

Почвоведение, 2020, № 8, стр. 948-958
Влияние уксусной кислоты и ацетат-ионов на сорбцию–десорбцию смеси фенольных кислот модифицированным каолинитом
А. Г. Заварзина a, b, *, М. С. Ермолин a, c, В. В. Демин a, П. С. Федотов a, c
a МГУ им. М.В. Ломоносова
119191 Москва, Ленинские горы, 1, Россия
b Палеонтологический институт им. А.А. Борисяка РАН
117647 Москва, ул. Профсоюзная, 23, Россия
c Институт геохимии и аналитической химии им. В.И. Вернадского
119991 Москва, ул. Косыгина, 19, Россия
* E-mail: zavarzina@mail.ru
Поступила в редакцию 05.11.2019
После доработки 11.12.2019
Принята к публикации 13.12.2019
Аннотация
Исследована сорбция и десорбция смеси фенольных кислот каолинитом, модифицированным гидроксидом алюминия, в присутствии компонентов ацетатного буферного раствора. Эксперименты проводили в проточной микроколонке объемом 1 мл в 5 и 50 мМ Na-ацетатном буферном растворе (рН 4.5) при скорости потока 0.5 мл/мин. Общая концентрация раствора кислот составляла 0.01 мг/мл (0.06 мМ), концентрация каждой кислоты – 0.01 мМ. Десорбцию проводили 5 и 50 мМ Na-ацетатным буфером (рН 4.5), а также 50 мМ Na-ацетатным буфером в присутствии 0.1 мМ щавелевой кислоты. Концентрацию фенольных кислот определяли методом обращеннофазовой жидкостной хроматографии высокого давления. Установлен следующий порядок сорбции: галловая > протокатеховая $ \gg $ п-гидроксибензойная ~ ванилиновая ~ феруловая ~ сиреневая кислоты. Сорбция кислот в 50 мМ буфере составила 18–35% от их сорбции в 5 мМ буфере, что свидетельствует о конкуренции ацетат-ионов и фенольных кислот за центры связывания на минерале. Ряд по десорбции кислот был противоположен ряду по сорбции и в целом коррелировал с константами устойчивости комплексов кислот с гидроксидом алюминия. Все кислоты, кроме галловой и протокатеховой, слабо связывались с минералом и почти полностью (на 88–98%) десорбировались 5 мМ ацетатным буфером. Общая десорбция галловой и протокатеховой кислот всеми элюентами составила соответственно 25 и 45% от их сорбированного количества, и была значительной только в 50 мМ ацетатном буфере (12 и 23%) и в том же буфере с добавлением 0.1 мМ щавелевой кислоты (10 и 15%). Таким образом, показано, что распределение фенольных кислот в системе твердая фаза–раствор в значительной степени определяется присутствием конкурирующих алифатических соединений. На основе экспериментов по десорбции рассмотрены предполагаемые типы образующихся поверхностных комплексов.
ВВЕДЕНИЕ
Фенольные кислоты – лабильные ароматические соединения, поступающие в почву в виде микробных и растительных метаболитов и продуктов деструкции компонентов растительных тканей, в первую очередь, лигнина [12]. Обладая высокой реакционной способностью, они участвуют в реакциях комплексообразования с ионами металлов, в свободнорадикальной конденсации и в сорбционных взаимодействиях с органо-минеральными фазами почв [21]. Сорбция фенольных кислот почвенными минералами является важным фактором стабилизации и накопления ароматического углерода в почвах [20, 22, 26]. Этот процесс контролирует распределение фенольных кислот между жидкой и твердой фазами почв, их участие в окислительных реакциях [19], а также биодоступность как агентов аллелопатических взаимодействий [30].
В модельных лабораторных экспериментах установлена быстрая и эффективная сорбция фенольных кислот глинистыми минералами и гидроксидами Fe и Al [11, 25, 30]. Сорбция может быть одной из причин низких концентраций фенольных кислот в почвенных растворах и водных вытяжках из почв – от долей до десятков микромолей в литре [4, 6, 24]. В природных условиях на сорбционные взаимодействия влияет множество факторов – не только рН раствора, структурные особенности минеральных фаз и самих кислот [3, 14], но и конкуренция фенольных кислот за центры связывания. Фенольные кислоты конкурируют как между собой [14, 25, 30], так и с другими органическими и неорганическими соединениями. Установлено, что аминокислоты в смеси с фенольными кислотами снижают сорбцию последних [14], на сорбцию фенольных кислот влияют пленки природного органического вещества на поверхности минералов [15, 17]. Таким образом, не только совместная сорбция, но и “кондиционирование” поверхности одним видом соединений влияет на сорбцию других [14, 15].
