавления многих соединений углерода с металлами существенно превышает температуру верхней мантии (1300–1600°C) и укладывается в диапазон 1000–4000°C. Следовательно, в практически “сухой” мантии соединения карбидов металлов могут находиться сколь угодно долго в устойчивом равновесном состоянии и сохранять геохимические маркеры своего экзогенного происхождения.
Рис. 2.
Геохимия гидротермальных процессов и механизмы генерации соединений углерода в рифтовой зоне срединно-океанического хребта: 1 – осадки; 2 – базальты (подушечные лавы); 3 – долеритовые дайки (комплекс дайка в дайке); 4 – серпентинитовый слой; 5 – подкоровый слой литосферы; 6 – мантия: а – магматический очаг под гребнем срединно-океанического хребта, б – астеносфера; 7 – постройки черных (а) и белых (б) “курильщиков”; 8 – направление движения океанических вод в толще океанической коры; 9 – критический уровень устойчивости воды; 10 – карбиды металлов и закапсулированные твердые минеральные соединения и газово-жидкие включения деградированных осадочных комплексов, перенесенных из зоны субдукции; 11 – область разложения карбидов металлов; 12 – дегазация продуктов распада карбидов металлов.
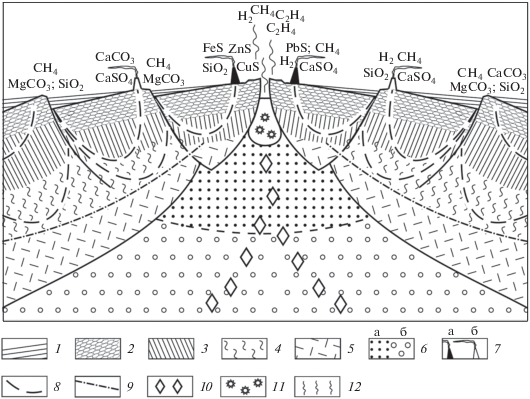
Попадая в приповерхностные зоны рифтов и подвергаясь процессам гидратации, карбиды кальция и натрия разлагаются с выделением ацителена [13]:
(22)
$\begin{gathered} {\text{Са}}{{{\text{С}}}_{2}} + 2{{{\text{Н}}}_{2}}{\text{О}} \to {\text{Са(ОН}}{{{\text{)}}}_{2}} + {{{\text{С}}}_{2}}{{{\text{Н}}}_{2}}{\kern 1pt} \uparrow \\ {\text{и}}\,\,{\text{N}}{{{\text{a}}}_{{\text{2}}}}{{{\text{C}}}_{2}} + 2{{{\text{H}}}_{2}}{\text{O}} \to 2{\text{NaOH}} + {{{\text{C}}}_{2}}{{{\text{H}}}_{2}}{\kern 1pt} \uparrow {\kern 1pt} . \\ \end{gathered} $(23)
${{{\text{C}}}_{2}}{{{\text{H}}}_{2}} + {{{\text{H}}}_{2}} \to {{{\text{C}}}_{2}}{{{\text{H}}}_{4}} + {{{\text{H}}}_{2}} \to {{{\text{C}}}_{2}}{{{\text{H}}}_{6}}{\kern 1pt} \uparrow {\kern 1pt} .$Следует отметить, что в обычных условиях процесс гидролиза карбидов щелочных металлов реализуется исключительно бурно и приводит к взрыву в случае его быстрого попадания в большое количество воды. В геологической же системе данные процессы протекают исключительно медленно (сотни тысяч и миллионы лет), в субсолидусной среде, при относительно высоких давлениях (несколько кбар) и в присутствии незначительных количеств свободной воды. Данный факт позволяет предположить, что процессы гидратации карбидов щелочных металлов в сплошной среде происходят без катастрофических последствий, хотя и могут влиять на общий фон проявления микросейсмичности в рифтовой зоне.
