

Петрология, 2021, T. 29, № 6, стр. 588-602
Рассеянный углерод в базальтах измененной океанической коры: изотопный состав и механизмы формирования
В. Б. Поляков a, b, *, С. Н. Шилобреева a, **
a Институт геохимии и аналитической химии им. В.И. Вернадского РАН
Москва, Россия
b Институт экспериментальной минералогии им. Д.С. Коржинского РАН
Москва, Черноголовка, Московская обл., Россия
* E-mail: vpolyakov@mail.ru
** E-mail: shilobre@mail.ru
Поступила в редакцию 30.04.2021
После доработки 14.05.2021
Принята к публикации 15.05.2021
Аннотация
Проведен сравнительный анализ данных по содержанию углерода и его изотопного состава для базальтового матрикса разновозрастной океанической коры в зоне Восточно-Тихоокеанского поднятия. В образцах базальтового матрикса древней океанической коры (~270 млн лет, скв. 801C ODP), в которых обнаружена карбонатная фаза, изотопный состав окисленной компоненты углерода (δ13С = ±1.5‰) свидетельствует о ее образовании за счет осаждения растворенного в морской воде неорганического углерода. В образцах, где карбонатная фаза отсутствует, низкая концентрация (<0.1 мас. % CO2) окисленного рассеянного углерода и его изотопный состав (δ13С < –7‰) лежат в области значений, характерных для углерода, растворенного в нераскристаллизованных базальтовых стеклах. Это позволяет связать окисленный рассеянный углерод с остаточным углеродом, растворенным в магматическом расплаве после дегазации CO2. Осаждение растворенного неорганического углерода морской воды, наряду с образованием карбонатной фазы, ведет к положительной корреляции между концентрацией углерода и значениями величины δ13С. Применение этого критерия к образцам базальтового матрикса молодой коры (~15 млн лет, скв. 1256D ODP) показало, что окисленная компонента рассеянного углерода в молодой океанической коре не образована за счет осаждения растворенного неорганического углерода из морской воды, вопреки общепринятой парадигме. Постоянство концентрации и значений δ13С восстановленной компоненты рассеянного углерода в базальтовом матриксе молодой и древней океанической коры, в том числе в зонах, где микробиальная активность не зафиксирована, приводит к модели абиогенного формирования восстановленного рассеянного углерода вблизи оси хребта при высокой температуре. Синтез Фишера–Тропша и/или реакция Белла–Будуара являются возможной основой для абиогенной модели. Реакция Белла–Будуара 2CO = C + CO2 приводит к формированию адсорбированного слоя элементарного углерода на свежеобразованных поверхностях минералов в ходе изменения океанической базальтовой коры. Газофазное равновесие CO2–CO обеспечивает необходимое обеднение образующегося элементарного углерода изотопом 13С до значений δ13С < –20‰. Абиогенные модели не требуют присутствия в магматическом расплаве рассеянного углерода с низкими значениями δ13С для формирования в базальтовом матриксе рассеянного углерода, обогащенного легким изотопом 12C.
ВВЕДЕНИЕ
Происхождение рассеянного углерода в изверженных и измененных породах, а также его связь с углеродом в мантии является фундаментальной проблемой геохимии углерода. Ее решение невозможно без привлечения изотопных данных и понимания механизмов формирования изотопного состава рассеянного углерода. Эта проблема исследуется в течение многих лет, начиная с классических трудов (Трофимов, 1950, 1952; Craig, 1953; Виноградов, 1958, 1959; Галимов, 1968), и не утратила своей актуальности в наши дни. Рассеянный углерод характеризуется низкой концентрацией (до 1000 ppm C) и обычно не образует углеродсодержащих фаз (за возможным исключением нанофаз). Изотопный состав рассеянного углерода в базальтах изменяется в широких пределах (δ13С = –30…–3‰) (Галимов, 1968; Sakai et al., 1984; Pineau et al., 2004; Delacour et al., 2008; Alt et al., 2013). Столь большие вариации значений δ13С в рассеянном углероде ставят вопрос о возможном наличии в мантии Земли источников углерода с различным изотопным составом, который напрямую примыкает к проблеме изотопной неоднородности углерода в мантии Земли. Таким образом, решение проблемы происхождения рассеянного углерода и механизмов формирования его изотопного состава имеет принципиальное значение для понимания геохимического цикла углерода Земли.
Гипотезы формирования в интрузивных породах рассеянного углерода, обогащенного легким изотопом (δ13C < –20‰), можно разделить на две группы: первая группа связывает его наличие с присутствием в мантии Земли углерода с низкими значениями δ13C; вторая группа объясняет легкий изотопный состав рассеянного углерода в изверженных породах процессами изотопного фракционирования, проходящими в ходе формирования пород (кристаллизация и дегазация магм и др.). К первой группе относятся, в частности, гипотезы, связывающие наличие легкого рассеянного углерода с реликтовым углеродом метеоритного вещества, попавшим в мантию в процессе аккреции Земли (Галимов, 1968); с органическим веществом, поступившим в мантию в результате субдукции (Cartigny, 2005; Cartigny et al., 2014); с процессами рэлеевской дистилляции при окислении метана до СО2 в мантийных условиях (Ga-limov, 1991). Ко второй группе относятся модели формирования восстановленного рассеянного углерода по реакции Фишера–Тропша с образованием органических молекул (Shilobreeva et al., 2011); модели образования углеродных и/или карбидных пленок на свежеобразованных поверхностях минералов (Tingle, Hochella, 1993; Mathez et al., 2008); модели, связывающие образование восстановленного рассеянного углерода с бактериальными процессами (Menez et al., 2012; Alt, Shank, 2011). В середине 80-х годов ХХ столетия в литературе развернулась дискуссия о наличии углерода в атомарном состоянии в кристаллах оксидов и породообразующих минералах. Основанием для дискуссии явились работы Ф. Фреунда с соавторами (Freund, 1981, 1987; Freund et al., 1977, 1978, 1980; Wengeler et al., 1982; Kathrein et al., 1983), свидетельствующие о способности углерода в значительных количествах (до 1000 ppm) растворяться в кристаллической решетке оксидов и силикатов. В последние годы появились сведения о возможности растворения углерода только в небольших количествах (1–50 ppm) в мантийных минералах (Tingle, Green, 1987; Tingle et al., 1988; Keppler et al., 2003; Ni, Keppler, 2011). Столь большая разница в содержании растворенного углерода между ранними и более поздними работами группы Ф. Фреунда, по-видимому, связана с методическими трудностями, в частности с определением контаминационного углерода. Современное представление о форме растворенного углерода в силикатных минералах предполагает нахождение его в виде C4+ при изоморфном замещении кремния в кремнекислородных тетраэдрах, а не в восстановленной форме (Shcheka et al., 2006). В расплавах также превалирует окисленная форма углерода; восстановленный углерод может растворяться только в при очень низких значениях fО2, соответствующих буферу железо–вюстит (Kadik et al., 2004). Основываясь на результатах группы Ф. Фреунда (Freund, 1981; Freund et al., 1977, 1978, 1980; Wengeler et al., 1982; Kathrein et al., 1983), Э.М. Галимов (1984) предположил, что обогащение восстановленного рассеянного углерода легким изотопом 12С связанно с его растворением в кристалле в атомарной нуль-валентной форме, которой соответствует бета-фактор, равный единице. Это предположение, очевидно, не согласуется c моделью растворения углерода в мантийных минералах в виде C4+, который при анализе выделяется в окисленной компоненте рассеянного углерода.
Интенсивный обмен веществом между корой и мантией проходит в зоне океанических хребтов/поднятий при формировании океанической коры в процессе спрединга. Изучение рассеянного углерода в базальтах океанической коры и механизмов формирования его изотопного состава, с одной стороны, дает информацию о рассеянном углероде в мантии Земли, а с другой – является необходимым элементом для понимания глобального геохимического цикла углерода. В последние десятилетия, благодаря бурению океанической коры, массив данных о концентрации углерода и его изотопном составе в океанических базальтах значительно вырос, что обеспечило лучшее понимание физико-химических процессов, связанных с изменением океанической коры (см., например, Alt et al., 2003; Früh-Green et al., 2004; Delacour et al., 2008; Shilobreeva et al., 2011). Изменение океанической коры вызывает химические и минералогические преобразования вещества, содержащего углерод. Общий углерод в измененной океанической коре включает окисленную и восстановленную компоненты, однако происхождение этих компонентов все еще не совсем ясно. С точки зрения изучения рассеянного углерода, первоочередной интерес представляет углерод в базальтовом матриксе, в отличие от углерода, сосредоточенного в жилах, гало, межзерновых полостях и т.п., где он существенно контаминирован. Именно такие образцы базальтовой измененной коры разного возраста были исследованы в (Shilobreeva et al., 2011; Martinez et al., 2021). В настоящей статье проведено сравнительное рассмотрение результатов, полученных в этих работах, в свете происхождения рассеянного углерода и возможных моделей формирования его изотопного состава в базальтах измененной океанической коры.
