

Почвоведение, 2021, № 12, стр. 1449-1480
Гуминовые вещества – гипотезы и реальность (обзор)
А. Г. Заварзина a, b, *, Н. Н. Данченко c, В. В. Демин a, З. С. Артемьева c, Б. М. Когут c
a Факультет почвоведения МГУ им. М.В. Ломоносова
119991 Москва, Ленинские горы, 1, Россия
b Палеонтологический институт им. А.А. Борисяка
117647 Москва, ул. Профсоюзная, 123, Россия
c Почвенный институт им. В.В. Докучаева
119017 Москва, Пыжевский пер., 7, Россия
* E-mail: zavarzina@mail.ru
Поступила в редакцию 02.06.2021
После доработки 19.07.2021
Принята к публикации 22.07.2021
Аннотация
Гуминовыми веществами (ГВ) называют совокупность гетерогенных темноокрашенных полидисперсных веществ, распространенных в почвах, торфах, природных водах и донных осадках. Согласно гипотезам, ГВ образуются в результате деградации и трансформации биополимеров органических остатков и реакций свободнорадикальной конденсации (процесс называется гумификацией). ГВ принято относить к особой категории специфических природных соединений, которые не имеют аналогов в живых организмах, отличны от биомолекул растительных и микробных тканей и обладают устойчивостью к биодеструкции. При этом ГВ выделяют из природных объектов щелочами и на основе растворимости делят на гуминовые кислоты (ГК, растворимы, осаждаются при рН < 2), фульвокислоты (ФК, растворимы при всех значениях рН) и гумин (нерастворимый остаток). В обзоре дан критический анализ гуминовой терминологии, номенклатуры, способа извлечения ГВ из природных объектов, а также гипотез, описывающих их образование. Показана неоднозначность и двойственность понятия ГВ (специфические соединения и сумма операционных фракций), а также условность разделения органического вещества (ОВ) на темноокрашенные соединения неопределенной структуры (ГВ) и вещества известного строения (не-ГВ). Трактовка ГВ как специфических продуктов вторичного синтеза требует пересмотра. Возможность протекания свободнорадикальных внеклеточных реакций в почвах высока, но количественный вклад продуктов этого процесса в природное ОВ пока не установлен. Традиционную щелочную экстракцию следует рассматривать как способ извлечения гидрофильных полярных веществ, осаждение кислотой – как их концентрирование для дальнейшего изучения. Исторические названия гумусовых фракций (ГК, ФК, гумин) следует сохранить как устоявшиеся групповые понятия и названия препаратов, полученных определенным способом, не придавая этим фракциям значения особых специфических веществ. Соотношения Cгк/Cфк (или Сгк/Сорг) являются простым и удобным показателем типов гумуса, отражающим биоклиматические условия его образования.
ВВЕДЕНИЕ
Почвы являются главным резервуаром Сорг в наземных экосистемах. В почвенном гумусе сосредоточено около 1500 Гт С в слое 0–1 м, это втрое больше, чем в наземной биомассе (550 Гт С) [44]. Почвы служат долговременным стоком атмосферного СО2, что во многом сформировало и определяет современный климат. Вещества гумуса обусловливают плодородие почв и их биосферные функции. Гумус является основой наземной жизни, это “есть продукт живого вещества и его источник” (А. Тэер, цит. по [3]).
История изучения веществ гумуса насчитывает более 200 лет, но до сих пор нет консенсуса об их природе, происхождении, механизмах накопления в почвах. Не утихают споры о гуминовых веществах (ГВ) как специфических и массовых компонентах гумуса. Дискуссия носит цикличный характер. Принципиальные расхождения во взглядах, характерные для первых двух десятилетий нынешнего века [49, 58, 64, 81, 83, 92, 97, 108], были присущи и первым трем десятилетиям XX в. “Одни исследователи стояли на позиции признания гумусовых [гуминовых] веществ как группы своеобразных соединений, …образование которых обязано сложным процессам превращения органических остатков… и настойчиво продолжали изучение их свойств, происхождения и механизмов образования. Другие считали гумус смесью органических веществ, являющихся продуктами разложения остатков растительного и животного происхождения, гумусовые же вещества рассматривались ими как искусственные продукты, образующиеся в процессе выделения их из почвы щелочными растворами” [15], 16]. В 1930-х гг. XX в. яркими выразителями первой точки зрения были Тюрин и Шпрингер [15, 16], а второй – Ваксман [3].
Согласно концепции ГВ, гумус на 80–90% состоит из ГВ-специфических темноокрашенных соединений, образующихся в результате разложения биополимеров органических остатков и реакций свободнорадикальной конденсации (вторичного синтеза). ГВ отличны по структуре от веществ известного строения (биомолекул), обладают высокой молекулярной массой и устойчивостью к биодеструкции. На основе растворимости в щелочах ГВ делят на группы гуминовых кислот (ГК, растворимы, осаждаются при рН < 2), фульвокислот (ФК, растворимы при всех значениях рН) и гумина (нерастворимый остаток). ГВ-ориентированная парадигма получила широкое развитие во второй половине XX в. [15, 16, 22, 21, 32, 36, 157, 158 ]. Этому способствовал и тот факт, что лидер “оппозиции” микробиолог С. Ваксман, переключился с гумуса на исследование антибиотиков (Нобелевская премия по физиологии и медицине (1952 г.) за открытие стрептомицина). Взгляды C. Ваксмана и его предшественников, скептически относившихся к специфической природе гуминовых веществ (например, Шрайнер и Шори, 1908–1930, А.Г. Трусов, 1915, 1916) в значительной мере утратили значение и были преданы забвению [15]. Происходящее в настоящее время возрождение этих идей [97, 108, 170] во многом связано с внедрением в практику почвенных исследований масс-спектрометрии высокого разрешения: пиролитической масс-спектрометрии (Pi-FIMS), масс-спектрометрии вторичных ионов (Nano-SIMS), масс-спектрометрии ионного циклотронного резонанса (MS-ICR); а также недеструктивных методов анализа органического вещества (ОВ) почв, таких как спектроскопия ядерного магнитного резонанса (ЯМР), метод прикраевой структуры спектров рентгеновского поглощения (XANES). Эти методы позволили получить информацию о структурных фрагментах [92], компонентном составе, брутто-формулах [56, 89] и пространственной организации веществ гумуса, в том числе in situ [105, 107]. Интерпретация полученных данных возродила представления о том, что гумус представлен не особыми “химически уникальными” [58] соединениями, образующимися вне живых организмов за счет реакций деградации и конденсации [15, 31, 157, 158], а смесью микробных и растительных биомолекул на разных стадиях деструкции [49, 92, 169]. Накопление этих веществ в почвах обеспечивается не молекулярной структурой, а физико-химическими, биологическими и экологическими условиями, ограничивающими скорость деструкции [142]. Важную роль играет взаимодействие с минеральными компонентами почв [103, 169].
В 2015 г. в журнале “Nature” была опубликована статья, фактически отрицающая существование ГВ как особого класса природных соединений [108]. Ее авторами предложена “модель почвенного континуума” (soil continuum model, SCM) согласно которой ОВ почв представляет собой континуум биомолекул (от лат. continuum – сплошной, непрерывный), деструкция которых идет в сторону уменьшения молекулярных масс и увеличения растворимости. Авторы модели SCM отрицают новообразование высокомолекулярных и устойчивых “ГВ” и любые реакции внеклеточного вторичного синтеза, характерную темную окраску экстрактов из почв связывают со щелочной обработкой или же присутствием пигментов (в том числе меланинов) и продуктов абиотических реакций [97, 108]. Критике подвергается вся гуминовая терминология и щелочная экстракция как метод извлечения и изучения природного ОВ, создающий артефакты [97, 108]. Статья вызвала большой резонанс и породила новую волну исторического спора о гумусе. Опубликована серия обзоров в защиту ГВ [58, 64, 81, 83], щелочной экстракции как метода изучения ОВ природных объектов [121] и реакций вторичного синтеза как приводящих к образованию новых структур, изначально отсутствующих в живых организмах [55].
В проблеме ГВ можно выделить три основных вопроса:
1) Терминологический – что именно называть гуминовыми веществами, насколько обосновано разделение гумуса на ГВ и не-ГВ, сохранять ли термин ГВ для характеристики ОВ природных объектов,
2) Фундаментальный – что представляют собой вещества, составляющие гумус почв и гумифицированное ОВ других природных объектов, образуются ли путем внеклеточного синтеза соединения, изначально отсутствующие в тканях живых организмов и каков их вклад в природное ОВ,
3) Практический – для изучения каких свойств природного ОВ применима традиционная щелочная экстракция.
Цель обзора – критически проанализировать перечисленные вопросы гуминовой проблематики, не ставя перед собой задачу изложить весь относящийся к проблеме материал, который с различной степенью подробности уже освещался в недавней как зарубежной [45, 55, 58, 64, 81, 83, 121, 140], так и отечественной литературе [12, 29, 30]. В настоящей работе рассмотрены проблемы терминологии и номенклатуры, в том числе в историческом аспекте, реакции вторичного синтеза, представления об устойчивости ГВ и их молекулярной организации, поскольку эти составляющие лежат в основе выделения ГВ в особый класс геомолекул. Обсуждение будет в основном касаться веществ, растворимых в щелочных условиях, так как эти вещества чаще всего и объединяют под термином “ГВ” [121].
КОНЦЕПЦИЯ ГУМИНОВЫХ ВЕЩЕСТВ И ГИПОТЕЗЫ ГУМИФИКАЦИИ
Прежде, чем обсуждать спорные вопросы гумусовой проблематики, стоит остановиться на концепции ГВ в ее традиционном виде [15, 16, 22, 21, 31, 157, 158 ].
Согласно гуминовой номенклатуре (рис. 1), в составе органического вещества (ОВ) почв выделяют органические остатки и гумус – темноокрашенное тонкодисперсное и аморфное ОВ или “совокупность всех органических соединений в почве, за исключением входящих в состав живых организмов и тканей, сохраняющих анатомическое строение” [21, 158]. Гумус далее делят на неспецифические соединения – вещества известных в органической химии классов (лигнин, белки, липиды и др.) и соединения неопределенной структуры – ГВ [15, 22, 21, 31, 157, 158 ]. ГВ по принципу растворимости разделяют на группы или фракции ГК, ФК и гумина [21, 81, 157]. Из фракции ГК далее могут быть выделены гиматомелановые кислоты экстракцией этанолом, а из фракции ФК – “истинные ФК” адсорбцией на активированном угле [26] или смолах XAD-8 [163] и DAX-8 [128]. В водной среде присутствуют только ГК и ФК (www.humic-substances.org).
Рис. 1.
Традиционная номенклатурная схема ОВ почв c ориентировочным содержанием компонентов в почвах. Показано несоответствие между отнесением ГК, ФК, гумина к особой категории веществ, не имеющих аналогов в живых организмах – ГВ, и щелочной экстракцией, которая применяется к гумусу в целом. В щелочной экстракт переходят как биомолекулы (вещества известной химической структуры), так и продукты их трансформации. Доля веществ известного строения от С гумуса может быть значительно выше, чем 10–15% [15] с учетом данных о высоком вкладе идентифицируемых веществ гидрофобной природы в состав гумина [80, 82].