Гидратация карбида алюминия и марганца протекает с выделением метана:
(24)
$\begin{gathered} {\text{A}}{{{\text{l}}}_{4}}{{{\text{C}}}_{3}} + 12{{{\text{H}}}_{2}}{\text{O}} \to 4{\text{Al(OH}}{{{\text{)}}}_{3}} + 3{\text{C}}{{{\text{H}}}_{4}}{\kern 1pt} \uparrow \\ {\text{и}}\,\,{\text{M}}{{{\text{n}}}_{3}}{\text{C}} + 3{{{\text{H}}}_{2}}{\text{O}} \to 3{\text{Mn(OH}}{{{\text{)}}}_{2}} + {\text{C}}{{{\text{H}}}_{4}} + {{{\text{H}}}_{2}}{\kern 1pt} \uparrow {\kern 1pt} . \\ \end{gathered} $Аналогичным образом идут реакции распада карбида бериллия (BeC2) и лития (Li2C2). Образующийся гидроксид марганца легко окисляется в присутствии кислорода, который в достаточных количествах присутствует в воде. Реакция происходит по двухступенчатой схеме с получением пиролюзита:
(25)
$\begin{gathered} 2{\text{Mn}}{{({\text{OH}})}_{2}} + {{{\text{O}}}_{2}} + {{{\text{H}}}_{2}}{\text{O}} \to \\ \to {\text{MnO}}{{({\text{OH}})}_{2}} \to {\text{Mn}}{{{\text{O}}}_{2}}. \\ \end{gathered} $Распад карбида железа сопровождается выделением этилена, однако данная реакция, скорее всего, не имеет широкого распространения в природе ввиду того, что основная масса железа стремится погрузиться в ядро Земли и не формирует ощутимого количества карбидов, которые затем попадают в рифтовые зоны океана:
(26)
$2{\text{FeC}} + 3{{{\text{H}}}_{2}}{\text{O}} \to {\text{F}}{{{\text{e}}}_{2}}{{{\text{O}}}_{3}} + {{{\text{C}}}_{2}}{{{\text{H}}}_{4}}{\kern 1pt} \uparrow {\kern 1pt} .$(27)
$\begin{gathered} \mathop {4{\text{M}}{{{\text{g}}}_{{\text{2}}}}{\text{Si}}{{{\text{O}}}_{4}}}\limits_{{\text{форстерит}}} \, + 4{{{\text{H}}}_{2}}{\text{O}} + 2{\text{C}}{{{\text{O}}}_{2}} \to \\ \to \mathop {{\text{M}}{{{\text{g}}}_{6}}[{\text{S}}{{{\text{i}}}_{4}}{{{\text{O}}}_{{10}}}]{{{({\text{OH}})}}_{8}}}\limits_{{\text{серпентин}}} \, + \,\mathop {2{\text{MgC}}{{{\text{O}}}_{3}}}\limits_{{\text{магнезит}}} \, + {{T}^{^\circ }}{\text{C}}, \\ \end{gathered} $(28)
$\begin{gathered} \mathop {2{\text{CaA}}{{{\text{l}}}_{{\text{2}}}}{\text{S}}{{{\text{i}}}_{{\text{2}}}}{{{\text{O}}}_{8}}}\limits_{{\text{анортит}}} \, + 4{{{\text{H}}}_{2}}{\text{O}} + 2{\text{C}}{{{\text{O}}}_{2}} \to \\ \to \mathop {{\text{A}}{{{\text{l}}}_{4}}[{\text{S}}{{{\text{i}}}_{4}}{{{\text{O}}}_{{10}}}]{{{({\text{OH}})}}_{8}}}\limits_{{\text{каолин}}} \, + \mathop {2{\text{CaC}}{{{\text{O}}}_{3}}}\limits_{{\text{кальцит}}} \, + {{T}^{^\circ }}{\text{C}}. \\ \end{gathered} $Благодаря этим реакциям в океан постоянно привносится исходный материал для нормальной жизнедеятельности скелетных организмов (кораллов, моллюсков фораминифер и кокколитофорид), которые преобразуют растворенные хемогенные карбонаты в породы биогенного происхождения.