ОТБОР ОБРАЗЦОВ И МЕТОДИКА ЭКСПЕРИМЕНТА
Образцы базальтового матрикса из скважин 1256D и 801C, пробуренных в рамках проекта ODP (Ocean Drilling Program), были исследованы в работах (Shilobreeva et al., 2011; Martinez et al., 2021). Скважина 1256D (6°44.20′ с.ш., 91°56.10′ з.д.) пробурена на восточном склоне Восточно-Тихоокеанского поднятия в базальтовой коре, возраст которой составляет ~15 млн лет. Литостратиграфия измененной океанической коры, вскрытой этой скважиной, включает (Teagle et al., 2006): 1) 250-метровую толщу осадков; 2) вулканическую (лавовую) зону на глубинах от 250 до 1004 м, отсчитываемых от морского дна (Meters Below Sea Floor – mbsf); 3) переходную зону (1004–1061 mbsf); 4) комплекс слоистых даек (1061–1407 mbsf); 5) плутонический комплекс (1407–1507 mbsf), состоящий из двух тел габбро мощностью 52 и 24 м, которые разделены гранобластовой дайкой, мощностью 24 м. Нижнее габбровое тело до конца не пройдено.
Скважина 801C (18°38.538′ с.ш., 156°21.588′ в.д.) пробурена в юрской базальтовой океанической коре, возраст которой 270 млн лет, в западной части Тихого океана, в акватории бассейна Пигафетта. Глубина морского дна – 5674 м, скважина прошла сквозь осадочный чехол (461.6 м) и 474 м вулканической (лавовой) зоны кристаллического фундамента. Литостратиграфия скв. 801С детально изучена (Lancelot et al., 1990; Plank et al., 2000; Alt, Teagle, 2003). Пройденный базальтовый фундамент разделен на восемь литологических единиц (блоков), отсчитываемых сверху вниз от границы осадков и кристаллического фундамента. Блок I представляет собой 60.2 м щелочных базальтовых силов, возраст которых составляет 157 млн лет. Блоки II и V – это гидротермальные отложения, обогащенные Si и Fe. Блок II (мощностью 10–20 м) разделяет щелочные и толеитовые базальты, а блок V (мощностью 1 м) находится в базальтовом фундаменте на глубине 150 м от его верхней границы. Пройденная 400 м толща толеитовых базальтов представлена массивными, слоистыми или подушечными лавами (блоки III, IV, VI и VIII) и включает 10-метровый слой брекчии на глубине 360 м от верхней границы базальтового фундамента. В целом литология вулканической части базальтового фундамента, вскрытого скв. 801C, типична для океанической коры за исключением присутствия более молодых щелочных базальтов (блок I), образовавшихся вне оси поднятия, и гидротермальных отложений (блоки II и V), которые сформированы потоками низкотемпературных гидротермальных флюидов непосредственно на оси спрединга (Alt, Teagle, 2003; Alt, 2003). До 300-метровой глубины от верхней границы базальтового фундамента средняя пористость базальтов составляет 11 ± 4%; на бόльших глубинах – 7 ± 1% (Jarrard et al., 2003). Мы соотнесли глубины базальтового фундамента менее 300 м с верхней вулканической зоной (ВВЗ), а нижележащую пробуренную часть фундамента с нижней вулканической зоной (НВЗ).
Было исследовано 19 образцов из скв. 1256D, характеристика и локализация которых представлены в Tabl. 1 и 2 в статье (Shilobreeva et al., 2011). Образцы были взяты из всех литологических зон базальтового фундамента, в которых пробурена эта скважина. Из скв. 801C было исследовано 11 образцов толеитовых базальтов с глубин более 143.5 м от верхней границы фундамента (Tabl. 1, Martinez et al., 2021), шесть образцов из ВВЗ и 5 – из НВЗ (Tabl. 1, Martinez et al., 2021). В образцах из скважин 1256D и 801C тщательным образом отделяли от базальтового матрикса жилы, гало и области, содержащие полости. Основное внимание было уделено именно изучению образцов матрикса, хотя в отдельных случаях для сравнения анализировали изотопный состав углерода и кислорода в жилах (Shilobreeva et al., 2011; Martinez et al., 2021).
Таблица 1.
Средние значения концентрации и изотопного состава общего углерода в базальтовом матриксе молодой (~15 млн лет) океанической коры, вскрытой скв. 1256D ODP (рассчитано по данным (Shilobreeva et al., 2011))
| Литологическая единица | Мощность, км | Скважина 1256D | |
|---|---|---|---|
| концентрация углерода (CO2, мас. %) | δ13C, ‰ | ||
| Вулканическая зона | 0.75 | 0.153 ± 0.062 | –23.5 ± 2.0 |
| Переходная зона | 0.2 | 0.237 ± 0.010 | –23.6 ± 1.0 |
| Зона даек | 1.2 | 0.129 ± 0.073 | –20.9 ± 2.6 |
| Габбро | 0.1 | 0.076 ± 0.013 | –20.8 ± 0.5 |
| По разрезу в целом | 2.25 | 0.144 ± 0.061 | –22.2 ± 2.1 |
Методика исследования образцов из скважин 1256D и 801C была различна. Образцы из скв. 1256D последовательно подвергали термической обработке: 1) при 100°C в вакууме в течение одного часа для удаления поверхностной контаминации; 2) при 250°C в течение одного часа при парциальном давлении кислорода 4 мбар для окисления и удаления органических соединений; 3) при 900°C в течение одного часа при давлении кислорода ~5 × 10–4 мбар; 4) при 1170°C в течение одного часа при давлении кислорода ~5 × 10–4 мбар; 5) при 1170oC в течение 30 мин и давлении кислорода 4 мбар (Shilobreeva et al., 2011). Выделяющиеся газы CO2 и H2O вымораживали, измеряли их количество и изотопный состав углерода и водорода. Суммарное количество и изотопный состав углерода, выделившегося при температурах 900 и 1170°C, принимали за общее количество углерода в базальтовом матриксе (Shilobreeva et al., 2011). Разделить окисленный и восстановленный углерод таким образом не удалось.
Образцы из скв. 801С анализировали по новой схеме (Martinez et al., 2021). Образцы измельчали до размера <150 мкм и прогревали при 100°C в вакууме в течение одного часа. После этого каждый измельченный образец делили на три части. Первую часть использовали для измерения общего количества и изотопного состава углерода в образце. Это осуществляли путем прогрева не менее 100 мг образца при температуре 1100°C в течение одного часа при давлении кислорода 4 мбар. В результате этой процедуры весь выделившийся углерод окислялся до CO2, количество и изотопный состав которого измеряли методом, описанным ранее в работе (Javoy, Pineau, 1991). Погрешности измерений составляли 5% и 0.2‰, соответственно. Выделившийся водород также окисляли до H2O, далее вымораживали, а затем измеряли его количество и изотопный состав (погрешность измерения изотопного состава водорода ~2‰). Вторую часть образца использовали для измерения изотопного состава восстановленного углерода, для чего ее предварительно обрабатывали 6-нормальным раствором HCl с целью удаления окисленной компоненты углерода. После обработки эту часть образца промывали свободной от CO2 деионизованной водой, а затем высушивали при 50°C в течение одной недели. После этого проводили термическую обработку и измерение количества выделяющегося CO2 и изотопного состава углерода аналогично тому, как это было сделано для первой части образца. Третью часть образца использовали для измерения количества и изотопного состава окисленной части углерода. Для этого проводили обработку фосфорной кислотой в соответствии с методикой, предложенной в (McCrea, 1950). Эту обработку проводили при комнатной (25°C) и повышенной (80°C) температурах. При комнатной температуре преимущественно разлагался CaCO3, а при повышенной – другие карбонаты ((Ca,Mg,Fe)CO3). Погрешности измерений составляли ±0.1‰ для δ13C, ±0.5‰ для δ18O. Относительная погрешность содержания окисленной части составляла 10%. Подробное описание методики измерений можно найти в (Martinez et al., 2021).