В почвах широко распространены алифатические кислоты: уксусная, лимонная, щавелевая и др. – которые продуцируются как микробные и растительные метаболиты. Концентрация этих соединений в почвенных растворах сравнима или превышает концентрацию фенольных кислот, составляя от 0.1–100 мкM до 1–10 мМ [5, 8] и может быть очень высокой вблизи корней или грибных гифов [29]. Поэтому сорбция фенольных кислот в присутствии алифатических органических соединений является характерным для почв явлением. Установлено, что органо-минеральные соединения в почвах обеднены ароматическими структурами по сравнению с их содержанием в растительной биомассе, а алкильный и O/N алкильный углерод (алифатические кислоты) является важным компонентом органического вещества в комплексах с гидроксидами и оксидами Fe в кислых почвах [22]. Эти данные свидетельствуют о высокой конкуренции ароматических и алифатических кислот за центры связывания в почвах. Однако влияние алифатических карбоновых кислот на сорбцию ароматических соединений при природных соотношениях веществ остается малоизученным. Кроме того, большинство экспериментов по изучению совместной или индивидуальной сорбции органических соединений минералами проведено в статических условиях, при длительном времени взаимодействия и концентрациях веществ, превышающих таковые в почвенных растворах [11, 13, 30]. В то же время в почвах процессы протекают при постоянном обновлении жидкой фазы за счет миграции растворов. Нами ранее показано, что использование в сорбционных экспериментах проточных микроколонок позволяет имитировать почвенные условия и исследовать конкурентную сорбцию смесей фенольных кислот при их очень низких концентрациях [2].
Целью настоящей работы было изучение влияния уксусной кислоты и ацетат-ионов на сорбцию-десорбцию смеси фенольных кислот модифицированным каолинитом в динамических проточных условиях. Выбор минеральной фазы, использованной нами ранее [2], обусловлен тем, что каолинит и гидроксиды Al (например, гиббсит) типичны для кислых почв, а осаждение гидроксида Al на глинистых минералах является широко распространенным в почвах явлением [21, 28]. Конкурентное взаимодействие ароматических и алифатических кислот и их анионов с комплексом каолинит– гидроксид алюминия может служить адекватной моделью протекающих в почвах процессов.
ОБЪЕКТЫ И МЕТОДЫ
Минерал. В работе использовали каолинит (Просяновское месторождение, Украина), модифицированный аморфной гидроокисью алюминия (каол-Al(OH)x) и содержащий 2.5 ммоль Al/г глины. Некоторые физико-химические свойства минерала описаны ранее [2]. Площадь поверхности минерала, определенная по адсорбции паров воды, составляет 47.8 м2/г [2].
Фенольные кислоты. В экспериментах использовали галловую (Gal), протокатеховую (PCat), п‑гидроксибензойную (HDB), ванилиновую (Van), феруловую (Fer) и сиреневую (Syr) кислоты (Sigma-Aldrich, США). Некоторые свойства кислот приведены в табл. 1. Готовили исходный раствор кислот с общей концентрацией 0.5 мг/мл в 5 мМ или 50 мМ Na-ацетатном буфере (рН 4.5). Для растворения кислот использовали обработку ультразвуком (2 раза по 10 мин) на ультразвуковой бане (Bandelin Sonorex DT 50, Германия). Рабочий раствор кислот с общей концентрацией 0.01 мг/мл (0.06 мМ) и концентрацией каждой кислоты 0.01 мМ готовили из исходного раствора путем разведения соответствующим буфером.
Таблица 1.
Некоторые свойства кислот, использованных в работе
| Кислота | Обозна-чение | Формула | рКа СООН-группы |
αCOOH*, % (рН 4.5) |
Sp.a**, Å min–max |
lgβ1*** по [stepn] |
|---|---|---|---|---|---|---|
| Уксусная |
Ac |
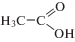 |
4.76 |
35 |
– |
1.57 |
| Щавелевая | Ox | 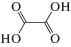 |
1.25 4.14 |
100 70 |
– | 6.10 |
| Галловая | Gal | 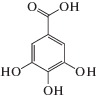 |
4.38 | 57 | 26.5–31.6 | 14.24 |
| Протокатеховая | PCat | 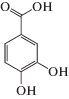 |
4.16 | 69 | 23.5–49.1 | 15.33 |
| п-Гидроксибензойная | HDB |  |
3.92 | 79 | 22.6–47.1 | 1.66 |
| Ванилиновая | Van | 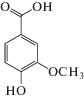 |
4.16 | 69 | 26.3–53.8 | – |
| Сиреневая | Syr |  |
3.93 | 79 | 35.4–62.2 | – |
| Феруловая | Fer | 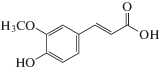 |
3.58 | 89 | 27.3–64.1 | – |
* Степень диссоциации карбоксильных групп, рассчитанная по формуле α = 10pH-pKa/(1 + 10pH-pKa). ** Проекционная поверхность (www.chemicalize.org). *** Константа устойчивости комплексов кислот с гидроксокомлексами Al для реакции Al + L = AlLβ = [AlL]/[Al][L] (цит. по [29]), соотношение металл : лиганд 1 : 1; прочерк означает отсутствие данных.
Проточная микроколонка. В экспериментах использовали тефлоновую микроколонку со следующими параметрами: внутренний диаметр 7 мм, высота 28 мм, объем 1 мл. Принципиальная схема колонки приведена в работе [1]. От колонки, использованной ранее [2], настоящая модификация отличается соотношением диаметра к высоте, равным 1 : 4, которое считается оптимальным в почвенных исследованиях [23]. Кроме того, предполагается, что диаметр входного отверстия, равный диаметру колонки, обеспечивает лучшее взаимодействие подвижной фазы с образцом. Для удерживания образца минерала в колонке использовали фильтры “синяя лента” (Апекслаб, Россия).