При гидратации оливинов в рифтовых зонах в процессе окисления двухвалентного силикатного железа до трехвалентного состояния в присутствии углекислого газа образуется абиогенный метан:
(29)
$\begin{gathered} \mathop {4{\text{F}}{{{\text{e}}}_{2}}{\text{Si}}{{{\text{O}}}_{4}}}\limits_{{\text{фаялит}}} \, + \mathop {12{\text{M}}{{{\text{g}}}_{{\text{2}}}}{\text{Si}}{{{\text{O}}}_{4}}}\limits_{{\text{форстерит}}} \, + 18{{{\text{H}}}_{2}}{\text{O}} + {\text{C}}{{{\text{O}}}_{2}} \to \\ \to \mathop {4{\text{M}}{{{\text{g}}}_{6}}[{\text{S}}{{{\text{i}}}_{{\text{4}}}}{{{\text{O}}}_{{10}}}]{{{({\text{OH}})}}_{8}}}\limits_{{\text{серпентин}}} \, + \mathop {4{\text{F}}{{{\text{e}}}_{2}}{{{\text{O}}}_{3}}}\limits_{{\text{гематит}}} \, + \mathop {{\text{C}}{{{\text{H}}}_{4}}{\kern 1pt} \uparrow }\limits_{{\text{метан}}} \, + {{T}^{^\circ }}{\text{C}}. \\ \end{gathered} $Большая часть образованного метана окисляется (служит пищевой базой) метан-потребляющими бактериями, которые принимают участие в формировании органического вещества. Часть метана переносится в атмосферу, а некоторое количество этих летучих соединений может сохраняться в океанических осадках и формировать в них углеводородные залежи газогидратов [1].
ИЗОТОПНАЯ ГЕОХИМИЯ УГЛЕРОДА
Рассматривая закономерности геохимических преобразований соединений углерода не стоит забывать, что все они имеют экзогенное происхождение, а их изотопный состав зависит от составов участвующих в химических реакциях соединений.
В рифтовых системах и зонах поддвига плит изотопы углерода имеют свои генетические особенности и вариативность сдвигов. Исходя из этого, изотопно-геохимические преобразования углерода целесообразно описать последовательно от конвергентных к дивергентным границам океанических литосферных плит.
Определение изотопных сдвигов углерода, образованого по реакции (9) из абиогенного метана (Смет) и карбонатного углерода (Скарб) из зон субдукции, можно следующими выражениями [29]:
(30)
${{\delta }^{{{\text{13}}}}}{{{\text{С}}}_{{{\text{алм}}}}} = \frac{{{{\delta }^{{{\text{13}}}}}{{{\text{С}}}_{{{\text{мет}}}}} + {{\delta }^{{{\text{13}}}}}{{{\text{С}}}_{{{\text{карб}}}}}}}{{\text{2}}},$(31)
${{\delta }^{{{\text{13}}}}}{{{\text{С}}}_{{{\text{алм}}}}} = \frac{{{{\delta }^{{{\text{13}}}}}{{{\text{С}}}_{{{\text{мет}}}}} + {\text{2}}{{\delta }^{{{\text{13}}}}}{{{\text{С}}}_{{{\text{карб}}}}}}}{{\text{3}}}.$Изотопные сдвиги углерода в алмазах, которые сформировались по реакции (10) с участием органического углерода (Сорг) из углеводородов широкого спектра CnH2n ± k можно определить выражением
(32)
${{\delta }^{{{\text{13}}}}}{{{\text{C}}}_{{{\text{алм}}}}} = \frac{{{\text{4}}n{{\delta }^{{{\text{13}}}}}{{{\text{C}}}_{{{\text{орг}}}}} + {\text{(2}}n \pm k{\text{)}}{{\delta }^{{{\text{13}}}}}{{{\text{С}}}_{{{\text{карб}}}}}}}{{{\text{6}}n \pm k}}.$(33)
${{\delta }^{{13}}}{{{\text{C}}}_{{{\text{алм}}}}} \approx {{\delta }^{{13}}}{{{\text{C}}}_{{{\text{карб}}}}}.$Известно, что изотопный сдвиг углерода в абиогенном метане, который образуется в срединно-океанических хребтах, приблизительно равен –13…–14‰ [27]. Отклонение изотопной сигнатуры органического вещества чаще всего составляет δ13Cорг ≈ –15 до –50‰, и в среднем равен –25‰ [30]. В связи с тем, что возраст образования большинства кристаллов алмазов составляет около 2.3–2 млрд лет, следует учитывать, что тогда для карбонатного углерода наблюдалась положительная изотопная аномалия со сдвигом до +13‰ [18]. Следовательно, используя уравнение (30) при среднем значении δ13Cмет ≈ –25‰, можно определить, что δ13Cалм ≈ –6‰. Подобные распределения изотопных сдвигов по выражению (31) расположены в пределах от +0.3 до –6.3‰ [29].