СРАВНИТЕЛЬНЫЕ РЕЗУЛЬТАТЫ ПО СКВАЖИНАМ 801C И 1256D И ИХ ОБСУЖДЕНИЕ
На рис. 1 представлены зависимости содержания углерода и его изотопного состава от глубины в базальтовом фундаменте скважин 801C и 1256D по данным (Martinez et al., 2021; Shilobreeva et al., 2011). Как видно из рис. 1, профили скважин 801C и 1256D отличаются друг от друга. Для образцов базальтового матрикса из скв. 801C, в которых обнаружена карбонатная фаза, в основном из ВВЗ характерны сравнительно высокие содержания углерода и обогащенность тяжелым изотопом 13C. Для образцов базальтов из скважин 801C и 1256D, в которых не обнаружена карбонатная фаза, характерны низкие содержания углерода и низкие значения δ13C < –15‰ (рис. 1а, 1б). По содержанию углерода и отсутствию наблюдаемых карбонатных фаз именно эти образцы и следует рассматривать как рассеянный углерод. Дальнейшее понимание природы рассеянного углерода может быть получено при анализе результатов исследования окисленной и восстановленной компонент углерода в образцах базальтового матрикса из скв. 801С.
Рис. 1.
Зависимость концентрации общего углерода и его изотопного состава от глубины для базальтового матрикса измененной океанической коры по данным (Martinez et al., 2021; Shilobreeva et al., 2011). Образцы из скв. 801C, пробуренной в древней океанической коре (~270 млн лет), образцы из скв. 1256D – в молодой коре (~15 млн лет). Концентрация углерода и значения величины δ13С для образцов базальтового матрикса, в которых обнаружена (залитые значки) и не обнаружена (незалитые значки) карбонатная фаза, различны. При этом концентрация и изотопный состав общего углерода в образцах, в которых не обнаружена карбонатная фаза, находятся в одном диапазоне значений для молодой и древней океанической коры. Образцы, в которых обнаружена карбонатная фаза, показывают закономерное изменение с глубиной.

Окисленная компонента рассеянного углерода в базальтах измененной океанической коры
Вышеописанная схема, примененная при анализе образцов из скв. 801С (Martinez et al., 2021), дает значительно больше информации, чем методика, использованная для скв. 1256D (Shilobreeva et al., 2011), и позволяет понять происхождение и особенности поведения углерода в базальтах измененной океанической коры. На рис. 2 показаны профили содержания и изотопного состава окисленной и восстановленной компонент углерода в образцах скв. 801C. Там же для сравнения показаны содержание и изотопный состав углерода в жилах. Как видно из рис. 2, профили этих компонент различны. Для окисленной компоненты в образцах, в которых обнаружена карбонатная фаза, наблюдается отчетливый тренд изменения содержания углерода и значений δ13C с глубиной. В то же время для образцов, в которых таковая фаза не обнаружена, отсутствует какая-либо корреляция указанных параметров и глубины их локализации в базальтовом фундаменте. Следует также отметить, что изотопный состав окисленного углерода в образцах матрикса, содержащих и не содержащих карбонатную фазу, существенно различается. Если значения δ13C (–0.4…+1.5‰) для окисленной части образцов, в которых наблюдается карбонатная фаза, лежат в том же диапазоне значений δ13C, что и окисленный углерод в карбонатных жилах (рис. 2), и характерны для изотопного состава углерода в кальцитах, осаждающихся из морской воды (Hoefs, 2018), то изотопный состав окисленного рассеянного углерода образов, в которых карбонатная фаза не наблюдается (δ13C = –7.1…–10.7‰), не может быть объяснен осаждением карбонатного углерода (рис. 3). Столь низкие значения δ13C для карбонатного углерода ранее были обнаружены в жилах на глубинах, меньших 260 м от границы осадочного слоя и фундамента, и объяснялись окислением органического углерода (Alt, Teagle, 2003). Однако в данном случае все образцы базальтового матрикса, содержащие окисленный углерод с низкими значениями δ13C, приурочены к бóльшим глубинам. Значения δ13C окисленного рассеянного углерода в силикатных жилах (в пределах погрешности измерений) совпадают с соответствующими значениями окисленного рассеянного углерода в базальтовом матриксе. Для выяснения генезиса наблюдаемого окисленного рассеянного углерода в базальтовом матриксе, мы сравнили данные по изотопному составу и содержанию углерода в скв. 801C (рис. 2а, 2б; Martinez et al., 2021) с соответствующими данными в базальтовых стеклах (рис. 3). Как показано на рис. 3, содержание и изотопный состав окисленного рассеянного углерода в базальтовом матриксе в изученных образцах из скв. 801С (Martinez, 2021), в которых не обнаружена карбонатная фаза, находятся в тех же диапазонах значений, что и соответствующие величины в базальтовых стеклах срединно-океанических хребтов, океанических островов, островных дуг и активных континентальных окраин (Des Marais, Moore, 1984; Mattey et al., 1984; 1989; Sakai et al., 1984; Blank et al., 1993; Pineau et al., 2004; Aubaud et al., 2004). Значения величины δ13C рассеянного углерода из силикатных жил в скв. 801C находятся в той же области координатной плоскости “содержание углерода–изотопный состав” (рис. 3). Это обстоятельство свидетельствует об общем генезисе рассеянного углерода в указанных объектах. Рассеянный углерод, растворенный в базальтовых стеклах, представляет остаточный магматический углерод, сохранившийся в магматическом расплаве в виде ${\text{CO}}_{3}^{{2 - }}$ групп после дегазации CO2 (Javoy et al., 1982; Pineau et al., 2004).
Рис. 2.
Зависимость концентрации окисленного и восстановленного углерода (a) и их изотопного состава (б) от глубины для базальтового матрикса древней измененной океанической коры (скв. 801C) по данным (Martinez et al., 2021). Образцы базальтового матрикса, в которых обнаружена карбонатная фаза (залитые треугольники), отличаются от образцов, в которых карбонатная фаза не обнаружена (незалитые треугольники), не только по концентрации окисленного углерода, но и по его изотопному составу. Изотопный состав окисленного углерода в образцах базальтового матрикса при наличии карбонатной фазы находится в том же диапазоне значений, что и изотопный состав окисленного углерода в карбонатных жилах (залитые ромбы). Концентрация и изотопный состав окисленного углерода в образцах матрикса, где карбонатная фаза отсутствует, совпадают (в пределах ошибки измерений) с концентрацией и изотопным составом окисленного углерода в силикатных жилах (незалитые ромбы). В образце, в котором присутствовали и силикатная, и карбонатная жилы, наблюдаются промежуточные значения концентрации и δ13С. Концентрация и изотопный состав восстановленного углерода в базальтовом матрице (залитые и незалитые квадраты) отличаются постоянством по глубине и не зависят от наличия/отсутствия в образце карбонатной фазы.
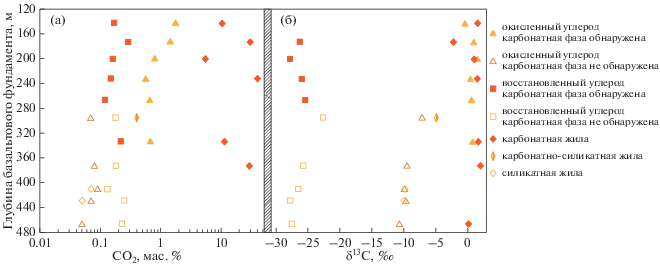
Рис. 3.
Анализ генезиса окисленного и восстановленного углерода в образцах (скв. 801C) базальтового матрикса древней измененной океанической коры (модифицированный рисунок из работы (Martinez et al., 2021)). Значения δ13С для окисленного углерода в образцах матрикса при наличии карбонатной фазы соответствуют диапазону, который характерен для высадившего в виде карбонатов растворенного в морской воде неорганического углерода, что свойственно и для окисленного углерода карбонатных жил. Окисленный углерод в образцах матрикса, в которых карбонатная фаза отсутствует, находится в том же диапазоне значений концентрации и δ13С, что и углерод, растворенный в базальтовых стеклах, что свидетельствует об их общем источнике. Этот же диапазон характерен и для окисленного углерода в силикатных жилах. Восстановленный углерод находится в том же диапазоне значений δ13С, что и углерод органических соединений, имеющих биогенное или абиогенное (синтез Фишера–Тропша) происхождение. В этом же диапазоне лежат значения изотопного состава и концентрации углерода в стеклах, в которых наблюдается крипто- и микрокристалличность. Это обстоятельство является аргументом в пользу образования восстановленного элементарного углерода на свежеобразованных поверхностях минералов по реакции Белла–Будуара (см. текст).