Эксперименты по динамической сорбции фенольных кислот в присутствии уксусной кислоты и ацетат-ионов. Эксперименты по сорбции фенольных кислот проводили в 5 или 50 мМ Na-ацетатном буфере (рН 4.5). Установлено, что концентрация буфера 5 мМ была достаточной для поддержания рН раствора на постоянном уровне в ходе экспериментов. Образец минерала массой 20 мг помещали в колонку и прокачивали через нее в направлении снизу вверх 5 мл элюирующего Na-ацетатного буфера (5 или 50 мМ) с помощью перистальтического насоса (Watson Marlow 120U/DV, Великобритания) для того, чтобы уравновесить минерал с элюентом. Затем остатки буфера откачивали и прокачивали через колонку 50 мл раствора кислот в 5 или 50 мМ Na-ацетатном буфере (pH 4.5) при скорости потока 0.5 мл/мин. Общее количество каждой кислоты, пропущенной через колонку, составляло 0.49 мкмоль (в 50 мл раствора). Раствор на выходе из колонки собирали фракциями по 2–5 мл.
Десорбция фенольных кислот. Эксперимент по десорбции проводили сразу после опыта по сорбции в 5 мМ Na-ацетатном буфере (рН 4.5). Минерал после опыта по сорбции оставляли в колонке, откачивали раствор фенольных кислот, затем прокачивали 25 мл 5 мМ Na-ацетатного буфера (pH 4.5), 15 мл 50 мМ Na-ацетатного буфера (pH 4.5) и, наконец, 15 мл 50 мМ Na-ацетатного буфера (pH 4.5) с добавлением 0.1 мМ щавелевой кислоты. На выходе собирали фракции по 3–5 мл. Известно, что щавелевая кислота является сильным комплексообразователем, присутствие ее в почвенных растворах приводит к мобилизации ионов Al, входящих в состав аморфных гидроксидов [29]. Нами установлено (данные ICP-MS), что существенного изменения минеральной фазы в присутствии 0.1 мМ щавелевой кислоты не происходило и процессом растворения гидроксида Al можно пренебречь.
Анализ фенольных кислот. Кислоты анализировали методом обращеннофазовой жидкостной хроматографии на хроматографе Agilent 1100 с диодно-матричным детектором, насосом (QuatPump), термостатом для колонок и инжектором (Rheodyne, Cotati, США). Для разделения фенольных кислот использовали колонку SunergiHydro-RP (150 × 4.6 мм, 4 мкм, Phenomenex, США). Кислоты определяли как описано ранее [2].
Параметры сорбции–десорбции фенольных кислот. Для количественного описания сорбции кислот минералом использовали следующие параметры:
1) количество сорбированных кислот (мкмоль/г) при минимальном (0.02 мкмоль) и максимальном (0.49 мкмоль) количестве вещества, пропущенного через колонку.
2) количество каждой кислоты, сорбированное минералом в процентах от ее общего внесенного количества (0.49 мкмоль).
3) доля поверхности минерала, занятая молекулами каждой кислоты (Sуд(с), %) при максимальном количестве внесенного вещества, рассчитанная, как описано ранее [2] с учетом средней проекционной поверхности молекул (табл. 1).
4) абсолютное количество кислот, десорбированное различными элюентами (мкмоль/г, мкмоль/л), а также десорбция кислот в процентах от их сорбированного количества.
РЕЗУЛЬТАТЫ
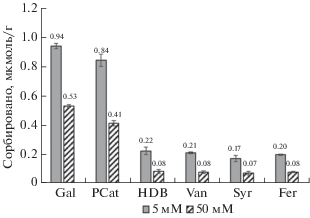
Сорбция фенольных кислот модифицированным каолинитом в присутствии компонентов ацетатного буфера. Установлены существенные различия в сорбции фенольных кислот в присутствии различных концентраций Na-ацетатного буфера. Общее количество кислот, сорбированных в присутствии 50 мМ Na-ацетатного буфера, составляло около 30% от количества кислот, сорбированных в присутствии 5 мМ Na-ацетатного буфера (рис. 1, табл. 2). При этом с увеличением степени заполнения активных центров на минерале конкурирующее влияние компонентов ацетатного буфера увеличивалось. При минимальном количестве веществ, пропущенных через колонку, сорбция фенольных кислот в 50 мМ ацетатном буфере составляла 37–59% от сорбции в 5 мМ ацетатном буфере (рис. 2), а при максимальном – 18–35%. Присутствие ацетат-ионов оказывало наибольшее влияние на сорбцию моногидроксибензойных кислот: п-гидроксибензойной, ванилиновой, сиреневой и феруловой. При 10-кратном увеличении концентрации буфера их сорбция снижалась примерно на 60 и 80%, а сорбция галловой и протокатеховой кислот – на 45 и 65% (при минимальном и максимальном количестве вещества, пропущенного через колонку соответственно).
Рис. 1.
Сорбция фенольных кислот: А – в 5 мМ Na-ацетатном буфере (pH 4.5), В – в 50 мМ Na-ацетатном буфере (pH 4.5), Б – в 5 мМ ацетатном буфере, % от максимального количества каждой кислоты, сорбированной минералом, Г – то же в 50 мМ буфере: 1 – галловая кислота, 2 – протокатеховая кислота, 3 – п-гидроксибензойная кислота, 4 – ванилиновая кислота, 5 – сиреневая кислота, 6 – феруловая кислота.