Выполненные теоретические оценки изотопных отношений в алмазах, в общем, соответствуют имеющимся экспериментальным данным. Так, в работе [41] приводятся результаты послойного анализа вариаций изотопных отношений углерода в индивидуальных кристаллах алмазов, которые показывают, что в большинстве случаев наблюдается закономерный тренд изменения изотопных отношений углерода от центра кристаллов к периферии. Ядра кристаллов всегда обогащены легким изотопом 12С, а внешние оболочки “утяжеляются” изотопом 13С. При этом общее смещение изотопных составов достигает 4‰ и в среднем меняется от –11.01‰ в центре алмазов до –7.32‰ на их поверхности.
Скорее всего, выявленные изменения изотопного состава углерода в алмазах свидетельствуют о его генетической вариативности. Так, скорее всего, начальный рост кристаллов происходил за счет биогенного углерода, а по мере погружения литосферной плиты в зону субдукции их обрастание шло с участием глубинного хемогенного углерода. Исходя из этого, мы можем заключить, что большинство кристаллов алмазов образуются из смеси биогенного и абиогенного метана, а так же продуктов разложения карбонатов различного генезиса.
Не менее интересную особенность распределения изотопов в алмазах разного парагенезиса выявили Соболев, Галимов с коллегами [20]. При изучении зерен алмазов из эклогитовых и перидотитовых ксенолитов они обнаружили, что широкое распределение изотопных сдвигов, описанное выше, относится к кристаллам, образовавшимся в кимберлитовой матрице и эклогитах. При этом в алмазах из перидотитовых ксенолитов наблюдаются сравнительно узкие границы распределения δ13Салм (от –2 до –8‰ со средним значением –6‰). По-видимому, данная особенность связана с тем, что в кимберлитах и эклогитах алмазы формировались из экзогенного углерода, содержащего биогенные карбонаты и углеводороды. В противоположность этому, в кристаллы перидотитового парагенезиса, углерод мог поступать только из хемогенных карбонатов, образовавшихся еще на стадии гидратации пород бывшей океанской коры, например по реакциям (27 и 28), и хемогенного метана, генерирующегося по реакции (29).
Как уже отмечалось выше, весь углерод в рифтовых зонах формируется двумя основными способами. Первый предполагает его перенос из зон субдукции верхнемантийными конвективными течениями, а второй за счет гидратации основных и ультраосновных пород океанической литосферы. В процессе переноса рассеянного углерода, карбидов металлов и закапсулированных частиц корового вещества из зон поддвига плит его изотопные сдвиги отвечают тем показателям, которые сформировались в родоначальной геодинамической обстановке. Следовательно, в рифтовой зоне происходит наложение спектров изотопных сдвигов углерода, характерных для зон субдукции и сформированных “in situ” за счет гидратации пород океанической литосферы. Это существенно затрудняет процесс идентификации генетической принадлежности соединений углерода и увеличивает разброс значений изотопных сдвигов.