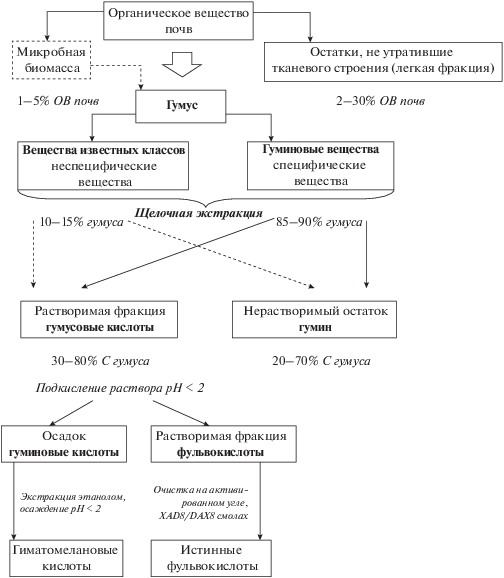
Общепринятого определения ГВ не существует. По Орлову с соавт. [25] это “совокупность веществ, образующихся в процессе разложения и трансформации растительных и микробных остатков, не имеющих аналогов в живых организмах и отличающихся темной окраской, полидисперсностью, высокими молекулярными массами и высокой биотермодинамической устойчивостью”. Сходные определения ГВ как общей категории относительно высокомолекулярных и устойчивых соединений от желтой до темной окраски, отличных от веществ известного строения, приводят и другие авторы (табл. 1). Считалось, что ГВ составляют 80–90% гумуса (остальная часть приходится на вещества известного строения), обусловливают плодородие почв и их биосферные функции [15, 20, 157]. Устойчивость почвенных ГВ связывали с их сложной конденсированной структурой [15, 22, 31, 36], а также со стабилизацией в результате взаимодействия с минеральными компонентами почв [1, 22, 158]. Предполагали, что накоплением ГВ в составе гумуса обусловлено длительное (сотни–тысячи лет) время пребывания Сорг в почвах [21, 158].
Таблица 1.
Терминология, используемая в химии гумуса
| Термин | Определение | Автор | Ссылка |
|---|---|---|---|
| Природное органическое вещество (Natural organic matter, NOM) |
Сложная смесь тысяч органических веществ в природных водах, почвах и осадках, образованных естественным путем из остатков растений, микроорганизмов и животных на разных стадиях процесса их деструкции. В почвах NOM представлено органическим веществом почв (SOM), в поверхностных водах растворенным (DOM) и взвешенным ОВ (POM) | Knicker et al., 2018 (p. 1209) |
[101] |
| Органическое вещество почв (Soil organic matter, SOM) |
Вся совокупность соединений, присутствующих в почвах в свободном состоянии или в форме органо-минеральных соединений, исключая только вещества, которые входят в состав живых организмов; включает как органические остатки (ткани растений и животных, частично сохранившие исходное анатомическое строение), так и отдельные органические соединения специфической и неспецифической природы. Четко делится на две большие группы: органические остатки и гумус | Орлов, 1992 (с. 185, 186, 188) | [24] |
| Вся совокупность органических материалов в почвах, включая подстилку, легкую фракцию, микробную биомассу, водорастворимое ОВ и стабилизированное ОВ (гумус) | Stevenson, 1994 (p. 6) | [158] | |
| Все органические соединения в почвах: 1) живая биомасса (ненарушенные ткани животных и растений, а также микроорганизмы); 2) мертвые корни и другие различимые растительные остатки или лесная подстилка; 3) в основном аморфная и коллоидная смесь сложных органических веществ, не идентифицируемая более как ткани. Третья категория сложных органических материалов является собственно почвенным гумусом. | Brady and Weil, 2008 (p. 510) |
[48] | |
| Сумма всех природных и термически измененных органических материалов биологического происхождения, находящихся в почве или на ее поверхности, независимо от их источника, состояния (живое или мертвое ОВ), стадии деструкции, но исключая надземные части живых растений | Baldock and Broos, 2011 | [43] | |
| Многокомпонентный, гетерогенный и полифункциональный континуум отдельных частиц и ансамблей биомолекул частично и полностью трансформировавшихся остатков биоты, которые отличаются по размеру, массе, химической структуре, возрасту, питательной ценности и доступности микроорганизмам, имеют разную природу и прочность внутренних и внешних химических связей, характеризуются объемной конфигурацией и пространственной неравномерностью расположения в конгломерате минеральных частиц | Семенов, Когут, 2015 | [29] | |
| Живое, отмершее и разрушающееся вещество в почвах | Kleber and Lehmann, 2019 | [96] | |
| Гетерогенная смесь всех органических компонентов в почвах, которые могут быть разделены на две группы с различными морфологическими и химическими характеристиками: 1) неизмененные материалы, включающие свежий детрит и нетрансформированные компоненты более старых остатков, 2) трансформированные продукты или гумус, который не имеет морфологического сходства со структурами, из которых он образовался. Эти трансформированные продукты относятся к гумифицированным продуктам. Состоят из гуминовых и не-гуминовых веществ | Hayes and Swift, 2020 | [83] | |
| Синоним термина гумус | Stevenson, 1994 (p. 6, 33), Piccolo, 1996 (p. 225), Huang and Hardie, 2009 (p. 43) | [132, 158] [88] |
|
| Гумус (Humus) | Органическое вещество почвы в целом (неразложившиеся органические остатки исключаются из почвенного органического вещества) | Ваксман, 1937 | [3] |
| Часть органических веществ почвы, которая потеряла анатомическое строение исходных растительных остатков, подверглась в почве процессам гумификации и формирует гумусовые горизонты, равномерно прокрашивая минеральную массу их в темный цвет | Александрова, 1980 (с. 34) |
[1] | |
| Совокупность всех органических соединений, находящихся в почве, но не входящих в состав живых организмов или образований, сохраняющих анатомическое строение (тканей живых организмов). Гумус составляют специфические гуминовые вещества, неспецифические органические соединения и промежуточные продукты распада и гумификации, находящиеся в свободном состоянии или в форме органо-минеральных соединений | Орлов, 1992 (с. 188) |
[24] | |
| Термин в узком смысле относится к гуминовым веществам и продуктам ресинтеза микроорганизмов, которые стабилизированы и стали интегральной частью почвы | Stevenson, 1994 (p. 32) | [158] | |
| Совокупность органических соединений в почве за исключением неразложившихся растительных и животных тканей, продуктов их частичной деструкции (легкая фракция) и почвенной биомассы | Stevenson, 1994 (p. 33) | [158] | |
| Аморфная и коллоидная смесь органических веществ, не идентифицируемая более как ткани | Brady and Weil, 2008 (p. 510) |
[48] | |
| Органические материалы размером <53 мкм, остающиеся после удаления взвешенного органического вещества (particulate organic matter) и растворенного органического вещества | Baldock and Broos, 2011 | [43] | |
| Подсистема почвенного органического вещества, сформированная из органических материалов и соединений растительного, животного и микробного происхождения, прошедших гумификационные и негумификационные стадии стабилизации со временем полного разложения составляющих компонентов свыше 10 лет | Когут с соавт., 2021 | [14] | |
| Неспецифические соединения (nonhumic substances) | Вещества известного строения, индивидуальные соединения | Орлов, 1992, Stevenson, 1994 (p. 33) |
[24] [158] |
| Гуминовые вещества (Humic substances) |
Комплекс органических соединений коричневого, бурого или желтого цвета, выделяемых растворами щелочей, нейтральных солей или органическими растворителями | Кононова, 1963 | [15] |
| Общая категория природных биогенных гетерогенных органических веществ, которые характеризуются окраской от желтой до темной, высокой молекулярной массой и устойчивостью | Aiken et al., 1985 (p. 4) | [36] | |
| Более или менее темноокрашенные, азотсодержащие высокомолекулярные соединения, характерные специфические продукты почвообразования, гумификации, представлены гумусовыми кислотами (наиболее характерные вещества), прогуминовыми веществами (типа “молодых” гуминоподобных продуктов, образующихся при лабораторных опытах в культуральных средах) и гумином | Орлов, 1992 (с. 189) | [24] | |
| Совокупность веществ, образующихся в процессе разложения и трансформации растительных и микробных остатков, не имеющих аналогов в живых организмах и отличающихся темной окраской, полидисперсностью, высокими молекулярными массами и высокой биотермодинамической устойчивостью | Орлов с соавт., 1996 | [25] | |
| Серия относительно высокомолекулярных веществ от коричневой до черной окраски, образующихся за счет реакций вторичного синтеза: термин используется как общее название для описания окрашенных материалов, получаемых на основе растворимости, эти материалы характерны для почв (или геологических осадков) тем, что они отличны от микробных и растительных биополимеров (включая лигнин) | Stevenson, 1994 (p. 32–33) | [158] | |
| Гуминовые вещества состоят из огромных молекул переменной структуры и состава. В целом это темноокрашенные, аморфные вещества высокой молекулярной массы, варьирующей от 20 000 до 300 000 г/моль. Из-за сложности структуры они устойчивы к деструкции микроорганизмами | Brady and Weil, 2008 (p. 511–513) | [48] | |
| Темноокрашенные, устойчивые к биодеструкции вещества, образующиеся в результате микробного метаболизма, отличны от биомолекул, присутствующих в гумусе из-за их длительного времени пребывания и молекулярной архитектуры. Образуются в результате медленного процесса биодеструкции, окисления и конденсации как характерные органические смеси, имеющие два фундаментальных свойства: 1) супрамолекулярную ассоциацию (самоорганизующиеся ансамбли небольших молекул), 2) биомолекулярный источник – идентифицируемые биополимерные фрагменты, которые образуют неотъемлемую часть лабильной молекулярной архитектуры и которые управляют конформационным поведением и химической реакционной способностью | Sposito, 2008 (p. 70) | [155] | |
| Гуминовые вещества (Humic substances) |
Биогенный, химически активный и устойчивый в природе гетерогенный континуум биомолекул, образующих полимерно-супрамолекулярные ансамбли посредством случайных химических преобразований и невалентных взаимодействий разнообразных молекул-предшественников | Семенов с соавт., 2013 | [30] |
| Серия относительно высокомолекулярных веществ от желтой до темной окраски, образующихся в результате вторичного синтеза в почвах | Soil Science Society of America, 2019 | https:// www.soils.org/ | |
| Специфические вещества, образующиеся в процессе гумификации, которые можно выделить и затем фракционировать различными способами… Аморфные, темноокрашенные гуминовые компоненты, которые разделяют на основе растворимости на гуминовые кислоты (ГК), фульвокислоты (ФК) и гумин | Hayes and Swift, 2020 (p. 4) | [83] | |
| Серия обладающих повышенной кислотностью относительно высокомолекулярных веществ от желтой до темной окраски, образующихся в результате биохимических и химических реакций в процессе разложения и трансформации растительных и микробных остатков (процесс называется гумификация) | IHSS (определение на сайте 30.12.2019, см. Dou et al., 2020) | [58] | |
| Сложная и гетерогенная смесь полидисперсных материалов, формирующихся в почвах, осадках и природных водах в результате биохимических и химических реакций в процессе разложения и трансформации растительных и микробных остатков (процесс называется гумификацией) | IHSS (определение на сайте 01.06.2021) | [90] | |
| Гумусовые кислоты (Humus acids) |
Особый класс органических соединений, образующихся в процессе гумификации органических остатков | Александрова, 1980 (с. 35) | [1] |
| Особый класс органических соединений, главные и специфические продукты гумификации органических остатков в почвах (с. 208). Представляют собой азотсодержащие высокомолекулярные оксикарбоновые кислоты с интенсивной темно-бурой или красновато-бурой окраской… (с. 190), гетерогенны и полидисперсны (с. 232). Истинная полидисперсность обусловлена присутствием в их составе молекул различных размеров, вторичная – способностью образовывать ассоциаты молекул за счет водородных связей или межмолекулярного взаимодействия (с. 233). Гумусовые кислоты экстрагируют из почвы растворами щелочей, а затем по растворимости разделяют на гуминовые кислоты, гематомелановые кислоты и фульвокислоты (с. 190) | Орлов, 1992 | [24] | |