Таким образом, в образцах базальтового матрикса из скв. 801С присутствует окисленный углерод двух типов: (1) углерод, высадившийся в виде карбонатной фазы из морской воды, и (2) остаточный (после дегазации CO2) магматический углерод, не образующий отдельной фазы. Именно этот углерод может рассматриваться как рассеянный углерод. Углерод первого типа в основном приурочен к ВВЗ (глубины менее 300 м от верхней границы фундмента), его источником является растворенный в океанической воде неорганический углерод. В принятой парадигме именно этот углерод считается единственным источником окисленного углерода в образцах измененной базальтовой коры (см., например, Delacour et al., 2008; Shilobreeva et al., 2011; Alt et al., 2013). Очевидно, что различное содержание и изотопный состав окисленного углерода в ВВЗ и НВЗ при малых вариациях восстановленного углерода (рис. 2) обуславливают изменение концентрации и изотопного состава общего углерода в скв. 801С с глубиной (рис. 1). Осаждение карбоната из морской воды приводит к корреляции между изотопным составом общего углерода и его содержанием (рис. 4). Так, для образцов из скв. 801С, где проходило осаждение растворенного неорганического углерода в виде карбонатов, наблюдается положительная корреляция между содержанием общего углерода и его изотопным составом (коэффициент корреляции R = 0.862). В то время как для образцов, в которых окисленный углерод представлен остаточным магматическим углеродом и карбонатная фаза не наблюдается, сколь-либо заметная корреляция между содержанием общего углерода и его изотопным составом отсутствует (R = 0.124). Таким образом, вхождение осажденного из морской воды карбонатного углерода в базальтовый матрикс в ходе изменения базальтовой океанической коры приводит к появлению в матрице карбонатной фазы и положительной корреляции между содержанием общего углерода и его изотопным составом. Если применить эти критерии к изученным (Shilobreeva et al., 2011) образцам базальтового матрикса из скв. 1256D (рис. 4), в которых не наблюдается карбонатная фаза и отсутствует корреляция между содержанием общего углерода и его изотопным составом (R = 0.100), то следует признать, что сколь-либо значимая инфильтрация карбонатного углерода из морской воды в базальтовый матрикс за пределы гало в этой скважине не имела места. Этот вывод согласуется с результатами исследования обр. 123 (Tabl. 4 в статье Shilobreeva et al., 2011) по методике, аналогичной использованной при изучении образцов из скв. 801С (Martinez et al., 2021). Содержание окисленного углерода в этом образце составляет 0.039 мас. % CO2, а δ13C = –15.2‰ и разительно отличается от окисленного углерода в содержащем кальцит обр. 149 (6.7 мас. % CO2; δ13C = –4.5‰), данные по которому представлены там же (Tabl. 4 в статье (Shilobreeva et al., 2011)). Проведенный анализ происхождения окисленного углерода ставит под сомнение общепринятую парадигму, согласно которой окисленная компонента углерода в базальтах измененной океанической коры представлена исключительно углеродом, высадившимся из морской воды.
Рис. 4.
Анализ данных по общему углероду в образцах базальтового матрикса измененной океанической коры. Для образцов базальтового матрикса, в которых обнаружена карбонатная фаза (залитые кружки), наблюдается отчетливая положительная корреляция между концентрацией углерода и значением δ13С (коэффициент корреляции R = 0.862), в то время как для образцов, в которых карбонатная фаза не наблюдалась, такая корреляция отсутствует. Наличие карбонатной фазы и положительная корреляция между концентрацией и значением δ13С общего углерода может служить критерием того, что окисленный углерод в базальтовом матриксе сформирован за счет осаждения карбонатов из морской воды.

Восстановленная компонета рассеянного углерода в базальтах измененной океанической коры
В отличие от окисленного углерода вариации концентрации и изотопного состава восстановленного углерода в базальтовом матриксе в скв. 801С малы (рис. 2). Средние значения этих величин в ВВЗ (0.18 ± 0.07 мас. % CO2; δ13C = –25.7 ± ± 1.7‰) и НВЗ (0.20 ± 0.05 мас. % CO2; δ13C = = ‒27.0 ± 0.9‰) совпадают в пределах погрешности оценок (Martinez et al., 2021). Для скв. 1256D доступны данные по общему углероду, которые также отличаются высоким постоянством по всему разрезу (рис. 1). С учетом того, что восстановленный углерод доминирует над окисленным в образцах базальтового матрикса из скв. 1256D (Shilobreeva et al., 2011), можно заключить, что вариации концентрации и изотопного состава восстановленного углерода также малы11.
В качестве возможных источников восстановленного углерода могут быть (1) привнесенный с морской водой растворенный органический углерод; (2) углерод, являющийся результатом микробиальной активности, включая сами бактерии; (3) восстановленный углерод, формирующийся в результате абиогенных процессов. Постоянство концентрации и изотопного состава восстановленного углерода в базальтовом матриксе по всему разрезу скважин 801C и 1256D свидетельствует в пользу единого источника восстановленного углерода в каждой из скважин. Как показывают численные оценки (Martinez et al., 2021), для скв. 801C наблюдаемая концентрация углерода (среднее значение CO2 = 0.19 ± 0.06 мас. %) может быть достигнута при высоком отношении вода/порода (>860), сохраняющемся постоянным по всему разрезу скважины. Однако, как отмечено в (Martinez et al., 2021), это не согласуется с большими вариациями потоков воды в различных зонах базальтового фундамента (Alt, Teagle, 2003). Именно это обстоятельство делает маловероятным образование восстановленного углерода в базальтовом матриксе в образцах из скв. 801С за счет внедрения растворенного в морской воде органического углерода (Martinez et al., 2021). Аналогичные соображения, основанные на постоянстве концентрации восстановленного углерода по разрезу скв. 1256D, позволяют исключить растворенный в морской воде органический углерод из источников образования востановленного рассеянного углерода в образцах базальтового матрикса из скв. 1256D (Shilobreeva et al., 2011).
Вопрос о микробиальной составляющей в восстановленном углероде в образцах базальтового матрикса требует более внимательного рассмотрения. Микробиальная активность в образцах из скв. 1256D была оценена по изотопному составу серы (δ33S, δ34S) вторичных сульфидов, образовавшихся в ходе изменения океанической коры (Rouxel et al., 2008). Исходя из того, что сера, являющаяся результатом бактериальной сульфатредукции, характеризуется низкими значениями δ34S (<–30‰), в то время как первичная сера в базальтах имеет изотопный состав δ34S ~ 0‰, авторы этой работы пришли к выводу, что бактериальная сульфатредукция имеет место по всему разрезу скв. 801C даже на глубине базальтового фундамента 340 м. При анализе образцов, относящихся к базальтовому матриксу (basalt groundmass) и являющихся аналогами образцов из скв. 801C, в которых измерены концентрация и изотопный состав восстановленного углерода (Martinez et al., 2021), следует отметить, что на глубинах базальтового фундамента, меньших 260 м, для пирита δ34Sср = –27.1 ± 16.3‰, в то время как эта величина для глубин, бόльших 260 м, равна ‒5.1 ± 4.4‰22. Таким образом, микробиальная активность, оцененная по изотопному составу серы в пирите в образцах базальтового матрикса, уменьшается с глубиной. Поскольку при этом изотопный состав восстановленного углерода остается практически постоянным, то можно заключить, что микробиальный углерод не является существенным источником для восстановленного углерода в образцах базальтового матрикса из скв. 801C. Еще более явственно этот вывод следует для скв. 1256D. В образцах из этой скважины был также исследован изотопный состав серы и установлено, что микробиальная активность наблюдалась только в вулканической части разреза и затухала по достижению границы вулканической и переходной зон на глубине базальтового фундамента 750 м (Alt, Shank, 2011). Концентрация общего углерода в базальтовом матриксе в вулканической зоне (0.153 ± 0.062 мас. % CO2) с учетом погрешности совпадает с концентрацией углерода (0.142 ± ± 0.077 мас. % CO2) в нижележащих переходной и дайковой зонах (Shilobreeva et al., 2011). Поскольку общий углерод в базальтовом матриксе содержит в основном восстановленную компоненту, то совпадение концентрации в 750-метровой вулканической зоне и последующей 400-метровой толще (сумма переходной и дайковой зон) базальтового фундамента убедительно свидетельствуют о незначительном вкладе микробиальной составляющей в восстановленный углерод в скв. 1256D.