Учитывая принцип Ле Шателье, получается, что химическая реакция, протекающая с выделением тепла, всегда развивается по пути наибольшего снижения внутренней энергии (энтальпии). Следовательно, из углекислого газа со смесью легких и тяжелых изотопов углерода в реакции образования метана (СН4) должны участвовать преимущественно атомы легкого изотопа 12С, т.к. тепловой эффект реакции фракционирования изотопа 12С, в сравнении с изотопом 13С, достигает 0.412 ккал/г [28]. Данный эффект работает в сторону “облегчения” углерода в образующемся метане. Таким образом, обменная изотопная реакция имеет вид:
(34)
$^{{12}}{\text{С}}{{{\text{О}}}_{2}} + {{\,}^{{13}}}{\kern 1pt} {\text{С}}{{{\text{Н}}}_{4}} \to {{\,}^{{13}}}{\text{С}}{{{\text{О}}}_{2}} + {{\,}^{{12}}}{\text{С}}{{{\text{Н}}}_{4}} + {{T}^{^\circ }}{\text{C}}$Отклонение изотопной сигнатуры метана в черных курильщиках обычно составляет δ13C ≈ ≈ ‒13…–14‰, а составы растворенных в океанических водах ${\text{HCO}}_{3}^{ - }$ и СО2 характеризуются значениями, близкими к δ13C = –5.5‰ [27]. Отсюда следует, что реакция изотопного обмена между углекислым газом и метаном, согласно реакции (34), происходит в направлении снижения δ13C в метане. В дальнейшем, в процессе переработки метана специфическими бактериальными сообществами происходит дополнительное облегчение состава углерода в формирующемся органическом веществе (Сорг). Вследствие этого углерод в углеводородных газах приобретает экстремально низкие значения сдвигов δ13Cорг до –50‰ и даже до δ13Cорг≈ –80‰. Этим явлением можно объяснять и возникновение минимумов в распределении δ13Cорг в болотных газах четвертичных отложений и в залежах сланцевого газа, который образуется за счет захороненной в древних осадочных толщах органики [22–24].
ЗАКЛЮЧЕНИЕ
Изучая процессы коромантийного цикла углерода, нельзя не затронуть проблему его содержания в мантии Земли. Данный вопрос не может иметь однозначного ответа и его решение лежит в плоскости анализа косвенных признаков, позволяющих нам в той или иной степени достоверности на него ответить. Так, в изверженных породах основного состава рассеянный углерод характеризуется очень малыми концентрациями от 10 до 100 г/т и дефицитом тяжелого изотопа δ13C = –22 до –27‰. При этом углерод, содержащийся в земной коре, более тяжелый δ13C = –3 до –8‰ [6], а приведенные значения характерны и для показателей изотопных сдвигов в алмазах.
По данным Ронова и Ярошевского [15], в карбонатах земной коры связано около 3.91 × 1023 г СО2 и около 1.95 × 1022 г органического углерода (Сорг). Существенная часть этого вещества в виде осадков отлагается на морском дне и склонах континентов и участвует в конвейерном процессе коромантийного цикла углерода совместно с дрейфом литосферных плит и формированием конвергентных и дивергентных структур на их границах. По оценкам [16], в гидросфере Земли сконцентрировано около 1.0 × 1018 г органического углерода в трех формах существования: живом, взвешенном и растворенном.
В толеитовых базальтах океанических рифтовых зон содержится от 20 до 170 г/т углерода с изотопными сдвигами около –5‰ [34]. При этом следует учитывать, что, с одной стороны, часть углерода находится там в атомарном состоянии и входит в кристаллическую решетку силикатов [42]. С другой стороны, определенная его доля является продуктом переноса конвективными течениями мантии из зон поддвига плит в рифтовые системы и участвует в коромантийном цикле экзогенного углерода. Следовательно, в мантии Земли может содержаться ничтожно малое количество этого элемента, а его суммарные концентрации не влияют на процессы изменения во времени массы задействованного в глобальном цикле углерода.