| Гуминовые кислоты (Humic acids) |
Растворимая в щелочах группа гуминовых веществ, которую отделяют от других компонентов щелочной вытяжки путем ее подкисления до рН 1–2. Наиболее характерные компоненты гумуса | Орлов, 1992 (с. 190) | [24] |
| Фракция гуминовых веществ, которая нерастворима в воде в кислых условиях (ниже рН 2), но растворима при более высоких значениях рН | Aiken, 1985 (p. 5) | [36] | |
| Темноокрашенный органический материал, который может быть экстрагирован из почвы разбавленной щелочью и другими растворителями и нерастворим в разбавленных кислотах | Stevenson, 1994 (p. 33) | [158] | |
| Гиматомелановые кислоты (Hymatomelanic acid) |
Вещества, которые переходят в раствор при обработке осадка гуминовых кислот этанолом,
образуют вишнево-красный раствор (с. 190) Спирторастворимая часть гуминовой кислоты. Растворима в щелочи, осаждается кислотой, растворяется в спирте |
Орлов, 1992 (с. 190) Stevenson, 1994 (p. 33) |
[24] [158] |
| Термин употребляется в двух значениях. С одной стороны, это сумма кислоторастворимых органических веществ, остающихся в растворе при осаждении гуминовых кислот. С другой стороны, “собственно фульвокислоты” – это гумусовые кислоты, растворимые в водных, щелочных и кислых растворах… которые выделяют адсорбцией на активированном угле | Орлов, 1992 (с. 190) | [24] | |
| Фракция гуминовых веществ, растворимая при всех значениях рН | Aiken, 1985 (p. 5) | [36] | |
| Фульвокислоты (Fulvic acids) |
Фракция фульвокислот – фракция органического вещества почв, которая растворима и в щелочах, и в кислотах. Собственно фульвокислота – окрашенный материал во фракции фульвокислот | Stevenson, 1994 (p. 32) | [158] |
| Гумин или негидролизуемый
остаток (Humin, nonhydrolyzable residue) |
Неоднородная группа органических соединений [почв], отличающаяся от других групп нерастворимостью в щелочах. Включает, видимо, гумусовые кислоты, прочно связанные с минеральной частью, декарбоксилированные гуминовые вещества, неспецифические и нерастворимые органические соединения | Орлов, 1992 (с. 189) | [24] |
| Фракция гуминовых веществ, которая не растворима в воде при всех значениях рН | Aiken, 1985 (p. 5) | [36] | |
| Нерастворимые в щелочах компоненты гумуса | Stevenson, 1994 (p. 197) | [158] | |
| Смесь в значительной степени идентифицируемых биологических молекул, полученных из растительных материалов, состоящих преимущественно из устойчивых неполярных составляющих, находящихся в тесных ассоциациях и защищающих некоторые биоразлагаемые биомолекулы, все в тесных ассоциациях с минеральными коллоидами почвы | Hayes et al., 2010 | [84] |
Согласно концепции ГВ, эти соединения распространены не только в почвах, но и в компостах, торфе, бурых углях, природных водах и донных осадках [20, 27, 36]. Компонентный состав отмерших тканей и условия их преобразования при этом различны. Так, основным исходным материалом для ГВ наземных экосистем служат лигнифицированные растительные остатки (отсюда высокая ароматичность ГК и ФК наземного происхождения), а процесс их деградации в основном аэробный [1, 3, 15, 22, 158]. ГВ донных осадков образуется при дефиците кислорода из остатков водных организмов, не содержащих лигнина. Морские ГК и ФК, составляющие 10–50% Сорг в морской воде и известные как ГВ морских вод, состоят из фосфолипидов, полиненасыщенных жирных кислот [75], образуются, согласно гипотезам, в процессе окислительных кросс-сшивок двух и более ненасыщенных липидов [76] и имеют мало общего с почвенными ГК и ФК. В результате термин “ГВ” объединяет крайне разнородные продукты переработки органических остатков, растворимые или не растворимые в щелочах.
Из всего многообразия ГВ наиболее изучена группа гумусовых кислот (ГК и ФК) наземного происхождения. Эти вещества обогащены ионизируемыми функциональными группами, в первую очередь карбоксильными и фенольными, содержат гетероциклический и амидный азот (Nобщ 3–6%) и считаются важнейшими и наиболее реакционноспособными представителями ГВ [21, 121]. В почвах сумма ГК и ФК составляет 40–70% Сорг [21, 158], в пресных водах – до 50% (www.humic-substances.org). По Тюрину, группа ГК является наиболее характерной по закономерному изменению относительного содержания в гумусе и абсолютного количества в почве, ГК составляют от 10 до 40% от общего содержания гумуса, причем максимальные значения соответствуют черноземам [31]. Соотношение Cгк/Cфк в почвах варьирует от <0.5 до >2 и отражает условия гумусообразования [15, 21, 32]. Что касается строения гумусовых кислот, то широкое распространение получило представление об этих веществах, как обогащенных азотом гетерополимерах с ароматическим каркасом и алифатической “периферией” [30]. Предложены гипотетические модели макромолекул гумусовых кислот, основанные на функциональных и структурных составляющих, полученных при гидролизе и окислительной деструкции веществ щелочных экстрактов [21, 158].
Гипотезы гумификации. Образование ГВ связывают с гумификацией. Это комплексный процесс разложения органических остатков, обусловленный деятельностью биоты [1, 15, 21, 31, 158]. Гумификация протекает во всех природных объектах [20] и во всех гидротермических условиях, кроме гляциальных [4]. Однако, процессы, сопровождающие преобразование исходных компонентов отмерших тканей в темноокрашенные ГВ точно неизвестны, и являются областью гипотез, относящихся к аэробной ветви деструкции [15, 21, 158].
Согласно традиционным представлениям, гумификация в почвах включает гидролитическую и окислительную деградацию и трансформацию биополимеров, а также реакции внеклеточной конденсации (вторичного синтеза), приводящие к образованию N-содержащих гетерополимеров. Реакции конденсации считаются специфическими при гумификации [15, 158]. К этим реакциям относят взаимодействие фенолов, окисленных до фенокси-радикалов и хинонов, с N-содержащими соединениями, а также сахаро-аминную конденсацию (реакция Майара) [158]. В первом случае в реакции может вступать окисленный лигнин, низкомолекулярные продукты его деструкции (полифенольная теория Кононовой, Фляйга) или низкомолекулярные метаболиты микроскопических грибов (работы Мартина и Хайдера) [140, 158]. Ключевую каталитическую роль в окислении фенолов играют внеклеточные фенолоксидазы и пероксидазы микроорганизмов [1, 15, 158], хотя возможны и абиотические каталитические реакции c участием Fe и Mn, входящих в состав минералов [88]. Согласно конденсационной гипотезе в ее первоначальном виде, процесс гумификации протекает в две стадии: сначала разложение растительных остатков до более простых соединений, а затем синтез веществ сложной природы (ГВ) (Вильямс, 1914, цит. по [16]). Такой подход (сначала распад, потом синтез) развит Кононовой [15, 16]: предполагалось, что ГВ представляют собой систему новообразованных макромолекул, образующихся при конденсации мономеров. Сначала образуются более низкомолекулярные ФК, а затем ГК.
Большее признание получили теории не конденсации мономеров, а деградации биополимеров. Высокий вклад процессов окислительной деградации органических остатков в формирование гумифицированного ОВ является предметом консенсуса и у сторонников [21, 143, 157], и у противников [108, 142] гуминовой концепции. К деградационным гипотезам относятся теории окислительной биодеградации лигнина (например, [1]) и других относительно устойчивых растительных (кутин, суберин) и микробных (меланины, парафиновые макромолекулы) макромолекул (Хэтчер и Спайкер, 1988, цит. по [158]). Согласно Александровой [1], в процессе гумификации происходит деметилирование лигнина, образование карбоксильных и фенольных групп (“окислительное кислотообразование”), а также деполимеризация с образованием сначала ГК, а потом ФК. Распад исходных веществ идет в сторону уменьшения молекулярных масс и сопровождается взаимодействием продуктов деградации с минеральными компонентами почв. Согласно теории Хэтчера и Спайкера (1988), устойчивые микробные и растительные биополимеры селективно накапливаются в почвах и служат предшественниками ГВ. Гидрофобные вещества накапливаются во фракции гумина, их окисление приводит к образованию ГК, а затем ФК, которые рассматривались как наиболее гумифицированная фракция ГВ (цит. по [158]). В целом считается, что реакции деградации и конденсации могут протекать одновременно, но для гумификации характерно одно общее направление – отбор наиболее устойчивых в данных биоклиматических условиях соединений [21, 31].
Таким образом, единого определения ГВ не существует, механизмы образования и структуры ГВ гипотетичны, а понятие ГВ прочно ассоциируется с реакциями вторичного синтеза [15, 108, 158, 170] и щелочной экстракцией, приводящей к артефактам [81, 97, 121]. Тем не менее, “разложение органических остатков ведет к образованию специфической группы темноокрашенных веществ” ([3], с. 101). Возникает вопрос, что представляют собой эти вещества, насколько обосновано их выделение в особую категорию геомолекул и разделение гумуса и гумифицированного ОВ на ГВ и не-ГВ?
ПРОБЛЕМЫ ТЕРМИНОЛОГИИ И НОМЕНКЛАТУРЫ
Проблема ГВ начинается со сложности терминологии. Как отмечено в критическом обзоре [97] “для конструктивной дискуссии необходима ясность в отношении предмета дискуссии”. Действительно, “ни в одной части химии не было столько путаницы, как в химии гумуса, вследствие чего приходится иногда тратить много усилий для точного определения употребляемых терминов” [3]. Происхождение понятий и вкладываемый в них смысл имеют прямое отношение к пониманию того, что представляют собой ГВ.
Гумус и гумифицированное ОВ. Для оперирования термином “ГВ” следует в первую очередь определиться с субстратом, для которого эти вещества характерны и из которого их выделяют. Термин “гуминовые вещества” происходит от лат. слова humus (земля, почва). Однако в понятие “гумус” разными исследователями вкладывается разный смысл. Чаще всего к гумусу относят ОВ почв за исключением микробной биомассы и остатков, сохраняющих тканевое строение [21, 158]. Вещества гумуса могут находиться в свободном состоянии или в виде органо-минеральных соединений [21]. Тюрин включал в состав гумуса и микробную биомассу [31], что вызывает возражения с точки зрения номенклатуры [1, 21, 29], но оправдано с практической стороны, так как отделить биомассу невозможно. Кроме того, углерод живой микробной биомассы существенного вклада в гумус не вносит, составляя менее 4% С гумуса (цит. по [58]).
Нет согласия о генезисе гумуса. Одни исследователи считают, что гумус – это сугубо почвенное образование [1], образующееся внутри почвы [29], другие – что он есть в природе везде, где органические остатки подвергаются аэробной и анаэробной биодеструкции – в почве, компостах, торфяных болотах и водных бассейнах [3]. Последняя точка зрения представляется более соответствующей понятию о ГВ как о веществах, повсеместно распространенных в биосфере [20, 27, 36].
Понятия “гумус” и “органическое вещество почв” часто отождествляют. Начало этому положил С. Ваксман: определив гумус как “сложный агрегат аморфных веществ, окрашенных в коричневый или темный цвет, получившийся путем разложения микроорганизмами животных и растительных остатков” (с. 21), он затем предложил употреблять это название для “обозначения органического вещества почвы как целого” (с.76, [3]). Вслед за этой рекомендацией понятия гумус и ОВ почв часто употребляются как синонимы [58, 88, 158]. Это внесло путаницу, так как под ОВ почв понимают всю совокупность органических соединений, присутствующих в почвах, включая подстилку, слаборазложившиеся органические остатки и гумус [1, 24, 97, 158], иногда относя сюда микробную биомассу [158] (рис. 1).