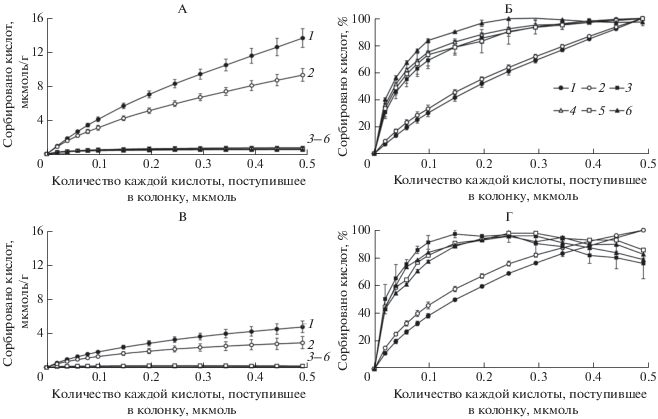
Таблица 2.
Сорбция фенольных кислот в 5 и 50 мМ Na-ацетатном буфере (рН 4.5)
| Кислота | 5 мМ Na-ацетатный буфер | 50 мМ Na-ацетатный буфер | |||||
|---|---|---|---|---|---|---|---|
| сорбировано, мкмоль/г | сорбировано, % от общего количества | S(уд)с, % | сорбировано, мкмоль/г | сорбировано, % от общего количества | сорбировано, % от 5 мМ буфера | S(уд)с, % | |
| Gal | 13.6 (1.09)* | 55.5** | 4.98 | 4.72 (0.12) | 19.3 | 35 | 1.73 |
| PCat | 9.26 (0.74) | 37.8 | 4.23 | 2.86 (0.19) | 11.7 | 31 | 1.31 |
| HDB | 0.74 (0.06) | 3.0 | 0.32 | 0.13 (0.02) | 0.5 | 18 | 0.06 |
| Van | 0.59 (0.02) | 2.4 | 0.30 | 0.15 (0.02) | 0.6 | 25 | 0.07 |
| Syr | 0.51 (0.04) | 2.1 | 0.32 | 0.14 (0.02) | 0.6 | 27 | 0.09 |
| Fer | 0.48 (0.01) | 2.0 | 0.28 | 0.15 (0.02) | 0.6 | 31 | 0.09 |
| Все кислоты | 25.18 | 17.1*** | 10.42 | 8.14 | 5.5 | 32 | 3.34 |
Рис. 2.
Сравнение сорбции фенольных кислот в 5 и 50 мМ Na-ацетатном буфере (рН 4.5) при минимальном количестве кислот, поступивших на колонку (0.02 мкмоль).
Независимо от концентрации ацетатного буфера, порядок сорбции кислот был следующий: галловая > протокатеховая $ \gg $ п-гидроксибензойная ~ ванилиновая ~ сиреневая ~ феруловая кислоты. Сорбция галловой и протокатеховой кислот более, чем на порядок превышала сорбцию остальных кислот и максимума не достигала (рис. 1А, 1В). В варианте с 5 мМ Na-ацетатным буфером кривая сорбции имела почти линейный вид (рис. 1Б). Моногидроксибензойные кислоты сорбировались примерно в равных количествах. Около 40–50% (50 мМ ацетатный буфер) и 30–40% (5 мМ ацетатный буфер) от общего количества п-гидроксибензойной, ванилиновой, сиреневой и феруловой кислот сорбировалось при пропускании первых 2 мл раствора (0.02 мкмолей каждой кислоты, рис. 1Б, 1В). Сорбция этих кислот достигала максимума при пропускании 0.25–0.35 мкмолей каждой кислоты (25–35 мл раствора) и далее в 50 мМ буфере наблюдали их десорбцию (рис. 1Г). В 5 мМ буфере вытеснения кислот не наблюдали. Площадь поверхности минерала, заполненная молекулами фенольных кислот, составляла 3 и 10% от общей площади минерала при сорбции в присутствии 50 и 5 мМ ацетатного буфера, соответственно.
Десорбция фенольных кислот, сорбированных в присутствии 5 мМ ацетатного буфера. Ряд по десорбции фенольных кислот: галловая < протокатеховая $ \ll $ п-гидроксибензойная ~ ванилиновая ~ ~ сиреневая ~ феруловая был противоположен ряду по их сорбции (табл. 3, рис. 3). Установлено, что п-гидроксибензойная кислота, ванилиновая, сиреневая и феруловая, помимо их низкой сорбции минералом, сорбируются непрочно, так как почти полностью (на 88–98%) смывались 5 мМ Na-ацетатным буфером. При этом десорбция первой порцией элюата (5 мл) составила около 60% от сорбированного количества для п-гидроксибензойной и ванилиновой кислот, и около 70% – для сиреневой и феруловой кислот (рис. 3Б). Остаточное количество моногидроксибензойных кислот (2–12%) было десорбировано 50 мМ Na-ацетатным буфером. Галловая и протокатеховая кислоты удерживались более прочно, десорбция 5 мМ Na-ацетатным буфером составила только 3 и 7%, соответственно, от их общего сорбированного количества. Однако абсолютные величины десорбции этих кислот 5 мМ Na-ацетатным буфером (мкмоль/г) были сходны с таковыми для моногидроксибензойных кислот. Увеличение концентрации элюирующего Na-ацетатного буфера в 10 раз резко увеличило десорбцию галловой и протокатеховой кислот – примерно в 20 раз в первых 3 мл элюата (рис. 3А). Введение в раствор 0.1 мМ щавелевой кислоты привело к двукратному увеличению десорбции этих кислот в первой порции элюата. Десорбция галловой и протокатеховой кислот всеми растворами составила, соответственно, 25 и 45% от сорбированного количества, из них 12 и 23% было десорбировано 50 мМ ацетатным буфером и 10, и 15% – тем же буфером с добавлением 0.1 мМ щавелевой кислоты.