В некоторых рифтовых зонах отмечен широкий спектр проявления различных составов углеводородных газов от метана (CH4) и этана (C2H6) до пропана (C3H8) и бутана (C4H10). Их генерация, по-видимому, происходит в приповерхностных и низкотемпературных условиях среды, а не за счет выноса из глубокой мантии, т.к. сложные углеводороды в свободном состоянии при высоких РТ условиях становятся неустойчивыми и разлагаются на более простые, вплоть до метана (CH4). Выявленное разнообразие составов углеводородных соединений в рифтовых зонах можно объяснить тем, что в условиях высоких температур и давлений сухой мантии карбиды металлов остаются устойчивыми, а их распад начинается только при достижении ими уровней гидратации пород, т.е. ниже 400○C.
Анализ изотопно-геохимических данных показывает, что в рифтовой зоне, наряду с широким спектром образования углеводородных газов наблюдается и эффект интенсивного фракционирования изотопов углерода, разброс значений которых может варьировать в очень больших пределах. Связано это с тем, что наряду с процессами “in situ” сюда конвективными течениями из зон субдукции привносится углерод, обладающий своими изотопными характеристиками и относящийся к другому генетическому типу. Например, это может приводить не только к искажению данных радиоуглеродного анализа, но и к получению не имеющих смысла значений возраста.
Исследованные нами процессы позволяют сделать вывод, что коромантийный цикл углерода связан с формированием данного элемента в одних геодинамических условиях и его переносом за счет мантийной конвекции и дрейфа литосферных плит в другие. В результате этого углерод подвергается многостадийному преобразованию из хемогенного состояния в биогенное и обратно, а также погружению в мантию на уровни ее конвективного перемешивания и выносу на поверхность через рифтовые зоны (рис. 1 ). Практически весь углерод при этом экзогенного происхождения. Данный процесс тесно связан с короатмосферным циклом углерода, т.к. первичным его поставщиком является углекислый газ и продукты преобразования углеродистого вещества (карбиды, карбонаты, углеводороды, органическое вещество). Вместе они формируют глобальный цикл углерода в природе.
Количество генерируемых описанными способами углеводородных газов абиогенного происхождения, скорее всего, не могут формировать крупных газовых и нефтегазовых месторождений, т.к. значительная их часть переносится в гидросферу, а затем и в атмосферу. Лишь некоторое количество углеводородных соединений может отлагаться в океанических осадках и формировать в них залежи газогидратов [1].
Одним из основных выводов проведенных исследований является фактор отсутствия необходимости нахождения большого количества воды и кислорода в мантии Земли для физико-химической трансформации корового вещества в ее астеносфере.
Приведенные в статье данные позволяют заключить, что предложенное впервые в 20-ые годы прошлого столетия академиком А.Е. Ферсманом понятие глобального цикла углерода следует расширить, включив в него процессы мантийного переноса углерода из зон субдукции в рифтовые системы. Следовательно, к традиционной ветви короатмосферного цикла углерода следует добавить коромантийную составляющую. Оценивая масштабы ее проявления, можно сделать вывод, что, скорее всего, глубинные процессы переноса экзогенного вещества не имеют широкого распространения. Попадающие в мантию многочисленные мелкие фрагменты и рассеянные частицы могут образовывать лишь устойчивый геохимический шлейф коровой направленности, распространяющийся в плоскости перемещения конвективных потоков. Объемы выноса растворенных углеродистых соединений в гидротермальных системах рифтовых зон земной коры могут также позволить оценить масштабы проявления данного процесса.
Источник финансирования. Работа выполнена в рамках государственных заказов № 0149-2019-0005 и № 0226-2019-0053.
Список литературы
Баланюк И.Е., Донгарян Л.Ш. Роль гидротермального метана в образовании газогидратных залежей // Геология, геофизика и разработка нефтяных месторождений. 1994. № 3. С. 22–28.
Богданов Ю.А., Леин А.Ю., Лисицын А.П. Полиметаллические руды в рифтах срединно-атлантического хребта (15–40° с.ш.): минералогия, геохимия, генезис. М.: ГЕОС, 2015. 256 с.
Богданов Ю.А., Лисицын А.П., Сагалевич А.М., Гурвич Е.Г. Гидротермальный рудогенез океанского дна. М.: Наука, 2006. 256 с.