В свете представлений о ГВ как о соединениях, повсеместно присутствующих в неживом ОВ природных объектов [20, 27, 36], важно понимать, что эти вещества являются характерной частью не ОВ почв, торфов, компостов в целом, а “гумифицированного” ОВ этих объектов, которое образуется при деструкции отмерших тканей и со временем теряет с ними визуальное структурное сходство. Выделение этой части необходимо и обосновано с точки зрения состава, функциональных и физико-химических свойств, существенно отличающихся от таковых неразложившихся остатков [21]. Как называть эту часть – “гумусом” в широком смысле по Ваксману (гумус почв, гумус торфов, гумус водных бассейнов) или “гумифицированным ОВ” почв, торфов и др., считая гумус только почвенным образованием, – открытый вопрос терминологии.
Гумификация. С термином “гумификация”, как и с остальной гуминовой терминологией, связано много путаницы. Первоначально под гумификацией понимали преобразование органических остатков в гумус [3, 82]. В результате все вещества гумуса – как относящиеся к известным классам веществ, так и ГВ рассматриваются как гумифицированные [82]. На это возникли возражения – не понятно, почему белки, ферменты, полисахариды (вещества известной структуры) становятся гумифицированными, всего лишь из-за того, что они находятся в составе гумуса (Оадес и Лэдд, 1977, цит. по [97]). Кроме того, если гумификация – это образование гумуса, то не вполне понятно, чем этот процесс отличается от гумусообразования [12, 28]. Возникают два термина для обозначения одного и того же явления. Под гумификацией понимают также образование только специфических веществ – ГВ в целом [20, 90], ГК и ФК [24] или только ГК [24]. Однако снова не вполне понятно, чем процесс преобразования органических остатков в ГВ отличается от процесса преобразования этих остатков в вещества гумуса в целом. Отсюда, видимо, возникла узкая трактовка гумификации как реакций вторичного синтеза, которые называют иначе “реакциями гумификации” [108, 170 ]. В классическом понимании (по [15]) это синтез высокомолекулярных и устойчивых веществ из низкомолекулярных продуктов распада. Такая трактовка термина приводит к отрицанию существования ГВ как количественно значимых продуктов этого процесса [97, 108].
Вероятно, имеет смысл понимать под “гумификацией” процесс преобразования тканей отмерших организмов в коллоидную темноокрашенную материю (гумифицированное ОВ) в различных природных объектах.
Гумусовые кислоты и гумин. В рамках дискуссии о гумусе возникает вопрос, почему понятие ГВ и их классификация настолько привязаны к щелочной экстракции? Стоит отметить, что понятия “гуминовые кислоты”, “фульвокислоты”, “гумин” первичны, а термин ГВ – вторичен. Начало исследованию ГВ было положено Ф. Ахардом, получившим в 1786 г. темноокрашенный раствор при обработке торфа раствором щелочи. Щелочной раствор был позднее назван гумусовой кислотой (Доберейнер, 1822), а темный осадок – гуминовой кислотой (Шпренгель, 1826) (цит. по [3]). Кислотами эти вещества назывались потому, что имели отрицательный заряд и реагировали с основаниями с образованием солей – “гуматов”. Темноокрашенные вещества, не растворимые в щелочах, были названы “гумином” (Берцелиус, 1839, цит. по [3]). Берцелиус же выделил из минеральных вод и болотного ила, богатых железом (1833), а также из гниющего вяза (1839) перегнойные вещества светло-желтого цвета, растворимые в воде и спирте и осаждаемые ацетатом меди – креновую (ключевую) и апокреновую кислоты. На смену последним, вслед работам Тюрина (1940 г.) пришел термин фульвокислоты (от лат. fulvus, желтый) введенный С. Оденом (1912–1919 гг.) для обозначения водорастворимых веществ торфа. Термин “гиматомелановая кислота”, введенный Хоппе-Зейлером (1889, цит. [3]), закрепился за фракцией, выделяемой спиртом из свежего осадка ГК [26, 157, 158].
Сам способ выделения ГВ показывает, что это не особые специфические соединения, а гетерогенные препараты. Однако, исследователи первой половины XIX в. считали выделяемые “кислоты” индивидуальными веществами, давая каждому свое собственное наименование в соответствии с источником выделения или способом получения (слизевая кислота, фуминовая кислота, ульминовая кислота и др. [3, 16]). Названия “кислот” употреблялись в единственном числе. Гумусообразование рассматривали с точки зрения химии как окисление и дегидратацию отдельных растительных веществ, поскольку микробиологии как науки тогда не существовало [3, 16]. Наряду с выделением природных ГВ предпринимались попытки получить их искусственным путем, в основном из сахаров при обработке концентрированными кислотами и щелочами (например, Герман, 1837, Браконно, 1819, Мульдер, 1839, цит. по [3]). Уже тогда получение темноокрашенных веществ при химических обработках послужило поводом к продолжающейся до сих пор дискуссии [97, 121], не являются ли природные ГВ артефактом щелочной экстракции.
В конце XIX в. вслед за развитием микробиологии и биохимии были получены данные о роли микроорганизмов в разложении органических остатков, выявлена неоднородность выделяемых щелочами веществ. Было показано, что важную часть группы веществ, растворимых в щелочах и осаждаемых кислотами, составляют ксилан и окисленные производные лигнина (Хоппе-Зейлер, 1889, цит. по [3]). Из гумусовых “кислот” было выделено более 40 индивидуальных соединений (Шрайнер и Шори, 1909–1913) и высказано мнение, что “мысль о превращении большого числа составных частей растений несколько таинственным путем в единую группу родственных тел, называемых гуминовыми кислотами, совершенно неправильна” (цит. по [3], с. 58). Таким образом, современные взгляды на вещества щелочных экстрактов как совокупность биомолекул на разных стадиях деструкции [92, 97] отнюдь не новы и высказывались более 100 лет назад.
С. Ваксман [3], подводя итог изучению гумуса в период с 1786 г. (работа Ахарда) предложил отбросить всю номенклатуру “многочисленных кислот, обозначающих не определенные химические соединения, а препараты, полученные особыми способами”. Эта рекомендация не привела к отмене привычных наименований, однако названия “кислот” стали употребляться во множественном числе как групповые понятия. По Тюрину, под термином “гуминовые кислоты” следовало “подразумевать целую группу высокомолекулярных соединений, имеющих несколько различный состав, но обладающих рядом общих свойств и …, известным общим типом строения” [31, с. 116]. Представление о гуминовых и фульвокислотах как группе родственных специфических соединений осталось центральным в гуминовой теории. Орловым [22] для “уверенного отнесения соединений к классу гумусовых кислот почв” предложено сочетание пяти признаков: 1) элементный состав (46–61% С в ГК, 36–45% С в ФК, 3–6% N), 2) обязательное присутствие в продуктах окисления бензолполикарбоновых кислот, для которых характерно наличие 3–6% “гетероциклического” азота, 3) наличие “негидролизуемого” азота (25–55% от общего), часть которого представлена азотом гетероциклов, 4) характер электронных спектров поглощения и коэффициенты экстинкции $\left( {E\frac{{0.001\% }}{{465\,\,{\text{\;нм}}}}} \right)$ порядка 0.01–0.2, 5) набор полос на ИК-спектрах. Однако общность усредненных свойств можно трактовать и по-другому – свойства веществ гуминовых фракций отражают изменения, происходящие с исходным ОВ со временем в процессе гумификации [121]. Они также отражают особенности веществ, составляющих гумифицированное ОВ, например, элементный состав, близкий к фракции ГК почв (55–58% С, 3–6% N), характерен и для гумуса в целом [3]. Бензолполикарбоновые кислоты характерны для ОВ пирогенного происхождения [68], азот гетероциклов может быть частью микробных метаболитов (DHN-меланины, [65, 120]) и меланоидинов [88], спектры в видимой области, сходные с гумусовыми кислотами, характерны для полифенолов, например, галловой кислоты [59] и получаются также в результате щелочной обработки смесей фенольных кислот (Заварзина и Демин, неопубликованные данные). Поэтому диагностические признаки гумусовых кислот не доказывают существования ГК и ФК как группы родственных соединений. Тем не менее, ГК – наиболее характерная и, пожалуй, единственная группа среди веществ гумуса, безоговорочно относимая к ГВ.
Что касается ФК, неоднократно высказывались мнения, что кислоторастворимая фракция, получаемая после осаждения ГК, представляет собой набор индивидуальных веществ и продукт частичного гидролиза разнообразных соединений, в том числе веществ фракции ГК [15, 23, 31]. “По мнению ряда авторов эти предполагаемые кислоты скорее относятся к группе промежуточных продуктов распада, а не к гуминовым веществам” [31]. Соотношение Cгк/Сфк, широко использовавшееся в отечественной практике почвенных исследований для характеристики типов гумуса [26, 31], было предложено заменить на Cгк/Сорг [23]. Тем не менее, термин ФК сохранился и используется двояко – для кислоторастворимых веществ по Тюрину и “истинных ФК” по Форситу (1947), который разработал метод очистки кислоторастворимой фракции от неспецифических примесей (углеводов, уроновых кислот, азотсодержащих соединений) путем сорбции кислого раствора на активированном угле. Этот метод долго использовался в практике почвенных исследований [26], однако в настоящее время принята очистка фракции ФК на DAX-8 смолах [90, 128]. Этот же метод применяется для выделения ГК и ФК из растворенного ОВ пресных вод [88].
Стоит также отметить дискуссию о гумине – относить ли его к ГВ или нет [15, 21, 83, 158]? По мнению Кононовой “накапливающийся материал … указывает на реальное присутствие в почве лишь двух групп: гуминовых и креновых (и апокреновых) кислот” [16, с. 94]. Действительно, в ранних работах к ГВ относили только ГК и ФК ([16], Шеффер и Ульрих, 1960, Стивенсон и Батлер, 1969, цит. по [158]), считая, что гумин – это ГК и ФК, прочно связанные с минеральной частью и поэтому плохо экстрагируемые [15, 31 ]. Затем было установлено, что гумин – это гетерогенная фракция, содержащая в том числе набор гидрофобных веществ, не извлекаемых водными растворителями [22, 136, 158]. После исчерпывающей экстракции фракций ГК и ФК из почв щелочным раствором с использованием 6М мочевины и растворения остатка в ДМСО было показано (на основе данных 13С-ЯМР), что природа гумина по большей части алифатическая и источниками составляющих его соединений являются липиды, воска, смолы, элементы растительных кутикул [83]. Поскольку по определению ГВ – это вещества, не являющиеся веществами известного строения, гумин к категории ГВ предложено не относить [83]. При этом, фракция гумина составляет 30–70% гумуса минеральных горизонтов почв [21, 136], имеет важное значение в связывании ксенобиотиков [136], а степень гумификационной трансформации входящих в него гидрофобных веществ – отдельный вопрос, требующий изучения. Тем не менее, когда говорят о ГВ, часто имеют в виду только гидрофильные полярные вещества, переходящие в щелочные экстракты [108, 121]. Все это не служит определенности понятия ГВ.
Выделение операционных фракций с целью снижения гетерогенности веществ гумуса прочно вошло в практику химии гумуса, несмотря на признаваемую условность такого подхода [15, 16]. Например, способность веществ, входящих во фракцию ГК, выпадать в осадок, зависит, помимо рН, от ионной силы раствора и степени насыщения ГК ионами металлов (зольности) [23]. Важно отметить, что процедура фракционирования относится не к ГВ [25, 81], а к гумусу [158] или гумифицированному ОВ компостов, торфов и др., так как объектами для щелочной экстракции являются почва, торф, бурый уголь, а не выделенные из них предварительно специфические вещества (рис. 1). Таким образом, ГК, ФК, гумин – не более, чем операционные части гетерогенной совокупности веществ, составляющих переработанное биотой ОВ почв и других природных объектов. По компонентному составу сумма фракций ГК, ФК, гумина соответствует веществам гумуса (или гумифицированного ОВ торфов, углей и др.) в целом. Эти фракции содержат как вещества известного строения, так и продукты их трансформации неизвестного состава. Поэтому щелочную экстракцию следует рассматривать как способ извлечения гидрофильных полярных веществ гумуса, осаждение кислотой – как их концентрирование для дальнейшего изучения, а не как способ селективного выделения специфических ГВ. Выделение ГК и ФК с помощью щелочей приводит к трансформации веществ экстрактов, о чем будет сказано ниже.