Таблица 3.
Десорбция фенольных кислот, сорбированных минералом в присутствии 5 мМ Na-ацетатного буфера (рН 4.5)
| Кис-лота | Сорбция, всего, мкмоль/г | Десорбция, мкмоль/г | Десорбция, % от общего сорбированного количества | ||||||
|---|---|---|---|---|---|---|---|---|---|
| всего | 5 мМ буфер | 50 мМ буфер и 0.1 мМ щавелевая кислота (15 мл) | всего | 5 мМ буфер | 50 мМ буфер и 0.1 мМ щавелевая кислота (15 мл) | ||||
| 25 мл | 15 мл | 25 мл | 15 мл | ||||||
| Gal | 13.6 (1.09) | 3.45 (0.09) | 0.43 (0.09) | 1.61 (0.30) | 1.42 (0.26) | 25 | 3 | 12 | 10 |
| PCat | 9.26 (0.74) | 4.18 (0.14) | 0.65 (0.14) | 2.16 (0.38) | 1.37 (0.26) | 45 | 7 | 23 | 15 |
| HDB | 0.74 (0.06) | 0.73 (0.07) | 0.65 (0.07) | 0.07 (0.02) | 0.01 | 99 | 88 | 10 | 1 |
| Van | 0.59 (0.02) | 0.59 (0.06) | 0.54 (0.07) | 0.05 (0.01) | 0.00 | 100 | 92 | 8 | 0 |
| Syr | 0.51 (0.04) | 0.51 (0.05) | 0.48 (0.05) | 0.03 | 0.00 | 100 | 94 | 6 | 0 |
| Fer | 0.48 (0.01) | 0.48 (0.02) | 0.47 (0.02) | 0.01 | 0.00 | 100 | 98 | 2 | 0 |
| Все кислоты | 25.18 | 9.94 | 3.22 | 3.93 | 2.79 | 39 | 13 | 16 | 11 |
Рис. 3.
Десорбция фенольных кислот: А – абсолютные количества, мкмоль/л; а – 5 мМ Na-ацетатный буфер (pH 4.5); б – 50 мМ Na-ацетатный буфер (pH 4.5); в – 50 мМ Na-ацетатный буфер (pH 4.5) с добавлением 0.1 мМ щавелевой кислоты. Б – доля от максимальной сорбции (мкмоль/г) каждой кислоты. 1 – галловая кислота, 2 – протокатеховая кислота, 3 – п-гидроксибензойная кислота, 4 – ванилиновая кислота, 5 – сиреневая кислота, 6 – феруловая кислота.

ОБСУЖДЕНИЕ
В динамических экспериментах по сорбции смеси фенольных кислот на модифицированном каолините в присутствии уксусной кислоты и ацетат ионов при близких к природным соотношениях и концентрациях веществ, показана конкуренция между алифатическими и ароматическими кислотами за центры связывания. Установлено, что высокие (~1000-кратные в случае 50 мМ ацетатного буфера) количества уксусной кислоты и ацетат-ионов существенно снижают сорбцию фенольных кислот, в первую очередь моногидроксибензойных. Сорбция кислот может происходить как на самом каолините, так и на осажденном на нем гидроксиде алюминия. Однако ранее нами показано, что количество галловой кислоты, сорбированное на “чистом” каолините при рН 4.5, составляет примерно 5% от сорбции на модифицированном минерале [2]. Таким образом, можно считать, что связывание органических кислот модифицированным каолинитом осуществляется в основном за счет гидроксида алюминия, который при рН 4.5 заряжен положительно (рН точки нулевого заряда составляет от 8.2 до 9.5) [16]. Помимо заряда, способствующего притяжению анионов органических кислот (например, за счет группировок [Al(OH)]2+ или [Al(OH)2]+), гидроксид алюминия существенно (в 3 раза) увеличивает площадь поверхности минерала [2], увеличивая тем самым и количество сорбционных центров.
Известно, что сорбция фенольных кислот группировками ${\text{Al(OH}})_{x}^{n}$ может осуществляется за счет водородных связей, электростатических взаимодействий и лигандного обмена с участием ОН-групп, в результате чего образуются внешнесферные и внутрисферные комплексы [21, 28, 29]. Тип образующейся связи определяет ее устойчивость в условиях конкурентной сорбции. Электростатические и водородные связи относятся к слабым взаимодействиям. Наиболее устойчивыми считаются хелатные комплексы с участием ОН- и СООН-групп в орто-положении друг к другу [7, 10, 13, 14, 18]. На рис. 4 представлены типы поверхностных комплексов фенольных кислот, образование которых возможно в условиях наших экспериментов. Электростатические взаимодействия могут осуществляться за счет карбоксильных групп, существенная часть которых при рН 4.5 ионизирована (57–89%). Возможно образование внешнесферных комплексов типа {[Al(OH)2]+}–OOC–R или {Al(OH)2${\text{OH}}_{2}^{ + }$}–OOC–R (рис. 4, структура 1). Образование водородных связей возможно за счет неионизированных СООН и ОН-групп (рис. 4, структуры 2, 4, 5). Реакции лигандного обмена возможны за счет карбоксильных групп. В результате образуются моно- и бидентатные комплексы (рис. 4, структуры 3, 7). Вероятны также взаимодействия при участии карбоксильной и фенольной групп не в орто-положении (рис. 4, структура 8) и образование хелатных комплексов за счет орто-фенольных групп (рис. 4, структуры 9, 10). При этом пятичленные циклы (структура 9) более устойчивы, чем семичленные “мостиковые” комплексы (рис. 4, структура 10). Образующиеся структуры могут быть дополнительно стабилизированы за счет водородных связей (рис. 4, структура 6).