Бурков В.Д., Крапивин В.Ф., Шалаев В.С. Сбалансированная модель глобального биохимического круговорот углерода // Лесной Вестник. 2012. № 9. С. 86‒93.
Галимов Э.М. Геохимия стабильных изотопов углерода. М.: Недра, 1968. 226 с.
Галимов Э.М. О возникновении и эволюции океана по данным об изменениях 18О/16О осадочной оболочки Земли в ходе геологического времени // Докл. СССР. 1988. Т. 299. № 4. С. 977–981.
Добрецов Н.Л., Кулаков И.Ю., Литасов К.Д., Кукарина Е.В. Значение геологии, экспериментальной петрологии и сейсмотомографии для комплексной оценки субдукционных процессов // Геология и геофизика. 2015. Т. 56. № 1–2. С. 21‒55.
Добрецов Н.Л., Шацкий А.Ф. Глубинный цикл углерода и глубинная геодинамика: роль ядра и карбонатитовых расплавов в нижней мантии // Геология и геофизика. 2012. Т. 53. № 11. С. 145‒147.
Доусон Дж. Кимберлиты и ксенолиты в них. М.: Мир, 1983. 300 с.
Жариков В.А. Основы физико-химической петрологии. М.: Изд-во МГУ, 1976. 420 с.
Каррер П. Курс органической химии. М.: Госхимиздат, 1962. 1216 с.
Крапивин В.Ф., Шалаев В.С., Бурков В.Д. Моделирование глобальных циклов углерода и метана // Лесной Вестник, № 5. 2015. С.170‒176.
Косолапова Т.Я. Карбиды. М.: Изд.-во Металлургия, 1968. 300 с.
Лисицын А.П. Гидротермальные системы Мирового океана – поставка эндогенного вещества // Гидротермальные системы и осадочные формации срединных океанических хребтов Атлантики. М.: Наука, 1993. С. 147–245.
Ронов А.Б., Ярошевский А.А. Химический состав земной коры и ее оболочек // Тектоносфера Земли. М.: Недра, 1978. С. 376–402.
Романкевич Е.А., Ветров А.А. Массы углерода в гидросфере Земли // Геохимия. 2013. № 6. С. 483–509.
Самсонов Г.В. Физико-химические свойства соединений тугоплавких металлов с бором, углеродом, азотом и особенности двойных сплавов // Укр. хим. журн. 1957, т XXIII. Вып. 3. 287 с.
Семихатов М.А., Раабен М.Е., Сергеев В.Н. и др. Биотические события и положительная изотопная аномалия карбонатного углерода 2.3–2.06 млрд лет назад // Стратиграфия. Геологическая корреляция. 1999. С. 3–27.
Соболев Н.В. Глубинные включения в кимберлитах и проблема состава верхней мантии. Новосибирск: Наука, 1974. 264 с.
Соболев Н.В., Галимов Э.М., Ивановская И.М., Ефимова Э.С. Изотопный состав углерода алмазов, содержащих кристаллические включения // Докл. АН СССР. 1979. Т. 249. № 6. С. 1217–1220.
Соболев Н.В., Добрецов Н.Л., Отани Э. и др. Проблемы, связанные с кристаллогенезисом и глубинным циклом углерода. // Геология и геофизика. 2015. Т. 56. № 1‒2. С. 5‒20.
Сорохтин И.О., Козлов Н.Е., Глазнев В.Н., Чикирёв И.В. Геология и потенциальная нефтегазоносность полуострова Рыбачий (Кольский полуостров) // Геология, геофизика и разработка нефтяных и газовых месторождений. 2011. № 5. С. 14‒19.
Сорохтин H.О., Козлов Н.Е., Глазнев В.Н., Чикирёв И.В. Потенциальная нефтегазоносность западной части арктического шельфа России и прогнозные критерии поиска УВ сырья в прибрежной зоне Кольского полуострова // Вестник Мурманского государственного технического университета. 2010. Т. 13. № 4-1. С. 736–750.