Гуминовые вещества. В русскоязычной литературе вплоть до 1980 г. для описания всех веществ, составляющих гумус, был принят термин “гумусовые вещества” [1, 15, 16, 22, 26, 31]. Для обозначения специфических веществ, отличных от веществ известного строения, употреблялся термин “собственно гумусовые вещества” [1, 15, 31, 26 ], замена которого на гуминовые вещества [21, 24] произошла, видимо, после выхода первого издания учебника Стивенсона [157].
Возникновение термина “ГВ” в его традиционной трактовке и отделение ГВ от веществ известного строения произошло в 1930-х гг. и принадлежит, видимо, Пейджу, 1930. Он предложил не употреблять термин “гумус” вследствие разных понятий, связываемых с этим словом, и заменить его на “гуминовое вещество” (humic matter), что можно также перевести как “гуминовая материя”. Под этим термином было предложено понимать “темноокрашенное, коллоидальное, высокомолекулярное органическое вещество, являющееся характерной составной частью почвы” [3, с. 70]. Видимо, отсюда впоследствии и возник термин “гуминовые вещества” (humic substances), как составляющие эту материю [31, 157]. Термин “не-гуминовое вещество” применяли для обозначения бесцветных органических веществ, хорошо растворимых и образующихся при разложении целлюлозы, лигнина или других компонентов тканей, а также самого “гуминового вещества” [3]. Такой подход сохранился и до настоящего времени.
Стоит признать, что разделение гумифицированного ОВ на темноокрашенные вещества неопределенной структуры (ГВ) и вещества известного строения (не-ГВ) вряд ли обосновано с точки зрения функционирования ОВ в почвах и других природных объектах, так как его компоненты составляют единый динамический ансамбль. Ваксман (1936) предложил употреблять термины “гуминовое вещество” (humic matter) или “гумусовые вещества” (humus substances) “для описания гумусовых комплексов как целого” (гумус по Ваксману есть не только в почвах) [3]. Однако такой подход практически сразу подвергся критике (Шпрингер, 1934, 1935, Тюрин, 1937, Лейн, 1940, Кононова, 1943, 1946, [15, 16]). По Тюрину, наличие темноокрашенных веществ “отличает неживое ОВ природных образований от неизмененного вещества растений, животных и микроорганизмов”, их “образование происходит вне живых клеток и идет в направлении отбора наиболее устойчивых соединений” и “никакой непонятной “таинственности”, о которой говорит Ваксман, в этом явлении нет” [31, с. 146]. В результате, выделение темноокрашенных веществ неопределенной структуры в отдельную категорию и обозначение их как “гуминовые вещества” стало общепринятым во второй половине XX в. [22, 157]. В 1981 г. в Денвере (Колорадо, США) было создано Международное гуминовое общество (IHSS), цель которого состояла в изучении ГВ (сейчас цели общества шире – “изучение природного органического вещества”).
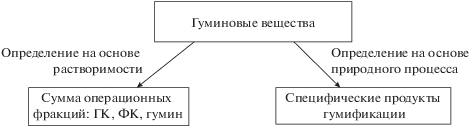
В настоящее время наряду с определением ГВ как специфических продуктов природного процесса, этот термин используется как групповое понятие для фракций ГК, ФК, гумин [157] (рис. 2). В результате возникает несоответствие теории и практики. С одной стороны ГВ – это темноокрашенные вещества, не имеющие аналогов в живых организмах [25, 81, 82], “химически уникальные соединения” [58], с другой стороны – сумма операционных фракций [58, 81, 158], содержащих и вещества известного строения, “очистка” от которых применяется в основном для фракции ФК и полностью отделить которые от веществ неизвестного строения невозможно и вряд ли целесообразно. Отнесение ГК, ФК, гумина к специфическим веществам [15, 22, 21, 157 ] было обосновано, видимо, ранними представлениями о невысоком (10–15%) вкладе веществ известного строения в эти фракции [15]. Примечательно, что во втором издании учебника Стивенсона, ГК, ФК и гумин называются фракциями гумуса, а определение ГВ “как относительно высокомолекулярных веществ от желтой до темной окраски, образующихся в результате реакций вторичного синтеза” существует как бы отдельно [158, табл. 2.3 , с. 33]. В результате термин ГВ превращается в некую теоретическую абстракцию.
Рис. 2.
Двойственность и неоднозначность понятия гуминовые вещества. С одной стороны – сумма операционных фракций, вместе составляющих всю совокупность веществ, входящих в гумус, с другой стороны – специфические соединения, не имеющие аналогов в живых организмах.
Проблемы идентификации ГВ в гумусе. Пожалуй, самый неоднозначный раздел гуминовой теории – это отделение веществ плохо охарактеризованной молекулярной структуры (ГВ) от веществ известного химического строения (биомолекул) [21, 158]. Следует признать, что такое подразделение возможно только на бумаге. Например, как в щелочном экстракте отличить и отделить ГВ от не-ГВ? Разнообразные определения ГВ (табл. 1) – это скорее описания, причем весьма неконкретные, и зависящие от текущего состояния знаний о структуре и свойствах входящих в экстракты веществ. Например, определение ГВ как высокомолекулярных веществ [90, 158] не соответствует теории о ГК как супрамолекулярных ассоциатах небольших молекул [130, 159, 174, 177]. Определение ГВ как устойчивых веществ также не корректно. Показано, что вне связи с минеральной матрицей высокомолекулярные компоненты ГК достаточно эффективно разрушаются в культурах грибов [7, 19, 70, 71, 185]. Описание ГК и ФК как темноокрашенных молекул [15, 21, 36] спорно, так как не все вещества в этих фракциях могут иметь темную окраску. Корректнее описывать ГВ как “темноокрашенную смесь молекул” [55], а не как “смесь темноокрашенных молекул”. Если исходить из определения ГВ как продуктов трансформации органических остатков (включая продукты внеклеточного ре-синтеза), которые отличны на функциональном и структурном уровне от исходных веществ [15, 21, 31, 158], то и здесь возникают проблемы. Химические критерии отличия отсутствуют. На основе каких признаков и как определить стадию биотрансформации исходных молекул, которая означает переход от не-ГВ к ГВ [110]? Считать ли идентифицируемые биомолекулы, связанные ковалентно с ГВ их неотъемлемой структурной частью [159] или пытаться отделить, применяя методы химической экстракции с разрушением эфирных связей (гумеомика Пикколо [117])? Как отделить и отличить продукты внеклеточного ре-синтеза (вторичной конденсации) от гетероструктур, исходно присутствующих в растительных и микробных тканях? Ткани живых организмов содержат как полимерные комплексы (лигнина с белками и полисахаридами, меланинов с белками, таннинов с белками [3, 49]), так и олигомерные растворимые комплексы фенольных соединений с сахарами и/или аминокислотами, называемые в физиологии растений конъюгатами [10]. Такие комплексы распространены в тканях высших растений и обнаружены в лишайниках – симбиотических организмах, доминирующих в напочвенном покрове тундр и олиготрофных лесов [9, 155]. В последнем случае они легко вымываются водой из живых талломов в окружающую среду. Перечисленные неопределенности эндемичны для области изучения ГВ и вносят вклад в “проблему гуминовых кислот” [110]. На основе существующих определений идентифицировать ГВ как некие новые вещества в составе уже сформировавшегося гумуса невозможно.
Следует признать сложность и неоднозначность термина “ГВ” в его классическом понимании и необходимость пересмотра этого понятия. Судя по всему, определенный шаг в этом направлении сделан, поводом к чему послужила статья [108], отрицающая существование ГВ как массовых продуктов вторичного синтеза, обладающих уникальными свойствами, и последовавшими за ней попытками “cпасти” это понятие [55, 58, 81, 83, 121]. В настоящее время на сайте IHSS ГВ определены как “сложная и гетерогенная смесь полидисперсных материалов, формирующихся в почвах, осадках и природных водах в результате биохимических и химических реакций в процессе разложения и трансформации растительных и микробных остатков (процесс называется гумификацией)” (www.humic-substances.org).
Современные тенденции и основания к сохранению гуминовой терминологии. Неопределенность понятия “гуминовые вещества” в его традиционной трактовке, невозможность идентификации этих веществ в составе гумуса, ассоциация понятия ГВ со щелочной экстракцией, создающей артефакты, и отсутствие корреляции между операционными фракциями ГВ и функциональными пулами ОВ в почвах [12] приводят к тому, что исследователи при изучении природного ОВ стараются пользоваться другими терминами – например, “non-living organic matter” (неживое ОВ) [85], SOM – soil organic matter (ОВ почв) [97, 140], NOM – natural organic matter (природное органическое вещество) [101, 118, 121]. Растворимую в щелочах фракцию гумуса предлагается называть “веществами щелочных экстрактов” (“substances of alkali extracts”) [108], а для описания темноокрашенных веществ неизвестного строения геохимиками предложен термин MUC (molecularly uncharacterized component of non-living organic matter) [43, 85].
Эти понятия не только не решают терминологической проблемы, а скорее, усугубляют ее. Например, термин SOM включает все ОВ почв – биомассу, органические остатки и гумус [97, 158]. Употребление этого термина автоматически включает в предмет изучения не только переработанное биотой ОВ (гумус), но и биоту и отмершие остатки. Все вместе, по Леманн и Клебер [108], и образует континуум веществ на разных стадиях деструкции. Однако становится непонятным, какую именно часть SOM исследуют, употребляя это понятие. Термин “NOM” создает проблемы с ОВ антропогенного происхождения, являющегося частью гумуса, а термин “неживое ОВ” – проблемы с микробной биомассой, которую отделить невозможно.
В составе неживого ОВ гумус почв и гумифицированное ОВ торфов, донных осадков и т.д. четко отличаются морфологически, химически и функционально от детрита – слаборазложенных органических остатков [24]. Поэтому необходимо краткое обозначение составляющих эту материю веществ. Термин ГВ, использующийся для описания “своеобразных” [15, 31], “специфических” [21, 31] и “химически уникальных” [58] веществ, образующихся в результате биотрансформации органических остатков и реакций внеклеточного синтеза [15, 158], на практике создает трудности с их идентификацией в сформировавшемся гумусе, так как эти вещества наследуют структурные составляющие исходных компонентов растительных тканей. Отделение этих веществ от веществ известного строения не менее условно, чем деление гумуса на ГК, ФК и гумин. Как вещества известного строения, так и их производные, образующиеся при переработке ОВ биотой, составляют динамический ансамбль – “гумус находится не в статическом, но скорее в динамическом состоянии, так как он постоянно образуется из растительных и животных остатков и дальше постоянно разлагается микроорганизмами” [3, с. 22]. Продукты распада биополимеров растительных и животных тканей включаются в состав микробной биомассы (что является вторичным синтезом в строгом смысле слова), подвергаются сополимеризации и конденсации [55] и вступают в надмолекулярные взаимодействия. В процессе трансформации в почвах происходит стабилизация этих соединений на минеральных фазах. Все эти соединения составляют единую систему коллоидной природы, для обозначения которой в широком смысле было бы уместно использовать термин “гуминовое вещество” (humic matter). Такая позиция соответствует предложению Ваксмана [3].