Рис. 4.
Возможные типы поверхностных комплексов фенольных кислот с группами Al(OH)x на поверхности модифицированного каолинита, подвергающиеся разрушению в присутствии: А – 5 мМ Na-ацетатного буфера (рН 4.5), Б – 50 мМ Na-ацетатного буфера (рН 4.5), В – 50 мМ Na-ацетатного буфера (рН 4.5) и 0.1 мМ щавелевой кислоты (буквы соответствуют обозначению элюентов на рис. 3). 1–3, 6, 7 – связывание за счет карбоксильной группы; 4, 5, 9, 10 – связывание за счет фенольных групп; 8 – связывание за счет карбоксильной и фенольной группы. 1 – внешнесферный комплекс (электростатические взаимодействия); 2, 4, 5 – связывание за счет водородных связей; 3 – монодентатный внутрисферный комплекс за счет лигандного обмена, 6 – то же и стабилизация водородной связью, 7 – бидентатный внутрисферный комплекс (4-членный цикл) за счет лигандного обмена, 8 – бидентатный внутрисферный мостиковый комплекс, 9 – бидентатный внутрисферный хелатный комплекс (5-членный цикл) за счет лигандного обмена, 10 – бидентатный внутрисферный хелатный мостиковый комплекс (7-членный цикл).

Проведенные эксперименты по десорбции фенольных кислот в присутствии различных концентраций ацетатного буфера, а также ацетатного буфера и щавелевой кислоты позволяют сделать выводы о преимущественном типе конкурентных взаимодействий изученных кислот с минералом и о прочности связи. Известно, что уксусная кислота и ацетат-ион сорбируются глинистыми минералами и гидроксидами за счет карбоксильной группы [9, 21]. Согласно Клеберу с соавт. [21], для уксусной кислоты и ее аниона в основном характерно образование слабых внешнесферных комплексов (примером может служить тип связи 1, рис. 4). При связывании с алюмогидроксильными группировками на поверхности каолинита показано также образование внутрисферных комплексов: монодентатных за счет –ОН карбоксильных групп и одного атома Al (рис. 4, тип связи 3), бидентатных с участием двух кислородов карбоксильной группы и одного атома Al (рис. 4, тип связи 7) и бидентатных “мостиковых” комплексов с участием двух соседних атомов Al [9]. При этом из трех вышеуказанных структур вклад первых в связывание ацетат-ионов, согласно [9], наиболее высокий. В целом, уксусная кислота и ацетат-ион образуют поверхностные комплексы с низкой энергией связывания. Монофенольные кислоты в наших экспериментах почти полностью десорбировались первыми порциями 5 мМ ацетатного буфера. Из этого следует, что связывание этих кислот группировками ${\text{Al(OH}})_{x}^{n}$ было непрочным и осуществлялось за счет тех же механизмов, что и уксусной кислоты. Действительно, спектральными методами показано, что карбоксильные группы играют ключевую роль в связывании фенольных кислот гидроксидом Al при низких значениях рН, если фенольные кислоты не содержат о-ОН групп [11, 18]. Наблюдаемое эффективное вытеснение монофенольных кислот (и их анионов) компонентами ацетатного буфера хорошо объясняется также близкими значениями констант устойчивости комплексов уксусной и п‑гидроксибензойной кислот с гидроксидом Al. Возможные типы конкурентных взаимодействий в присутствии 5 мМ ацетатного буфера показаны на рис. 4А (структуры 1–4). Небольшую долю (до 10%) в связывании монофенольных кислот могут составлять относительно более устойчивые структуры 6 и 7 (рис. 4), разрушение которых может требовать более высоких концентраций Na-ацетатного буфера. Образование поверхностных комплексов с низкой энергией связывания объясняет не только наблюдаемое явление конкуренции монофенольных кислот и уксусной кислоты за сорбционные центры, но и вытеснение монофенольных кислот орто-дифенольными при высоких концентрациях Na-ацетатного буфера (рис. 1В), показанное ранее [2].