Наблюдаемое постоянство концентрации и изотопного состава углерода в скв. 1256D хорошо согласуется с моделью формирования восстановленного углерода вблизи оси хребта при относительно высоких температурах (Shilobreeva et al., 2011). Для этой модели подходит абиогенный синтез восстановленного углерода. Обычно в качестве основы для абиогенного образования восстановленного углерода рассматривают процесс Фишера–Тропша (Shock, Schulte, 1998; Seewald et al., 2006; McCollom, Seewald, 2007)33:
(1а)
${\text{C}}{{{\text{O}}}_{2}} + {{{\text{H}}}_{2}} = {\text{CO}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}},$(1б)
${\text{CO}} + 3{{{\text{H}}}_{2}} \to {\text{C}}{{{\text{H}}}_{4}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}}.$Эти реакции в условиях QFM (кварц–фаялит–магнетит) буфера термодинамически разрешены до температур 400°C (McCollom, Seewald, 2007; McCollom, Bach, 2009), достигая максимальной интенсивности при температурах <250–300°C (Shock, 1990; Rhusdi, Simoneit 2001). Реакции Фишера–Тропша (1а и 1б) проходят с большим изотопным эффектом и приводят к обогащению образующихся углеводородов легким изотопом (McCollom, Seewald, 2006; Taran et al., 2007, 2010) до значений δ13C < –20‰, наблюдаемых для восстановленного углерода (Delacour et al., 2008; Shilobreeva et al., 2011; Martinez et al., 2021). Однако эффективность процесса Фишера–Тропша в условиях повышенных (>350°C) температур, характерных для дайковой и плутонической (габбро) зон скв. 1256D (Alt et al., 2010), представляется сомнительной. Кроме того, эффективность реакций Фишера–Тропша снижается в присутствии воды даже при больших значениях соотношения CO2/CO (Shock, 1990; Shock, Schutle, 1998; McCollom, Seewald, 2006, 2007).
Другой механизм формирования восстановленного углерода, основанный на реакции Белла–Будуара: 2CO → C + CO2, интенсивно исследовался экспериментально и теоретически (Mathez, Delaney, 1981; Mathez, 1984, 1987; Pineau, Mathez, 1990; Mathez, Mogk, 1998). Было показано, что этот механизм приводит к образованию восстановленного углеродистого вещества (графита, других форм элементарного углерода, карбинов и т.п.) на свежеобразованных поверхностях минералов и стенках пор, которые являются катализаторами реакции Белла–Будуара (Mathez, Delaney, 1981). В этом случае реакция Белла–Будуара может быть записана как (Mathez, Delaney, 1981):
(2)
${\text{C}}{{{\text{O}}}^{{{\text{ad}}}}} + {\text{C}}{{{\text{O}}}^{{\text{v}}}} \to {{{\text{C}}}^{{{\text{ad}}}}} + {\text{CO}}_{2}^{{\text{v}}},$где верхние индексы ad и v обозначают реагенты и продукты реакции, адсорбированные на поверхности и находящиеся в газовой фазе соответственно. Эта реакция смещена в сторону образования продуктов при температурах ниже 950°C и может протекать одновременно с реакциями Фишера–Тропша, причем присутствие воды смещает реакции (1а) и (1б) влево, что делает реакцию (2) более вероятной (Лапидус и др., 2011).
Несмотря на убедительность доказательств присутствия элементарного углерода и представленные соответствующие микрофотографии (Mathez, Delaney, 1981), механизм формирования восстановленного углерода в базальтовом матриксе не приобрел большой популярности. Это, по-видимому, связано с трудностью объяснения обеднения элементарного углерода изотопом 13C до значений δ13C < –20‰ на основе равновесной модели изотопного фракционирования. Действительно, равновесное фракционирование между графитом и CO2 не превышает 14‰ (рис. 5) и не может обеспечить наблюдаемый изотопный состав восстановленного углерода в образцах базальтового матрикса в скважинах 801C и 1256D. Чтобы обойти эту трудность, был предложен многостадийный механизм с использованием умножения изотопных эффектов на каждой из стадий (Pineau, Mathez 1990), который нельзя признать реалистичным. Между тем анализ различных механизмов реакции Белла–Будуара (2), учитывающих температуру, химический состав и структуру каталитических поверхностей, наличие примесей и водяных паров в газовой фазе и т.п. (Haas et al., 1968; Turkdogan, Vinters, 1974; Olsson, Turkdogan, 1974; Renshaw et al., 1970; Tsao et al., 1977; Лапидус и др., 2011), показывает, что все они включают стадию образования СО в равновесии с CO2, адсорбцию образовавшегося CO на каталитической поверхности и необратимую стадию диспропорционирования адсорбированного CO в элементарный углерод (графит) и CO2 по реакции Белла–Будуара (2). При этом важно отметить, что графит является крайне инертным веществом в отношении изотопного обмена, который между графитом и СО2 при температурах ниже 600°C практически не происходит (Kitchen, Valley, 1995; Polyakov, Kharlashina, 1995; Deines, Eggler, 2009). Эта принципиальная схема формирования изотопного состава углерода на основе реакции Белла–Будуара обеспечивает обеднение образующегося восстановленного углерода изотопом 13С. Действительно, согласно литературным данным, коэффициент равновесного фракционирования между СО2 и СО существенно превосходит таковой между CO2 и графитом (Bottinga, 1969; Richet et al., 1977; Chacko et al., 1991, 2001; Polyakov, Kharlashina, 1995) и в равновесии CO намного сильнее обеднен тяжелым изотопом 13С относительно углекислоты, чем графит (рис. 5). В интервале температур 500–200°C равновесный изотопный сдвиг между СО2 и СО изменяется от 18 до 40‰. Последующий кинетический изотопный эффект на стадии диспропорционирования CO может только несколько увеличить общее обеднение образующегося графита тяжелым изотопом 13С, однако основной эффект достигается за счет равновесия CO2–CO в газовой фазе. При температурах 500–300°C изотопный сдвиг между CO2 и CO изменяется от 18.0 до 29.6‰, что в предположении равновесия с магматическим CO2 (δ13C ≈ –6 ± 2‰) обеспечивает обеднение восстановленного углерода до наблюдаемых значений δ13C < –20‰ (рис. 2; Martinez et al., 2021), причем на нижнем температурном пределе даже при сравнительно высоком проценте конверсии СО2. Таким образом, механизм образования восстановленного углерода на основе реакции Белла–Будуара согласуется с предложенной (Shilobreeva et al., 2011) моделью, согласно которой это формирование происходит вблизи оси хребта на ранних стадиях формирования океанической коры и не сильно изменяется в ходе ее дальнейшего изменения.
Рис. 5.
Температурная зависимость равновесного фракционирования между CO2 и некоторыми углеродсодержащими соединениями. Равновесный сдвиг между углеродсодержащими соединениями рассчитан по значениям углеродных β‑факторов по формуле: ΔA–B (‰) ≈ 1000(lnβA – lnβB), где β – β-фактор, индексы A и B обозначают углеродсодержащие соединения, между которыми рассчитывается изотопный сдвиг. β-факторы CO2, кальцита и графита взяты из статьи (Mironenko et al., 2018), CO из (Richet et al., 1977).
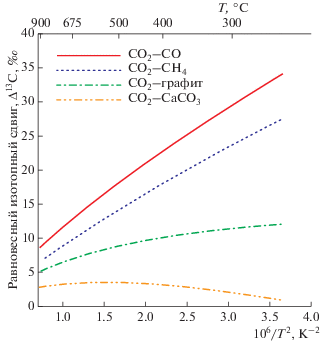
Абиогенное происхождение восстановленной компоненты рассеянного углерода наилучшим образом отвечает его наблюдаемым концентрациям и изотопному составу в базальтовом матриксе в скважинах 801C и 1256D. При высоких температурах (>350°C) механизм, основанный на реакции Белла–Будуара, является, по-видимому, доминирующим; при температурах ниже 250–300°C становится эффективным процесс Фишера–Тропша.