Что касается классификации, то по Ваксману “сумма современных знаний о генезисе и химии гумуса позволяет построить более логическую [чем растворимость веществ] схему классификации, целиком основанную на процессах и условиях его образования. А. Типы гумуса, образовавшегося в результате разложения растительных остатков в компостах и в почве при аэробных или только частично аэробных условиях. В. Типы гумуса, образовавшегося путем разложения растительных и животных остатков при анаэробных условиях. С. Типы гумуса, образовавшегося в водных бассейнах” [3, с. 24]. Эту классификацию можно отнести и к “гуминовому веществу” природных объектов. Вместе с тем, классификация по растворимости и термины “ГК”, “ФК” и “гумин” настолько давно используются, что предложения от них отказаться не оказались успешными ни в период, прошедший со времен С. Ваксмана, ни в период после выхода нашумевшей статьи в “Nature” (2015). Судя по данным Web of Science, количество публикаций, касающихся ГВ, и использующих соответствующую терминологию, не снижается [58]. Стивенсон (1994) отмечал, что “отмена этих понятий может привести к еще большей путанице”, а попытки замены устоявшихся терминов громоздкими описаниями, такими как: “растворимая в щелочах, не растворимая в кислотах фракция” (для ГК), вряд ли оправданы. Исторические названия гумусовых фракций (ГК, ФК, гумин) следует сохранить как краткие групповые понятия и названия препаратов, полученных определенным способом, не придавая этим фракциям значения особых специфических веществ. Основанием к сохранению гуминовых фракций является и то, что соотношение Сгк/Сорг (или Сгк/Cфк) является простым и удобным показателем типов гумуса, отражающим биоклиматические условия его образования.
ХИМИЧЕСКАЯ ПРИРОДА ВЕЩЕСТВ ГУМУСА И ВТОРИЧНЫЙ СИНТЕЗ КАК ЧАСТЬ ПРОЦЕССА ГУМИФИКАЦИИ
По Тюрину, “наиболее существенное расхождение, имеющее принципиальное значение для всей проблемы химической природы гумуса… – это является ли гумус сложным комплексом различных соединений, известных из химии растительных и животных веществ, или же в составе его присутствуют и являются для него характерными – специфические соединения, отсутствующие в списке веществ растительного и животного происхождения” [31, с. 116]. Этот вопрос является предметом обсуждения и в последнее время [49, 58, 81, 92, 108].
Ответ на фундаментальный вопрос очевиден, и был дан еще в 1927 г.: “по химическому составу гумус представлен некоторыми первоначальными составными частями растений, устойчивыми против разложения, веществами, подлежащими дальнейшему разложению, комплексами, получившимися в результате гидролиза, окисления или восстановления, и различными соединениями, синтезированными микроорганизмами” (Шрайнер и Доусон 1927, цит. по [3, с. 21–22]). Это описание дает достаточно точное представление о составе гумуса без употребления термина ГВ: 1) исходные вещества, 2) трансформированные биомолекулы – “вещества, подлежащие дальнейшему разложению и комплексы, получившиеся в результате гидролиза, окисления или восстановления” и 3) продукты внеклеточного синтеза – “различные соединения, синтезированные микроорганизмами”. Таким образом, внеклеточный вторичный синтез признавали даже противники гуминовой парадигмы (Шрайнер), не выделяя, однако продукты ресинтеза в отдельную категорию специфических веществ и называя именно их – ГВ [158].
Трансформированные биомолекулы в составе ГВ. Мнение о том, что извлекаемые щелочами вещества – это совокупность микробных и растительных биомолекул на разных стадиях трансформации, а не вновь синтезированные соединения, высказывались неоднократно еще в начале прошлого века. Возрождение этих идей во многом основано на данных ЯМР-исследований, не показавших дополнительных сигналов кроме тех, что могут дать смеси известных биополимеров [49, 77, 92, 111]. Стоит отметить, что совпадение сигналов на спектрах ЯМР гетерогенных смесей означает, что химическое окружение атомов одинаково, но не структура в целом [58]. Присутствие в спектрах ГК сигналов, присущих биомолекулам, не удивительно, так как гумифицированный материал наследует структурные фрагменты исходных веществ. Кроме того, любые спектральные методы имеют свои ограничения. Методом двумерного 1H-ЯМР [92] детектируются лишь протонированные С-атомы [51].
С помощью полуколичественной твердофазной 13С-ЯМР спектроскопии, детектирующей все атомы С независимо от молекулярной массы, и диполярного дефазирования (DD 13C-NMR), позволяющего уменьшить проблему перекрывания сигналов от различных атомов C, установлены существенные различия между спектрами ФК и ГК торфа и ГК чернозема (стандарты IHSS), спектрами биомолекул (лигнина, целлюлозы, белков) и спектрами растительных и микробных тканей [51]. Если для 13С-ЯМР спектров растительной и микробной биомассы характерно доминирование сигналов углеводов и белков соответственно, то ГК существенно отличаются от исследованных негумифицированных материалов доминированием сигналов непротонированной “ароматики”, которая не видна на спектрах 1H-ЯМР [92]. Ароматические атомы С, не связанные с атомами водорода или кислорода, составляют 28–33% в ГК против <10% в исследованных биомолекулах [51]. Часть их в ГК представлена конденсированными структурами. Спектры ГК отличаются от биомолекул и материала биомассы интенсивными сигналами атомов С, связанного с кислородом – СООН-групп, С=O кетонов, С–O алкильных групп. В спектрах ГК также присутствуют сигналы, соответствующие ароматическому С, связанному с тремя атомами С (область <125 ppm), а также алкильному С, связанному с тремя атомами С и одним атомом кислорода (около 85 ppm). Эти сигналы интерпретируются как результат образования новых С–С связей (поперечных сшивок) между ароматическими и алифатическими структурами при биотрансформации лигнина и других биополимеров в почвах и как свидетельство в пользу реакций вторичного синтеза [51]. Таким образом, данные 13C-ЯМР свидетельствуют о накоплении в процессе гумификации ароматических кислородзамещенных структур [51, 64], содержание которых невелико в исходном растительном материале и которые отсутствуют в исходных микробных тканях.
Тем не менее, анализ крайне гетерогенного гумуса и гумусовых вытяжек с помощью спектральных методов, для этого не вполне предназначенных, приводит порой к неожиданным выводам. Например, что почвенный гумус имеет алифатическую природу или в основном микробное, а не растительное происхождение (обзоры [104, 111] и ссылки в них). Так, методом твердофазной 13C-ЯМР с прямой поляризацией (13C DP/MAS NMR) было показано, что при анализе методом твердофазной 13С ЯМР с переносом поляризации с ядер 1Н на 13С (13C CP/MAS NMR) недоопределяется до 65% Сорг, в основном ароматического и карбонильного/карбоксильного [152]. Методом 13C DP/MAS NMR установлено, что содержание ароматического углерода в ГК варьирует от 33 до 55% [113]. Поэтому заключение об алифатической природе гумуса неверно. В широко цитируемой работе Симпсон с соавт. [149] на основе сравнения ЯМР спектров микробной биомассы и почвенного ОВ сделан вывод о том, что в некоторых почвах микробная биомасса составляет >50% фракций экстрагируемого ОВ, около 45% фракции гумина и >80% почвенного азота. Эти оценки явно завышены [40].
Таким образом, обогащенность окисленными ароматическими фрагментами является характерным признаком ОВ наземного происхождения [21, 158]. Основным источником этих структур в почвах современной биосферы является лигнин и другие полифенолы, например, гидролизуемые и конденсированные таннины [15, 21]. Высокий вклад ароматических структур лигнина в состав ОВ почв и природных вод показан во многих работах [60, 72, 115, 162, 188]. При окислении гумуса и ГК оксидом меди в щелочной среде, среди продуктов идентифицируются ванилиновая и сиреневая кислоты, соответствующие им альдегиды и кетоны, а также коричные кислоты: феруловая и п-кумаровая [60]. Эти структуры известны как биомаркеры лигнина [39, 86 ] и широко используются для определения содержания и изучения трансформации структурных компонентов этого полимера в почвах [13, 72, 162, 188]. В отличие от нативных растительных остатков, для гумуса и его щелочных экстрактов характерна обогащенность кислотами по сравнению с альдегидами и кетонами [60], что свидетельствует об окислительной трансформации исходных структур лигнина в почвах и согласуется с данными ЯМР-исследований ГК [51, 64].
В связи с компонентами тканей живых организмов, при отмирании вносящих вклад в темноокрашенное вещество гумуса, стоит упомянуть грибные пигменты меланины. Высказано мнение, что вещества фракции ГК – это и есть интактные меланины [11]. Действительно, эти пигменты имеют большое сходство с ГК почв по элементному составу, молекулярным массам, оптическим свойствам. Однако показано, что грибные меланины быстрее минерализуются, чем ГК, соответственно, ГК интактными меланинами не являются [187]. Вклад этих пигментов в формирование гумуса не известен.
Согласно новой модели SCM [108], процесс разложения ОВ направлен в сторону увеличения растворимости и уменьшения молекулярных масс подвергающихся деградации веществ, что способствует сорбции продуктов деструкции минеральными фазами. Действительно, в связи с процессами окисления растворимость в водных растворителях таких биополимеров, как лигнин и целлюлоза увеличивается. Однако увеличение растворимости может рассматриваться как переходный, но не конечный этап [55]. Например, процессы деградации ОВ в отсутствие минеральной составляющей (в компостах), приводят к снижению содержания водорастворимых гидрофильных компонентов по мере компостирования [53]. В процессе гумификации среди продуктов со временем накапливаются алкильные компоненты липидов, ароматические соединения, снижается содержание полисахаридов и аминокислот [189], увеличивается относительная гидрофобность, то есть уменьшается растворимость.
В целом в последние три десятилетия накоплен большой объем данных о структурных составляющих и компонентном составе гумифицированного ОВ различных объектов. Однако использование спектральных методов нарастающей сложности не приводит к принципиально новым данным о компонентном составе гумуса. Например, изучение состава фракций ГК и ФК высокоточным методом ионного циклотронного резонанса (ICR-MS) сводится к диаграммам Ван-Кревелена, представляющих собой композиционный состав вещества в координатах H/C–O/C [94] и к выделению на этих диаграммах областей, соответствующих предшественникам ГВ: лигнину, таннинам, терпеноидам, липидам, полисахаридам, белкам и пр. [151]. Методами нано-SIMS и NEXAFS получена ранее недоступная информация о кластерной организации компонентов гумуса in situ [105, 107]. Тем не менее, сложные спектральные методы анализа веществ гумуса не могут дать однозначный ответ на вопрос, присутствуют ли в гумусе продукты вторичного синтеза и каков их вклад. Эти вопросы могут быть решены только с биохимической точки зрения: “вместо того, чтобы все внимание обращать на уже образовавшийся гумус, логичнее начать со свежих растительных остатков и следить за ходом их изменений в процессе их превращения в гумус” [3, с. 105].
Продукты вторичного синтеза в составе гумуса. Этот раздел химии гумуса и есть главный предмет дискуссии. Если ранее считали, что основная масса веществ гумуса образуется при участии реакций свободнорадикальной конденсации [3, 15, 157], то в настоящее время вторичный синтез вовсе отрицают [108, 170]. Практически общепринятыми стали супрамолекулярные теории [130, 176], исключающие образование в природных условиях вторичных полимерных соединений. Согласно Пикколо [130], процесс гумификации заключается в накоплении небольших молекул (до 2000 Да), образующих надмолекулярные ассоциаты за счет нековалентных взаимодействий: сил Ван-дер-Ваальса, гидрофобных и водородных связей. Гидрофобные молекулы образуют оболочку для защиты гидрофильных молекул от деструкции [133]. Теория Пикколо является развитием гипотезы Вершау [176] о мицеллярном строении ГВ.