Сорбция галловой и протокатеховой кислот более, чем на порядок превышала сорбцию монофенольных кислот, что согласуется с нашими более ранними данными [2]. Кроме того, эти кислоты более прочно связывались с минералом. Отличительной структурной особенностью галловой и протокатеховой кислот является наличие о-ОН групп, за счет которых возможно образование хелатных комплексов (рис. 4, структуры 9, 10). Кроме того, возможно образование бидентатных хелатных комплексов этих кислот с участием фенольной и карбоксильной групп не в орто-положении (рис. 4, структура 8). Таким образом, для орто-дифенольных кислот наиболее широк спектр возможных взаимодействий, включающий все структуры, представленные на рис. 4. Это увеличивает вероятность связывания в условиях конкуренции за сорбционные центры. Абсолютное количество структур, неустойчивых в присутствии 5 мМ ацетатного буфера, было практически одинаковым для всех фенольных кислот, но относительный вклад их для монофенольных кислот составлял 88–98%, а для галловой и протокатеховой кислот всего 3–7%. Это свидетельствует о том, что основную роль в сорбции последних играли другие механизмы. Десорбция галловой и протокатеховой кислот 50 мМ Na-ацетатным буфером (вытесняющим 10–12% сорбированных кислот) может быть объяснена разрушением бидентатных комплексов этих кислот, образованных за счет фенольной и карбоксильной групп (рис. 4, структура 8). Наличие связей с участием о-ОH групп (рис. 4, структуры 9 и 10) подтверждают данные по десорбции галловой и протокатеховой кислот в присутствии щавелевой кислоты, образующей с гидроксидом алюминия внутрисферные комплексы аналогичного типа (рис. 4В): при введении в 50 мМ буфер щавелевой кислоты десорбция кислот увеличилось в 2 раза. Однако существенного эффекта вытеснения галловой и протокатеховой кислот в присутствии щавелевой кислоты не наблюдали. Это может указывать на прочность хелатных комплексов этих кислот c Al, что подтверждается литературными данными по величинам констант устойчивости (табл. 1).
В целом, установленные закономерности сорбции–десорбции исследованных фенольных кислот хорошо согласуются с константами устойчивости комплексов этих кислот с Al. Устойчивость гидроксокомплексов Al с орто-дифенольными кислотами (галловой и протокатеховой) намного выше, чем с монофенольными кислотами, например, п‑гидроксибензойной, которая образует комплексы, близкие по устойчивости к комплексам c уксусной кислотой. Слабое связывание п-гидроксибензойной, ванилиновой, сиреневой и феруловой кислот минеральными фазами и конкуренция с распространенными в почвах алифатическими кислотами могут объяснить присутствие этих фенольных кислот в вытяжках из минеральных горизонтов почв, тогда как орто-дифенольные кислоты в них не обнаруживаются [24].
ЗАКЛЮЧЕНИЕ
В динамических экспериментах, максимально приближенных к природным условиям по концентрациям и соотношениям веществ, показана конкуренция между алифатическими и ароматическими кислотами за центры связывания на модифицированном гидроксидом алюминия каолините. Сорбция всех исследованных фенольных кислот в 50 мМ ацетатном буфере составляла 30–40% от сорбции в 5 мМ ацетатном буфере. Наибольшее влияние компоненты ацетатного буфера оказывали на сорбцию п-гидроксибензойной, ванилиновой, сиреневой и феруловой кислот. Эти кислоты связывались непрочно и полностью десорбировались в присутствии 5 мМ ацетатного буфера. Установлено, что галловая и протокатеховая кислоты, помимо на порядок более эффективной сорбции, удерживались более прочно, общая десорбция всеми растворами, включая 0.1 мМ щавелевую кислоту, составила 25 и 45% соответственно. Закономерности сорбции–десорбции фенольных кислот на каолините, модифицированном Al(OH)3, хорошо согласуются с литературными данными по константам устойчивости комплексов этих кислот с гидроксокомплексами Al.
Список литературы
Ермолин М.С., Федюнина Н.Н., Карандашев В.К., Федотов П.С. Изучение подвижности наночастиц оксида церия в почвах с использованием динамического экстрагирования в микроколонке и вращающейся спиральной колонке // Журнал аналит. химии. 2019. Т. 74. № 8. С. 624–633. . https://doi.org/10.1134/S1061934819080070
Заварзина А.Г., Ермолин М.С., Демин В.В., Федотов П.С. Взаимодействие смеси фенольных кислот с модифицированным каолинитом в статических и динамических условиях // Почвоведение. 2018. № 8. С 1004–1013. https://doi.org/10.1134/S0032180X18080129
Караванова Е.И. Водорастворимые органические вещества: фракционный состав и возможности их сорбции твердой фазой лесных почв (обзор литературы) // Почвоведение. 2013. № 8. С. 924–936. https://doi.org/10.7868/S0032180X13080042
Малинина М.С., Иванилова С.В. Фенольные соединения в растворах различных типов почв Центрального лесного заповедника // Почвоведение. 2008. № 4. С. 421–430. https://doi.org/10.1134/S1064229308040030
Шамрикова Е.В., Пунегов В.В., Груздев И.В., Ванчикова Е.В., Ветошкина А.А. Индивидуальные органические соединения водных вытяжек из подзолистых почв Республики Коми // Почвоведение. 2012. № 10. С. 1068–1076. https://doi.org/10.1134/S1064229312100080
Шамрикова Е.В., Кубик О.С., Денева С.В., Пунегов В.В. Состав водорастворимой фракции почв побережья Баренцева моря: органический углерод и азот, низкомолекулярные компоненты // Почвоведение. 2019. № 11. С. 1322–1338. https://doi.org/10.1134/S0032180X19110108
Adams M.L., O’Sullivan B., Downard A.J., Powell K.J. Stability constants for Aluminum(III) Complexes with the 1,2-dihydroxyaryl ligands caffeic acid, chlorogenic acid, DHB, and DASA in aqueous solution // J. Chem. Eng. Data. 2002. V. 47. P. 289–296.