Применение к циклу углерода
Рассеянный углерод в базальтовом матриксе измененной океанической коры в скважинах 801C и 1256D имеет магматический генезис за исключением ВВЗ в древней океанической коре (скв. 801C). Окисленная компонента рассеянного углерода представляет собой остаточный после CO2-дегазации углерод, растворенный в магматическом расплаве. Восстановленная компонента рассеянного углерода представлена элементарным углеродом, формирующимся на свежеобразованных в ходе изменения пород кристаллических поверхностях по реакции Белла–Будуара из дегазированного CO2, и/или органическими соединениями в результате синтеза Фишера–Тропша. Рассмотренные данные по скв. 1256D позволяют оценить концентрацию и изотопный состав рассеянного углерода магматического происхождения в базальтовом фундаменте измененной океанической коры44. Результаты такой оценки для вскрытой части базальтового фундамента в скв. 1256D, сделанные по методике, предложенной в (Alt, Teagle, 1999), представлены в табл. 1. Концентрация магматического рассеянного углерода во вскрытом разрезе базальтового фундамента составляет 0.144 ± ± 0.061 мас. % CO2, а величина δ13C = –22.2 ± 2.1‰. Можно оценить среднюю по всей океанической коре концентрацию общего углерода для верхней части фундамента той же мощности, если воспользоваться данными по концентрации углерода для литологических зон (Alt, Teagle, 1999). Такая оценка дает значение 0.668 мас. % CO2. Таким образом, во вскрытой части базальтового фундамента на долю магматического рассеянного углерода приходится приблизительно 21.8 ± 8.1% общего углерода. Скв. 1256D вскрыла только первые 100 м зоны 5-километровой толщи зоны габбро. Поэтому нельзя использовать значения концентрации общего углерода и величины δ13C, приведенные в табл. 1, для всей зоны габбро. Если согласиться с оценкой 0.05 мас. % CO2 для концентрации рассеянного углерода в нижней части зоны габбро (Alt, Teagle, 1999), то средняя по всей измененной океанической коре концентрация магматического рассеянного углерода составит 0.059 ± 0.030 мас. % CO2. Сравнивая это значение с 0.21 мас. % CO2 для средней концентрации общего углерода в океанической коре (Alt, Teagle, 1999), получим, что магматический рассеянный углерод составляет 28 ± 14% от общего углерода, содержащегося в измененной океанической базальтовой коре. Очевидно, что доля магматического рассеянного углерода уменьшается по мере увеличения доли жил, гало, полостей и т.п. в общем объеме океанической базальтовой коры, инфильтрации карбонатного и органического углерода, растворенных с морской воде, в базальтовый матрикс и увеличения микробиальной активности. Очевидно, для древней коры эти процессы оказываются более развитыми, чем для молодой. По этой причине доля магматического рассеянного углерода в молодых скважинах может быть намного более значительной, чем приведенная средняя оценка.
Основываясь на сделанных оценках, можно сравнить поток магматического рассеянного углерода, поступающего в океаническую кору, с потоком углерода, поступающего в атмосферу через газовую фазу в зоне формирования базальтовой океанической коры. Скорость наращивания океанической коры составляет 6.0 ± 0.8 × 1016 г/год (Mottl, 2003). С учетом вышеприведенных оценок это дает 8.1 ± 4.2 × 1011 мольС/год. Оценки потока углерода через газовую фазу также варьируют в широких пределах от 2.0 × 1012 до 2.0 × 1012 мольС/год (Javoy et al., 1982; Des Marais, 1985; Javoy, Pineau, 1991; Marty, Tolstikhin, 1998; Cartigny et al., 2001, 2008). Столь большие вариации в оценке потока углерода через газовую фазу приводят, естественно, к большой неопределенности в доле, которую составляет рассеянный углерод в измененной базальтовой океанической коре, в общем потоке углерода, поступающего из магматического расплава в зоне формирования базальтовой океанической коры. Эта доля колеблется от 2 до 38%. Небольшая доля (~10–15%) потока магматического углерода через конденсированную фазу хорошо согласуется с обогащением восстановленного рассеянного углерода до значений δ13C < –20‰ за счет реакций Белла–Будуара и/или Фишера–Тропша.
ВЫВОДЫ
Проведенный анализ содержания и изотопного состава окисленного углерода в образцах базальтового матрикса из скв. 801С показал, что окисленный углерод в них имеет разный генезис. Окисленный углерод в образцах, в которых обнаружена карбонатная фаза, характеризуется изотопным составом δ13С = 0 ± 1.5‰ (рис. 2) и представляет собой высадившейся из морской воды растворенный в ней неорганический углерод.
Окисленный углерод в образцах, в которых карбонатная фаза отсутствует, отличается низким содержанием CO2 < 0.1 мас. % и значениями δ13С < –6‰. Концентрации и изотопный состав этого окисленного рассеянного углерода находятся в том же диапазоне значений, что и концентрации и изотопный состав углерода, растворенного в базальтовых стеклах океанических хребтов и океанических островов (рис. 3), что свидетельствуют об их общем генезисе. Окисленный углерод в этих образцах (как и в базальтовых стеклах) есть не что иное, как остаточный после CO2-дегазации углерод, растворенный в магматическом расплаве в виде ${\text{CO}}_{3}^{{2 - }}$ групп.
Показано, что положительная корреляция между концентрацией общего углерода и его изотопным составом при наличии карбонатной фазы в базальтовом матриксе является критерием инфильтрации в базальтовый матрикс высадившегося из морской воды карбонатного углерода. Применение этого критерия к образцам базальтового матрикса молодой океанической коры (~15 млн лет; скв. 1256D) показало что, инфильтрация карбонатного углерода не имела места и окисленная компонента рассеянного углерода является остаточной после CO2 дегазации магматическим углеродом.
Модели биогенного происхождения восстановленного рассеянного углерода не позволяют корректно описать особенности наблюдаемых в скважинах 801C и 1256D профилей концентрации углерода и его изотопного состава в базальтовом матриксе. Высокое постоянство концентрации и изотопного состава восстановленного рассеянного углерода с глубиной наиболее адекватно описывается моделью его абиогенного синтеза вблизи оси хребта при высоких температурах.
Показано, что предложенный ранее (Mathez, Delaney, 1981) механизм формирования восстановленного рассеянного углерода на свежеобразованных кристаллических поверхностях минералов по реакции Белла–Будуара обеспечивает обогащение образующегося элементарного углерода изотопом 12C до значений δ13С < –20‰ за счет изотопного фракционирования при равновесии CO2–CO в газовой фазе.
На основании имеющихся данных проведена оценка доли магматического углерода, содержащегося в измененной базальтовой океанической коре. Согласно этой оценке, 28 ± 14% общего углерода, запасенного в базальтовой океанической коре, составляет магматический углерод. Это дает 8.1 ± 4.2 × 1011 моль С/год для потока магматического углерода через конденсированную базальтовую кору, что составляет от 2 до 38% в общем потоке магматического углерода.
Представленные абиогенные модели формирования рассеянного углерода, обогащенного легким изотопом, в базальтовом матриксе не требуют присутствия углерода с низкими значениями δ13С в магматическом расплаве. Обогащение остаточного окисленного рассеянного углерода легким изотопом 12С (–15‰ < δ13С < –7‰) проходит в процессе дегазации мантийного расплава, а обогащение восстановленного рассеянного углерода изотопом 12С – в ходе постмагматических изменений базальтовой океанической коры на основе образования элементарного рассеянного углерода на свежих поверхностях минералов по реакции Белла–Будуара и/или синтеза органических соединений в процессе Фишера–Тропша.
Благодарности. Авторы признательны своим коллегам Dr. Isabelle Martinez, Prof. Jeffrey C. Alt и Prof. Pierre Agrinier за долгие, плодотворные дискуссии. С.Н. Шилобреева благодарит своих французских коллег Isabelle Martinez и Pierre Agrinier за приглашение в Université de Paris, Institut de physique du globe de Paris, CNRS и возможность проведения экспериментальных работ. Авторы благодарят чл.-корр. Е.О. Дубинину и д-ра геол.-мин. наук С.А. Силантьева за доброжелательную критику и комментарии, которые были чрезвычайно полезны и способствовали улучшению текста статьи.
Источники финансирования. Исследование выполнено в рамках Государственного задания ГЕОХИ РАН № 0137-2019-0015.
Список литературы
Виноградов А.П. Изотопный состав пород Земли и метеоритов // Атомная энергия. 1958. № 4. Вып. 5. С. 409–416.
Виноградов А.П. Химическая эволюция Земли. М.: Изд-во АН СССР, 1959. 49 с.
Галимов Э.М. Геохимия стабильных изотопов углерода М.: Недра, 1968. 226 с.
Галимов Э.М. Вариации изотопного состава алмазов и связь их с условиями алмазообразования // Геохимия. 1984. № 8. С. 1091–1117.
Лапидус А.Л., Елисеев О.Л., Крючков М.В. Реакции Белла–Будуара и водяного газа в условиях синтеза Фишера–Тропша // Химия твердого топлива. 2011. № 5. Р. 26–28. (Перевод: Lapidus A.L., Eliseev O.L., Kryuchkov M.V. Bell–Boudoir and Water Gas Shift Reactions under Conditions of the Fischer–Tropsch Synthesis // Solid Fuel Chem. 2011. V. 45. Р. 313–314. https://doi.org/10.3103/S0361521911050041)
Трофимов А.В. Соотношение изотопов углерода в метеоритах // Докл. АН СССР. 1950. Т. 72. № 4. С. 663–666.
Трофимов А.В. Изотопный состав углерода магматических пород // Докл. АН СССР. 1952. Т. 85. № 1. С. 169–172.