Сорохтин Н.О., Козлов Н.Е., Чикирев И.В. и др. Эволюция северо-западной части Тимано-Варангерского нефтегазоносного бассейна // Вестник КНЦ РАН. 2011. № 3. С. 3–20.
Сорохтин О.Г. Строение континентальных литосферных плит и происхождение кимберлитов // Проблемы теоретической геодинамики и тектоника литосферных плит. М.: Изд. Ин-та океанологии АН СССР, 1981. С. 161–168.
Сорохтин О.Г. Тектоника литосферных плит и происхождение алмазоносных кимберлитов // Общая и региональная геология, геологическое картирование. М.: ВИЭМС, 1985. 47 с.
Сорохтин О.Г., Леин А.Ю., Баланюк И.Е. Термодинамика океанических гидротермальных систем и абиогенная генерация метана // Океанология. 2001. Т. 41. № 6. С. 898–909.
Сорохтин О.Г., Митрофанов Ф.П., Сорохтин Н.О. Происхождение алмазов и Перспективы алмазоносности восточной части Балтийского щита // Апатиты: КНЦ РАН, 1996. 144 с.
Сорохтин О.Г., Чилингар Дж.В., Сорохтин Н.О. Теория развития Земли (происхождение, эволюция и трагическое будущее). М.: Ижевск: Изд-во “Институт компьютерных исследований”, 2010. 751 с.
Хефс Й. Геохимия стабильных изотопов. М.: Мир, 1983. 200 с.
Agte C., Moers K.Z. Anorg. allgem. Chem., 1931. B. 198. S. 233.
Cooper B.S., Coleman S.H., Barnard P.C., Butterworth J.S. Paleotemperatures in the northern North Sea Basin. Petrol. und Cont. Shelf North-West Europe // Geology. 1975. V. 1. P. 487–492.
Cruse A.M., Seewald J.S. Chemistry of low-molecular weight hydrothermal fluids from Middle Valley, Northen Juan de Fuca Ridge // Geochim. Cosmochim. Acta. 2006. V. 70. P. 2079–2092.
Exley R.A., Mattey D.P., Clague D.A., Pillinger C.T. Carbon isotope systematic of a mantle “hotspot”: a comparison of Loihi Seamount and MORB glasses // Earth and Planet. Sci. Lett., 1986. V. 78. P. 189–199.
Kampbelle J. The Vapor-Phase Deposition of Refractory Materials: I. General Conditions and Apparatus // Electrochem. Soc. 1949. V.96. №5. P. 318.
Kenney J.F., Kutcherov V.A., Bendeliani N.A., Alekseev V.A. The evolution of multicomponent system at high pressures: VI. The thermodynamic stability of the hydrogen–carbon system: The genesis of hydrocarbons and the origin of petroleum // Proceed. of the National Acad. of Sci. of the USA. 2002. V. 99. P. 10 976–10 981.
Kutcherov V.A., Bendeliani N.A., Alekseev V.A., Kenney J.F. Synthesis of hydrocarbons from minerals at pressures up to 5 GPa // Doklady phys. Chem. 2002. V. 387. P. 328–330.
Melton C.E., Giardini A.A. The composition and significance of gas released from natural diamonds from Africa and Brazil // Amer. Miner. 1974. V. 59. P. 775–782.
Proskurowski G., Lilley M.D., Seewald J.S. et al. Abiogenic hydrocarbon production at Lost City hydrothermal field // Science. 2008. V.319. P. 604–607.
Ringwood A.E., Major A. The system Mg2SiO4–Fe2SiO4 at high pressures and temperatures // Phys. Earth Planet. Int. 1970. V. 3. P. 89–108.
Swart P.K., Pillinger C.T., Milledge H.J., Seal M. Carbon isotopic variation within individual diamonds // Nature. 1983. V. 303, P. 793–795.
Watanabe S., Mishima K., Matsuo S. Isotopic ratios of carbonacens materials incorporated in olivine crystals from the Hualalai volcano Hawaii. An approach to mantle carbon // Geochim. J. 1983. V. 17. № 2. P. 95–104.
Дополнительные материалы отсутствуют.