Adeleke R., Nwangburuka C., Oboiriend B. Origins, roles and fate of organic acids in soils: A review // South Afr. J. Bot. 2016. https://doi.org/10.1016/j.sajb.2016.09.002
Alstadt V.J., Kubicki J.D., Freedman M.A. Competitive adsorption of acetic acid and water on kaolinite // J. Phys. Chem. A. 2016. V. 120. № 42. P. 8339–8346. https://doi.org/10.1021/acs.jpca.6b06968
Beneduci A., Furia E., Russo N, Marino T. Complexation behaviour of caffeic, ferulic and p-coumaric acids towards aluminium cations: a combined experimental and theoretical approach // New J. Chem. 2017. V. 41. P. 5182.
Cecchi A.M., Koskinen W.C., Cheng H.H., Haider K. Sorption-desorption of phenolic acids as affected by soil properties // Biol. Fert. Soils. 2004. V. 39. P. 235–242.
Croteau R., Kutchan T.M., Lewis N.G. Natural products (secondary metabolites) // Biochemistry and Molecular Biology of Plants / Eds. Buchanan B. et al. Rockville: American Society of Plant Physiologists, 2000. P. 1250–1318.
Evanko C., Dzombak, D. Influence of structural features on sorption of NOM-analogue organic acids to goethite // Environ. Sci. Technol. 1998. V. 32. P. 2846–2855.
Gao J., Jansen B., Cerli R., Helmus A., Mikutta R., Dultz S., Guggenberger G., Kalbitz K. Competition and surface conditioning alter the adsorption of phenolic and amino acids on soil minerals // Eur. J. Soil Sci. 2017. P. 1–11. https://doi.org/10.1111/ejss.12459
Gao J., Jansen B., Cerli C., Helmus R., Mikutta R., Dultz S., Guggenberger G., Vogel C., Kalbitz K. Organic matter coatings of soil minerals affect adsorptive interactions with phenolic and amino acids // Eur. J. Soil Sci. 2018. V. 69(4). P. 613–624. https://doi.org/10.1111/ejss.12562
Goldberg S., Davis J.A., Hem J.D. The surface chemistry of aluminum oxides and hydroxides. In: The Environmental Chemistry of Aluminum / Ed. Sposito G. CRC Press LLC, 1996. P. 271–332.
Gu B., Mehlhorn T.L., Liang L., McCarthy J.F. Competitive adsorption, displacement, and transport of organic matter on iron oxide: II. Displacement and transport // Geochim. Cosmochim. Acta. 1996. V. 60. № 16. P. 2977–2992.
Guan X.H., Shang C., Chen G.-H. ATR-FTIR investigation of the role of phenolic groups in the interaction of some NOM model compounds with aluminum hydroxide // Chemosphere. 2006. V. 65. P. 2074–2081.
Kaiser K., Guggenberger G. The role of DOM sorption to mineral surfaces in the preservation of organic matter in soils // Org. Geochem. 2000. V. 31. P. 711–725.
Kalbitz K., Schwesig D., Rethemeyer J., Matzner E. Stabilization of dissolved organic matter by sorption to the mineral soil // Soil Biol. Biochem. 2005. V. 37. P. 1319–1331.
Kleber M., Eusterhues K., Keiluweitk M., Mikutta C., Mikutta R., Nico P.S. Mineral-organic associations: formation, properties, and relevance in soil environments // Adv. Agron. 2015. V. 130. P. 1–140.
Kögel-Knabner I., Guggenberger G., Kleber M., Kandeler E., Kalbitz K., Scheu S. et al. Organo-mineral associations in temperate soils: integrating biology, mineralogy, and organic matter chemistry // J. Plant Nutr. Soil Sci. 2008. V. 171. P. 61–82.
Lewis J., Sjöstrom J. Optimizing the experimental design of soil columns in saturated and unsaturated transport experiments // J. Contam. Hydrol. 2010. V. 115. P. 1.
Olofsson M.A., Norström S.H., Bylund D. Evaluation of sampling and sample preparation procedures for the determination of aromatic acids and their distribution in a podzol soil using liquid chromatography-tandem mass spectrometry // Geoderma. 2014. V. 23. P. 373–380.
Polubesova T., Eldad S., Chefetz B. Adsorption and oxidative transformation of phenolic acids by Fe(III)-montmorillonite // Environ. Sci. Technol. 2010. V. 44. P. 4203–4209.
Schmidt M.W.I., Torn M.S., Abiven S., Dittmar T., Guggenberger G., Janssens I.A. et al. Persistence of soil organic matter as an ecosystem property // Nature. 2011. V. 478. P. 49–56.
Sposito G. (Ed.) The Environmental Chemistry of Aluminum. Boca Raton, Florida: CRC Press LLC, 1996.
Sposito G. The Chemistry of Soil. N.Y.: Oxford University Press, 2008.
Stepniewski W., Dudzinska R., Pawlowski L. Aluminium transport in soil with particular emphasis on the role of organic matter // J. Ecol. Chem. 1994. V. 3. № 3. P. 195–232.
Tharayil N., Bhowmik P.C., Xing B. Preferential sorption of phenolic phytotoxins to soil: implications for altering the availability of allelochemicals // J. Agric. Food Chem. 2006. V. 54. P. 3033–3040.
Дополнительные материалы отсутствуют.