Таким образом, основой дискуссии о гумусе является оспаривание “гуминовой парадигмы”, которую Клебер и Леманн [97], следуя за Вершау [177]) трактуют как представление о том, что “гумус состоит из конечных продуктов синтетических реакций, которые изменяют структуру биомолекул таким образом, что это сообщает этим вновь синтезированным материалам уникальные свойства, отличные от негумифицированного ОВ”. Фундаментальный вопрос, по Клебер и Леманн [97] состоит в следующем: “есть ли количественно значимый природный процесс вторичного синтеза, который существует независимо или в дополнение к процессам деградации и “собирает” продукты деградации в новые вещества, отличающиеся от исходных веществ на молекулярном и функциональном уровне”. Ранее, в обзоре Бардон [49], касающемся гипотетических структур ГК и ФК, был сделан вывод, что нет веских причин утверждать, что ГВ состоят из сложных новообразованных молекул, не встречающихся в растениях и микроорганизмах или не являющихся закономерными продуктами их деградации. Дискуссия в целом сводится к критике реакций конденсации (“реакций гумификации”), приводящих к образованию полимерных и устойчивых “ГВ” [108] и обеспечивающих стабилизацию Сорг в почвах [170].
Одним из аргументов против внеклеточного синтеза сложных гуминовых структур называют “эволюционный пресс”: микроорганизмы, тратящие свою энергию и ресурсы на производство трудноразлагаемых материалов, не выжили бы из-за межвидовой конкуренции [49]. Если сложные гуминовые структуры – нецелевой продукт спонтанного ре-синтеза, биохимическая “ошибка”, то вклад таких “ошибок” в природное ОВ должен быть мал [49]. Эти аргументы используются и в более поздней критике [96, 97, 108]. Стоит возразить, что вопрос эволюционного пресса вовсе не стоит, так как внеклеточные фенолоксидазы и пероксидазы, окисляющие фенольные субстраты, продуцируются микроорганизмами для их собственных метаболических целей, а процесс свободнорадикальной конденсации является спонтанным, не требующим энергетических затрат. Биокатализаторы конденсации, например, лакказа, тирозиназа, пероксидаза, широко распространены в почвах [50, 150], как и абиогенные катализаторы – ионы металлов с переменной валентностью (Fe3+, Mn4+), входящие в состав оксидов и гидроксидов [88].
Гораздо более существенным является вопрос – что понимают под вторичным синтезом: конденсацию из низкомолекулярных продуктов распада (по Кононовой) или к этим реакциям относится и сополимеризация (например, присоединение аминокислот к окисленному лигнину)?
Биохимические свободнорадикальные процессы при деградации полимеров. Эта часть гуминовой теории наименее дискуссионна. Возможность образования радикалов удобно рассмотреть на примере окисления лигнина – массового компонента древесины (10–30% в составе древесины) и основного источника реакционноспособных фенольных соединений в почвах современной биосферы. Биодеградация лигнина осуществляется в основном грибами (см. обзоры [28, 178, 182]). Грибы белой гнили древесины и родственные им базидиомицеты подстилок [124] разрушают лигнин до низкомолекулярных фрагментов за счет разрыва С–С связей как ароматического кольца, так и фенилпропановых звеньев [28, 95]. Ключевую роль в этом процессе играют неспецифические лигнолитические пероксидазы с высоким редокс-потенциалом – лигнин-пероксидаза (КФ 1.11.1.14, LP, обнаружена только в дереворазрушающих грибах), Mn-зависимая пероксидаза (КФ 1.11.1.3, MnP), универсальная пероксидаза (КФ 1.11.1.16, VP), а также фенолоксидаза лакказа (КФ 1.10.3.2) [178, 182]. Действие ферментов сводится к окислению субстрата молекулярным кислородом (лакказа), перекисью (VP, LP) или Mn3+ (MnP, VP) с образованием хинонов, семихинонов, арильных и фенокси-радикалов. Эти реакционноспособные частицы инициируют деградацию субстрата или вступают в спонтанные реакции конденсации. Деградации способствуют образующиеся в хинон/гидрохиноновых циклах активные формы кислорода [69]. Результатом является минерализация лигнина и его деполимеризация [87, 145], вплоть до мономеров [158]. Радикалы, карбонильные соединения и резонансные структуры, генерируемые ферментами в процессе деструкции лигнина [138] вступают в реакции конденсации и сополимеризации, присоединяя другие молекулы с нуклеофильными группами – аминокислоты, углеводы и др. Конденсации способствуют лакказы, одна из функций которых может быть связана с полимеризацией низкомолекулярных фенольных соединений, образующихся при деструкции лигнина, и токсичных для грибов-деструкторов [164]. В результате образуются темноокрашенные продукты различной молекулярной массы, что было показано при деструкции лигнина в культурах грибов [2, 179, 185]. Образование высокомолекулярных ГК-подобных соединений возможно также при частичной деструкции лигнина грибами-целлюлолитиками, которые не содержат лигнолитических пероксидаз. Базидиомицеты бурой гнили древесины способны к частичному окислению лигнина за счет активных форм кислорода, образующихся при реакции Фентона с сохранением его высокомолекулярной структуры и появлением бурой окраски [41]. Аскомицеты (грибы мягкой гнили древесины, почвенные микромицеты) могут частично деполимеризовывать лигнин за счет лакказ [28, 185]. Окисление лигнина лакказами сопровождается появлением темной окраски [123]. Результатом воздействия грибов-целлюлолитиков является деметилирование, деметоксилирование лигнина, обогащение ароматических колец карбоксильными группами. Показано, что трансформация веществ фракции ГК в ФК (и наоборот) осуществляется теми же неспецифическими ферментами грибов, которые участвуют в трансформации лигнина [19, 70, 71, 155, 185]. Перечисленные биохимические процессы, включая деградацию и сополимеризацию, лежат в основе деградационных теорий гумификации [1, 3, 62] и предполагают образование темноокрашенных гетероструктур различной молекулярной массы, отличных своими свойствами от исходных молекул. Например, окисленный лигнин и целлюлоза приобретают растворимость в щелочах (нативный лигнин и целлюлоза плохо растворимы).
Конденсация низкомолекулярных веществ. Низкомолекулярные фенольные соединения, образующиеся при деградации лигнина, а также фенольные метаболиты грибов могут подвергаться конденсации с образованием темноокрашенных гетероструктур, что является основой конденсационных гипотез гумификации [15, 62]. Однако масштабы этого процесса неоднократно подвергались сомнению с учетом низких концентраций низкомолекулярных предшественников в почвенных растворах [24]. Насколько далеко может идти полимеризация в природных условиях? Возможно ли образование макромолекул таким путем?
Большинство экспериментальных подтверждений вторичного синтеза получено для модельных замкнутых систем и/или высоких концентраций исходных веществ (1–10 мг/мл) – это реакции в гомогенных системах в присутствии фенолоксидаз и пероксидаз [15, 37, 47], реакции в гетерогенных каталитических системах с участием абиогенных катализаторов [88] или иммобилизованных тирозиназы [116] и лакказы [182, 184 ]. Показано образование темноокрашенных веществ в культурах миксобактерий [16], некоторые микроскопические грибы также продуцируют фенольные соединения, а затем окисляют их в присутствии различных аминокислот с образованием темноокрашенных, осаждаемых кислотой веществ (Хайдер и др., 1965, Хайдер и Мартин, 1967, Мартин и Хайдер, 1969 цит. по [140]). Образование гетерополимеров объясняют реакциями фенольных веществ с нуклеофилами, например, свободными аминогруппами лизина и тиольными группами цистеина с последующим дезаминированием и декарбоксилированием [15, 74].
В лабораторных условиях установлено, что синтезу темноокрашенных, макромолекулярных веществ способствует наличие биокатализатора и границы раздела фаз, “концентрирующей” как субстраты, так и продукты. В присутствии глинистых минералов сокращается время, требующееся на формирование микроскопическими грибами ГК-подобных конденсатов, и увеличивается их количество (Мартин с соавт., 1972, цит. по [140]); в присутствии глинистых минералов и иммобилизованной грибной лакказы образуются более высокомолекулярные продукты из фенольных кислот и аминокислот, чем в гомогенной системе [182, 184 ]; в погруженных культурах грибов показана возможность конденсации низкомолекулярных продуктов деструкции ГК на грибном мицелии [185]. В целом большой набор экспериментальных данных свидетельствует о возможности внеклеточного синтеза ГК-подобных гетероструктур, включая полимерные соединения, и о важной роли биокатализаторов и границы раздела фаз в этом процессе. Однако возможность образования таких структур при низких концентрациях субстратов (0.1–0.01 мг/мл) и в открытых динамических условиях, характерных для почв, дискуссионна и требует экспериментального подтверждения или опровержения. Нами показано, что пропускание смеси из шести фенольных кислот (0.01 мМ каждой) через микроколонку, содержащую глинистый минерал с иммобилизованной лакказой, не приводит к образованию полимерных веществ, однако, присутствие биокатализатора способствует связыванию отдельных фенольных кислот с минералом [182]. Возникает вопрос – так ли уж важно образование полимеров для стабилизации Сорг?
Существенным является вопрос о количественном вкладе конденсационных реакций в гумификацию в почвах современной биосферы, где ведущим процессом является трансформация растительных биополимеров. Вероятно, реакции конденсации с участием низкомолекулярных фенольных соединений имеют значение для первичного гумусообразования в отсутствие лигнина, например, на начальных стадиях заселения минеральных субстратов микробиотой. Большой вклад в исследование таких реакций и образование “грибного” гумуса внесли К. Хайдер и Дж. Мартин (1965–1978 гг.) [140]. Фенольные соединения, их водорастворимые коньюгаты с сахарами и аминокислотами [9], а также катализаторы гумификации – лакказы и тирозиназы, обнаружены в лишайниках – организмах, образующих сплошной напочвенный покров в тундрах и олиготрофных лесах [46]. Это предполагает потенциальное участие фенольных соединений и фенолоксидаз низших растений в первичном гумусообразовании. Существенно, что временной отрезок для таких реакций может составлять около 2 млрд лет до появления лигнина в девоне [186]. В истории кислородной биосферы (3 млрд лет) образование гумуса в лигнин-содержащих условиях составляет только 400 млн лет, и именно в это время сформировались современные почвы с гумусовыми горизонтами как корнеобитаемым слоем. Биохимические реакции свободнорадикальной конденсации мономеров должны быть в фокусе дальнейших исследований, чтобы разрешить вопрос значимости конденсационных процессов в стабилизации ОВ [140, 143] на разных этапах эволюции наземных экосистем.
В связи с биохимическими процессами образования ГВ в ранней биосфере стоит отметить потенциальную роль бактериальных лакказ. В бактериях существуют две формы лакказы – трехдоменная (характерная также для грибов, имеющая кислый рН-оптимум в окислении фенольных субстратов) и двухдоменная, эволюционно древняя форма фермента (см. ссылки в [106, 163]). Двухдоменная лакказа обладает щелочным рН-оптимумом при окислении фенольных субстратов, термостабильностью и устойчивостью к ингибиторам (азиду натрия). Недавно показано, что двухдоменные лакказы стрептомицетов способствуют полимеризации низкомолекулярных- и высокомолекулярных фракций гуминовых кислот, что приводит к образованию новых ковалентных связей в гуминовом ансамбле [109]. Показано участие двухдоменных лакказ в сополимеризации фенольных кислот и ГК, в результате происходит увеличение относительного содержания высокомолекулярных фракций в ГК [166]. Гумификация в щелочных условиях требует дальнейшего изучения.
В целом большой набор экспериментальных данных показывает безосновательность отрицания возможности вторичного синтеза в почвах. Эти реакции могут играть важную роль в молекулярно-массовой организации гумифицированного ОВ, а также в связывании органического углерода и азота в почвах.