Alt J.C. Stable isotopic composition of upper oceanic crust formed at a fast spreading ridge, ODP Site 801 // Geochem. Geophys. Geosyst. 2003. V. 4. № 5. Q8908. https://doi.org/10.1029/2002GC000400
Alt J.C., Shank W.C. Microbial sulfate reduction and the sulfur budget for a complete section of altered oceanic basalts, IODP Hole 1256D (eastern Pacific) // Earth Planet. Sci. Lett. 2011 V. 310. P. 73–83.
Alt J.C., Teagle D.A.H. The uptake of carbon during alteration of ocean crust // Geochim. Cosmochim. Acta. 1999. V. 63. P. 1527–1535.
Alt J.C., Teagle D.A.H. Hydrothermal alteration of upper oceanic crust formed at fast spreading ridge: mineral, chemical, and isotopic evidence from ODP Site 801 // Chem. Geol. 2003. V. 201. P. 191–211.
Alt J.C., Davidson G.J., Teagle D.A.H., Karson J.A. Isotopic composition of gypsum in the Macquarie Island ophiolite: Implications for the sulfur cycle and the subsurface biosphere in oceanic crust // Geology. 2003. V. 31. P. 549–552.
Alt J. C., Laverne C., Coggon R.M. et al. Subsurface structure of a submarine hydrothermal system in ocean crust formed at the East Pacific Rise, ODP/IODP Site 1256 // Geochem. Geophys. Geosyst. 2010. V. 11. Q10010.
Alt J.C., Schwarzenbach E.M., Fruh-Green G.L. et al. The role of serpentinites in cycling of carbon and sulfur: Seafloor serpentinization and subdurction metamorphism // Lithos. 2013. V. 178. P. 40–54. https://doi.org/10.1016/j.lithos.2012.12
Aubaud C., Pineau F., Jambon A., Javoy M. Kinetic disequilibrium of C, He, Ar and carbon isotopes during degassing of mid-ocean ridge basalts // Earth Planet. Sci. Lett. 2004. V. 222. P. 391–406.
Blank J.G., Delaney J.R., Des Marais D.J. The concentration and isotopic composition of carbon in basaltic glasses from the Juan de Fuca Ridge, Pacific Ocean // Geochim. Cosmochim. Acta. 1993. V. 57. P. 875–887.
Bottinga Y. Carbon isotope fractionation between graphite, diamond and carbon dioxide // Earth Planet. Sci. Lett. 1969. V. 5. P. 301–306.
Cartigny P. Stable isotopes and the origin of diamond // Ele-ments. 2005. V. 1. № 2. P. 79–84.
Cartigny P., Palot M., Thomassot E., Harris J.W. Diamond formation: A stable isotope perspective // Ann. Rev. Earth Planet. Sci. 2014. V. 42. P. 699–732.
Cartigny P., Pineau F., Aubaud C., Javoy M. Towards a consistent mantle carbon flux estimate: Insights from volatile systematics (H2O/Ce, δD, CO2/Nb) in the North Atlantic mantle (14° N and 34° N) // Earth Planet. Sci. Lett. 2008. V. 265. P. 672–685.
Cartigny P., Jendrzejewski N., Pineau F. et al. Volatile (C, N, Ar) variability in MORB and the respective roles of mantle source heterogeneity and degassing: The case of the Southwest Indian Ridge // Earth Planet. Sci. Lett. 2001. V. 194. P. 241–257.
Chacko T., Cole D.R., Horita J. Equilibrium oxygen, hydrogen and carbon isotope fractionation factors applicable to geologic systems // Rev. Mineral. Geochem. 2001. V. 43. P. 1–81.
Chacko T., Mayeda T.K., Clayton R.N., Goldsmith J.R. Oxygen and carbon isotope fractionation between CO2 and calcite // Geochim. Cosmochim. Acta. 1991. V. 55. P. 2867–2882.
Craig H. The geochemistry of the stable carbon isotopes // Geochim. Cosmochim. Acta. 1953. V. 3. P. 53–92.
Deines P., Eggler D.H. Experimental determination of carbon isotope fractionation between CaCO3 and graphite // Geochim. Cosmochim. Acta. 2009. V. 73. P. 7256–7274.
Delacour A., Früh-Green G.L., Bernasconi S.M. et al. Carbon geochemistry of serpentinites in the Lost City Hydrothermal System (30° N, MAR) // Geochim. Cosmochim. Acta. 2008. V. 72. P. 3681–3702.
Des Marais D.J. Carbon exchange between the mantle and the crust, and its effect upon the atmosphere: Today compared to Archean time // Eds. E.T. Sundquist, W.S. Broecker. The carbon cycle and atmospheric CO2: natural variations from Archean to present. AGU Geophysical Monograph. 1985. Series V. 32. P. 602–611. https://doi.org/10.1029/GM032p0602
Des Marais D.J. Moore J.G. Carbon and its isotopes in mid-oceanic basaltic glasses // Earth Planet. Sci. Lett. 1984. V. 5. P. 43–57.
Freund F. Hydrogen and carbon in solid solution in oxides and silicates // Phys. Chem. Mineral. 1987. V. 15. P. 1–18.
Freund F. Mechanism of the water and carbon dioxide solubility in oxides and silicates and the role of O− // Contrib. Mineral. Petrol. 1981. V.76. P. 474–482.
Freund F., Debras G., Demortier G. Carbon content of high-purity alkaline earth oxide single crystals grown by arc fusion // J. Amer. Ceram. Soc. 1978. V. 61. P. 429–434.
Freund F., Debras G., Demortier G. Carbon content of magnesium oxide single crystals grown by the arc fusion method // J. Cryst. Growth.1977. V. 38. P.277–80.
Freund F., Kathrein H.W., Knobel H.R., Heinen H.J. Carbon in solid solution in forsterite; a key to the untracable nature of reduced carbon in terrestrial and cosmogenic rocks // Geochim. Cosmochim. Acta. 1980. V. 44. P. 1319–1334.
Früh-Green G.L., Connolly J.A.D., Plas A. et al. Serpentinization of oceanic peridotites: Implications for geochemical cycles and biological activity // The subseafloor biosphere at MORs. AGU Geophysical Monograph Series. 2004. V. 144. P.119–136.
Galimov E.M. Isotope fractionation related to kimberlite magmatism and diamond formation // Geochim. Cosmochim. Acta. 1991. V. 55. P. 1697–1708.
Haas L.A., Khalafalla S.E., Weston, Jr, P.L. Kinetics of formation of carbon dioxide and carbon from carbon mono-xide in presence of iron pellets // U.S. Bur. Mines. Dep. Invest. 1968. P. 7064–7092.
Hoefs J. Stable isotope geochemistry. Springer International Publishing AG, part of Springer Nature. 2018. 437 p.
Jarrard R.D., Abrams L.J., Pockalny R. et al. Physical properties of upper oceanic crust: Ocean Drilling Program Hole 801C and the waning of hydrothermal circulation// J. Geophys. Res. 2003. V. 108. № B4. 2188.
Javoy M. Pineau F. The volatile record of a “popping” rock from the Mid-Atlantic Ridge at 14° N: Chemical and isotopic composition of gas trapped in the vesicles // Earth Planet. Sci. Lett. 1991. V. 107. P. 598–611.
Javoy M., Pineau F., Allègre C. The carbon geodynamic cycle // Nature. 1982. V. 300. P. 171–173.
Kadik A.A., Pineau F., Litvin Y.A. et al. Formation of carbon and hydrogen species in magmas at low oxygen fugacity during fluidabsent melting of carbonbearing mantle // J. Petrol. 2004. V. 45. P. 1297–1310.
Kathrein H, Gonska H., Freund F. Subsurface segregation and diffusion of carbon in magnesium oxide // Applied Physics. 1983. V. 30. P. 33–41.
Keppler H., Wiedenbeck M., Shcheka S.S. Carbon solubility in olivine and the mode of carbon storage in the Earth’s mantle // Nature. 2003. V. 424. P. 414–416.
Kitchen N.E., Valley J.M. Carbon isotope thermometry in marbles of the Andirondack mountains, New York // J. Metamorphic. Geol. 1995. V. 13. P. 577–594.
Lancelot Y., Larson R.L., Fisher A. Ocean Drilling Program Leg 129 preliminary report: Old Pacific Crust // Proc. Ocean Drill. Program Sci. Results. 1990. V. 129. P.1–28.
Martinez I., Shilobreeva S.N., Alt J. et al. The origin and fate of C during alteration of the oceanic crust // Insitut de France. Academie des Scienses. Les Comptes Rendus–Géoscience. 2021. V. 353 (in press).
Marty B., Tolstikhin I.N. CO2 fluxes from MORs, arcs and plumes // Chem. Geol. 1998. V. 145. P. 233–248.
Mathez E.A. Carbonaceous matter in mantle xenoliths: Composition and relevance to the isotopes // Geochim. Cosmochim. Acta. 1987. V. 51. P. 2339–2347.
Mathez E.A. Influence of degassing on oxidation states of basaltic magmas // Nature. 1984. V. 310. P. 371–375.
Mathez E.A., Delaney J.R. The nature and distribution of carbon in submarine basalts and peridotite nodules // Earth Planet. Sci. Lett. 1981. V. 56. P. 217–232.
Mathez E.A., Mogk D.M. Characterization of carbon compounds on a pyroxene surface from a gabbro xenolith in basalt by time-of-flight secondary ion mass spectrometry // Amer. Mineral. 1998. V. 83. P. 918–924.
Mathez E.A., Roberts J.J., Duba A.G. et al. Carbon deposition during brettle rock. Deformation: Changes in electrical propertes of fault zones and potential geoelectric phenomena during earthquakes // J. Geophys. Res. 2008. V. 113. B12201.
Mattey D.P., Carr R.H., Wright I.P., Pillinger C.T. Carbon isotopes in submarine basalts // Earth Planet. Sci. Lett. 1984. V. 70. P. 196–206.
Mattey D.P., Exley R.A., Pillinger C.T. Isotopic composition of CO2 and dissolved carbon species in basalt glass // Geochim. Cosmochim. Acta. 1989. V. 53. P. 2377–2386.
McCollom T., Bach W. Thermodynamic constraints on hydrogen generation during serpentinization of ultramafic rocks // Geochim. Cosmochim. Acta. 2009. V. 73. P. 856–875.
McCollom T., Seewald J.S. Abiotic synthesis of organic compounds in deep-sea hydrothermal environments // Chem. Rev. 2007. V. 107. P. 382–401.
McCollom T., Seewald J.S. Carbon isotope composition of organic compounds produced by abiotic synthesis under hydrothermal conditions // Earth Planet. Sci. Lett. 2006. V. 243. P. 74–84.
McCrea J.M. On the isotopic chemistry of carbonates and a paleotemperature scale // J. Chem. Phys. 1950. V. 18. P. 849–857.
Menez B., Pasini V., Brunelli D. Life in the hydrated suboceanic mantle // Nat. Geosci. 2012. V. 5. № 2. P. 133–137.
Mironenko M.V., Polyakov V.B., Alenina M.V. Simultaneous calculation of chemical and isotope equilibria using the GEOCHEQ_isotope software: Carbon isotopes // Geochem. Intern. 2018. V. 56. P. 1354–1367.
Mottl M.J. Partitioning of energy and mass fluxes between MOR axes at high and low temperature // Eds. P.E. Halbach et al. Energy and Mass Transfer in Marine Hydrothermal Systems. Dahlem University Press, 2003. P. 271–286.
Ni H., Keppler H. Carbon in silicate melts // Rev. Mineral. Geochem. 2013. V. 75. P. 251–287.
Olsson R.G., Turkdogan E.T. Catalytic effect of iron on decomposition of carbon monoxide, II. Effect of additions of H2, H2O, CO2, SO2 and H2S // Metall. Trans. 1974. V. 5. P. 21–26.
Pineau F., Mathez E.A. Carbon isotopes in xenoliths from the Hualalai Volcano, Hawaii, and the generation of isotopic variability // Geochim. Cosmochim. Acta. 1990. V. 54. P. 211–227.
Pineau F., Shilobreeva S., Hekinian R. et al. Deep-sea explosive activity on the Mid-Atlantic Ridge near 34°50′ N: A stable isotope (C, H, O) study // Chem. Geol. 2004. V. 211. P. 159–175.
Plank T., Ludden J.N., Escutia C. Leg 185: 3. Site 801 // Proceedings of the Ocean Drilling Program. Initial Reports. 2000. V. 185. https://doi.org/10.2973/odp.proc.ir.185.2000
Polyakov V.B., Kharlashina N.N. The use of heat capacity data to calculate carbon isotope fractionation between graphite, diamond, and carbon dioxide: A new approach // Geochim. Cosmochim. Acta. 1995. V. 59. P. 2561–2572.
Renshaw G.D., Roscoe C., Walker P.L., Jr. Disproportionation of CO: I. Over iron and silicon-iron single crystals // J. Catal. 1970. V. 18. P. 164–183.
Rhusdi A.I., Simoneit B.T., Lipid formation by aqueous Fischer-Tropsch-type synthesis over a temperature range of 100 to 400°C // Origins of Life and Evolution of the Biosphere. 2001. V. 31. P. 103–118.
Richet P., Bottinga Y., Javoy M. A review of hydrogen, carbon, nitrogen, oxygen, suphur and chlorine stable isotope fractionation among gaseous molecules // Ann. Rev. Earth Planet. Sci. 1977. V. 5. P. 65–110.
Rouxel O., Ono S., Alt J. et al. Sulfur isotope evidence for microbial sulfate reduction in alteredoceanic basalts at ODP Site 801 // Earth Planet. Sci. Lett. 2008. V. 268. P. 110–123.
Sakai H., Des Marais D. J., Ueda A., Moore J. G. Concentrations and isotope ratios of carbon, nitrogen and sulfur in ocean-floor basalts // Geochim. Cosmochim. Acta.1984. V. 48. P. 2433–2441.
Seewald J.S., Zolotov M.Y., McCollom T. Experimental investigation of single carbon compounds under hydrothermal conditions // Geochim. Cosmochim. Acta. 2006. V. 70. P. 446–460.
Shcheka S.S., Wiedenbeck M., Frost D., Keppler H. Carbon solubility in mantle minerals // Earth Planet. Sci. Lett. 2006. V. 245. P. 730–742.
Shilobreeva S., Martinez I., Busigny V. et al. Insights into C and H storage in the altered oceanic crust: Results from ODP/IODP Hole 1256D // Geochim. Cosmochim. Acta. 2011. V. 75. P. 2237–2255
Shipboard Scientific Party Site 1256 // Proc. ODP, Init. Repts., 206: College Station, TX (Ocean Drilling Program). Eds. D.S. Wilson, D.A.H. Teagle, G.D. Acton. 2003. https://doi.org/10.2973/odp.proc.ir.206.103.2003
Shock L.E. Geochemical constraints on the origin of orga-nic compounds in hydrothermal systems // Origins of Life and Evolution of the Biosphere. 1990 V. 20. P. 331–367.
Shock L.E., Schulte M.D. Organic synthesis during fluid mixing in hydrothermal systems // J. Geophys. Res. 1998. V. 103. P. 28513–28527.
Taran Y.A., Kliger G.A., Cienfuegos E., Shuykin A.N. Carbon and hydrogen isotopic compositions of products of open-system catalytic hydrogenation of CO2: Implications for abiogenic hydrocarbons in Earth’s crust // Geochim. Cosmochim. Acta. 2010. V. 74 P. 6112–6125.
Taran Y.A., Kliger G.A., Sevastianov V.S. Carbon isotope effects in the open-system Fischer–Tropsch synthesis // Geochim. Cosmochim. Acta. 2007. V. 71. P. 4474–4487.
Teagle D.A.H., Alt J.C., Umino S. et al. Superfast spreading rate crust 2 and 3 // Integrated Ocean Drilling Program Management International. 2006. V. 309/312. 50 p. https://doi.org/10.2204/iodp.-proc.309312.2006
Tingle T.N., Green H.W. Carbon solubility in olivine: Implications for upper mantle evolution // Geology. 1987. V. 15. P. 324–326.
Tingle T.N., Green H.W., Finnerty A.A. Experiments and observations bearing on the solubility and diffusivity of carbon in olivine // J. Geophys. Res. 1988. V. 93. P. 15289–15304.
Tingle T.N., Hochella M.F. Formation of reduced carbonaceous matter in basalts and xenoliths: Reaction of C–O–H gases on olivine crack surfaces // Geochim. Cosmochim. Acta. 1993. V. 57. P. 3245–3249.
Tsao T.C. Li K., Philbrook W.O. Kinetics of dissociation of carbon monoxide on α-iron // Can. Metall. Q. 1977. V. 16. P. 93–103.
Turkdogan E.T., Vinters J.V. Catalytic effect of iron on decomposition of carbon monoxide: I. Carbon deposition in H2-CO mixtures // Metall. Trans. 1974. V. 5. P. 11–20.
Wengeler H., Knobel R., Katherin H. et al. Atomic carbon in magnesium oxide single crystals – depth profiling? Temperature- and time-dependent behavior // J. Phys. Chem. Solids. 1982. V. 43. P. 5971.
Дополнительные материалы отсутствуют.