Устойчивость гуминовых веществ. Этот постулат является одним из главных в гуминовой теории: “совершенно ясно, что из разнообразных реакций превращения органического вещества вне живых организмов, приобретают значения только те реакции, которые приводят к образованию относительно устойчивых соединений, а такими соединениями являются гуминовые вещества” [31]. Это во многом объясняет и интерес к вторичному синтезу. Считалось, что эти реакции приводят к образованию сложных полимерных конденсированных гетероструктур, устойчивых к биодеструкции [15]. Особая устойчивость, приписываемая специфическим соединениям (наиболее яркими представителями которых являются ГК) долгое время служила главным основанием к выделению ГВ в особую категорию геомолекул [22, 158].
Однако к настоящему времени ошибочность этого представления можно считать доказанной. Целый ряд данных свидетельствует о том, что причиной длительного времени пребывания Сорг в почвах является не столько молекулярная устойчивость веществ гуминовых фракций, как считалось ранее [15, 158], сколько изоляция ОВ от деятельности микробиоты (движущей силы деструкции и синтеза). Сложность молекулярной структуры имеет значение лишь на ранних стадиях деструкции в органогенных горизонтах почв [104, 170]. Относительной первичной молекулярной устойчивостью (сложность структуры) обладают лигнин и липиды, тогда как углеводы и белки входят в лабильный пул [102]. Однако в присутствии легкоразлагаемых субстратов лигнин может также активно подвергаться деструкции, как и другие материалы растительного происхождения (Хайдер и Мартин, 1981, цит. по [39, 135, 140]. Относительно низкая молекулярная устойчивость характерна и для веществ щелочных экстрактов (ГК) при их инкубировании в культурах грибов. Вне связи с минеральной матрицей ГК эффективно обесцвечиваются и деполимеризуются как лигнолитическими [70, 182], так и целлюлолитическими грибами [8, 19, 71].
В связи с устойчивостью веществ гумуса стоит упомянуть проблему пирогенного углерода. Для ГК характерно наличие конденсированных ароматических структур [51]. Противники гуминовой концепции связывают это с присутствием в ОВ почв веществ пирогенного происхождения (black carbon, BC) [141]. Высказано мнение [68, 141, 142], что BC является основным носителем ароматического углерода в почве, обусловливает темную окраску черноземов и обогащенность ОВ этих почв ароматическими фрагментами, показанную во многих работах [137]. Косвенным подтверждением этого служат данные исследования химической структуры ОВ в ненарушенных профилях почв методом 13С СP/MAS NMR: установлено, что с глубиной растет вклад нефенольного ароматического углерода в ОВ почв, а фенольного, наоборот, снижается [189]. Однако исследование образцов чернозема с опытных участков Центрально-черноземного государственного заповедника [167] показало, что содержание пирогенного С в 0–30 см слое почвы, определенное по бензолкарбоновым кислотам, составляет 3–4% от Сорг для некосимой степи и 5–7% для многолетнего чистого пара. Таким образом, углеподобные вещества вносят большой вклад в общее содержание ароматических соединений не во всех почвах. Кроме того, методы определения ВС в почвах, основанные на термическом и химическом анализе весьма условны [17]. Маркеры “BC”, например, бензолполикарбоновые кислоты [68] могут быть непирогенного происхождения [64] – образование BC-подобных веществ, а также алициклических кислот показано в ходе инициированной гидроксил-радикалами (реакция Фентона) деградации лигнина [171]. Что касается длительной сохранности ВС в почвах, то было установлено, что в среднем 42–66% пирогенного С приповерхностного слоя почвы уничтожается уже следующим пожаром [57].
Таким образом, молекулярная устойчивость не абсолютна, а структурно-молекулярное описание наиболее стабильных компонентов ОВ почв до сих пор не получено [102]. В длительной перспективе все вещества гумуса разлагаются до СO2 и H2O [91]. Дефицит кислорода (например, в гидроморфных почвах) не является препятствием к минерализации ОВ [114]. Вместе с тем среднее время пребывания, измеренное для различных идентифицированных индивидуальных соединений в ОВ почв, оказывается меньше, чем для ОВ почв в целом [39, 142]. Это может свидетельствовать о том, что устойчивость ОВ обеспечивается не структурой отдельных соединений, а ковалентными и нековалентными взаимодействиями в сложном комплексе неживого ОВ, адсорбционными взаимодействиями с минеральной составляющей и физической изоляцией внутри почвенных агрегатов [96, 103, 104, 142, 170]. Длительную консервацию ОВ обеспечивают также захоронение в осадках (бурые угли).
Молекулярно-массовая организация ГВ. Если ранее ГК считались относительно высокомолекулярными соединениями – 5–100 кДа [15, 158], образующими надмолекулярные ассоциаты [21, 122], то в настоящее время их принято считать надмолекулярными ансамблями небольших молекул (2–6 кДа) [130, 159, 175]. В связи с проблемой молекулярно-массовой организации ГВ стоит отметить, что исследователи гумуса в трактовке результатов всегда опирались на текущие тренды классической химии.
ГВ как система полимеров. Представление о ГВ как системе полимеров [15, 16] последовало за работами Штаудингера (Нобелевская премия по химии в 1953 г.), который сформулировал в 1922 г. представления о полимерах – соединениях, состоящих из больших молекул, атомы которых связаны между собой ковалентными связями. Для описания таких молекул он ввел в науку понятие “макромолекула” [35]. Макромолекулярная теория была вскоре приложена и к химии гумуса. Были предложены схемы синтеза ГВ из мономеров, являющихся продуктами распада биополимеров [16]. Чтобы “запустить” реакции синтеза ГВ, система биополимеров должна была деградировать практически до самого конца. При этом, не рассматривались такие факторы, как ограниченная диффузия мономеров к месту реакции, концентрация почвенных растворов и др. Высокомолекулярная теория ГВ доминировала в химии гумуса вплоть до 1990-х гг. [25, 158]. Конформационное поведение макромолекул ГК и ФК в растворах описывали на основе поведения белков, в связи с чем стоит упомянуть и модель стохастического клубка (random coil) [161]. Согласно этой модели ГК и ФК в щелочных условиях принимают вытянутую конформацию из-за электростатического отталкивания между отрицательно заряженными функциональными группами, при снижении рН и увеличении ионной силы раствора молекулы, составляющие ГК и ФК, становятся более компактными [161].
Несмотря на недостатки конденсационной гипотезы, присутствие в составе гумифицированного ОВ высокомолекулярных веществ очевидно хотя бы на основании исходного состава органических остатков и тех биохимических процессов, которые сопровождают деструкцию. Еще в 1970–1990 гг. Д.С. Орловым было сформулировано представление о ГВ как о совокупности молекул, обладающих: 1) истинной полидисперсностью за счет различных молекулярных масс входящих в их состав веществ и 2) вторичной полидисперсностью за счет образования надмолекулярных ассоциатов. О присутствии истинно макромолекулярных компонентов в ГК свидетельствуют и хроматографические данные. Методом гель-фильтрации на геле Toyo-Pearl для 77 образцов гуминовых материалов различного происхождения получены среднечисловые молекулярные массы (Mn) порядка 2.9–9.7 кДа, а средневесовые (Mw) – 4.7–30.4 кДа [127]. Близкие результаты получены методом высокоэффективной жидкостной хроматографии для 33 образцов почвенных ГК (Mn 1.3 ± 0.5 кДа, а Mw 13.2 ± 1.2 кДа, [63]). Для 130 образцов водных ГК и ФК получены значения Mn порядка 800–1700 Да [125]. Стоит отметить, что значения молекулярных масс ГВ, определенные экспериментально, весьма условны, так как зависят от типа используемых матриц и элюирующих буферов [55]. Например, гель Toyo-Pearl [127] имеет сродство к гидрофобным/высокомолекулярным компонентам ГК, которые сорбируются на геле, что приводит к заниженным значениям Mn и Mw. Еще одной проблемой является отсутствие молекулярно-массовых маркеров для ГК и ФК. Калибровку хроматографических колонок проводят по глобулярным белкам или полистиролсульфоновым кислотам. Конформация ГВ, определяющая их размерные параметры, может значительно отличаться от модельных полимеров. Тем не менее, о присутствии истинно макромолекулярных компонентов в ГК свидетельствуют биохимические исследования деполимеризации ГК в культурах грибов в присутствии ферментов, действующих на ковалентные связи в фенольных субстратах [19, 70, 185]. Методом диффузионно-упорядоченной 1H-ЯМР спектроскопии (DOSY – diffusion-ordered spectroscopy) исследована природа высокомолекулярных компонентов ГК [147, 148 ]. Обнаружены компоненты с химическими сдвигами, характерными для лигнинов, полисахаридов и пептидов, и диффузивностью, согласующейся с молекулярными массами порядка 2500, 1000 и 200–600 Да соответственно. Эти данные подтверждают общее заключение, что в щелочных экстрактах присутствует совокупность компонентов на различных стадиях деструкции с широким диапазоном молекулярных масс и структур.
ГВ как надмолекулярные структуры. В 1990-х гг. Леном были сформулированы основные концепции супрамолекулярной химии [18] (Нобелевская премия в 1987 г. совместно с Педерсеном и Крамом), после чего широкое распространение получила надмолекулярная теория строения ГВ. Стоит отметить, что надмолекулярные теории организации природного ОВ обсуждались и до этого. Так, еще в 1972 г. [144] была приведена модель ГВ как ансамбля (рыхлой сетки) кислот и фенолов, которые захватывают другие соединения. О ГВ как полидисперсной смеси веществ и их надмолекулярных ассоциатов в 1975 г. писал Орлов с соавт. [22, 122]. В 1990-х гг. появилась мицеллярная теория строения ГВ по Вершау [176], развитая затем Пикколо [130]. Именно начиная с работ Пикколо надмолекулярная теория прочно закрепилась как в почвенной науке, так и в химии природных вод. Согласно Пикколо, ГК рассматриваются как надмолекулярные ассоциаты небольших молекул (до 2 кДа), стабилизированные слабыми дисперсионными взаимодействиями (силы Ван-дер-Ваальсовая, π – π, CH-π) и водородными связями. Вывод о надмолекулярной природе ГК сделан на основе изучения молекулярно-массовых распределений ГК методом гель-хроматографии в присутствии органических кислот, вызывающих дезагрегацию молекул [131]. Условия эксперимента не учитывали электростатических и других неэксклюзионных эффектов [126], а заключения о столь низкой молекулярной массе были сделаны на основании данных масс-спектрометрии, в которых не определялись соединения с массами более 2 кДа. Тем не менее, модель получила развитие [146, 159] и в настоящее время практически общепринята.
Наконец, появление универсальной теории водородных связей Джилли и Джилли [67], сила которых варьирует от слабых (<1 ккал/моль) до близких к ковалентным (30–45 ккал/моль) привело Уэллс к созданию в 2015–2019 гг. новой модели самоорганизации природного ОВ [61, 175]. В этой модели учитывается роль не только слабых (по Пикколо), но и сильных (близких к ковалентным) водородных связей в агрегации природного ОВ [174, 175]. Согласно модели [175], устойчивые к возмущениям (турбулентность, изменение рН и др.) первичные ассоциации ОВ размером <1 мкм, для которых введен термин “метахимический гидрогель”, образуются путем кросс-сшивки отдельных молекул посредством сильных водородных связей. Это так называемые “заряд-ассистированные” связи, образующиеся при распределении протона между двумя диссоциированными ароматическими карбоксильными или фенольными группами, “резонанс-ассистированные” связи между незаряженными группами, а также водородные связи между недиссоциированными фенольными группами (поляризационно стабилизированные). Метахимический гель не разрушается ни при щелочной экстракции, ни при десорбции ОВ щелочью с DAX-смол. Ассоциаты <1 мкм сохраняются в растворах ГВ и природном ОВ даже при pH 13 [99]. При благоприятных условиях частицы метахимического гидрогеля за счет более слабых водородных связей и дисперсионных взаимодействий объединяются в рыхлые объемные структуры размером >1 мкм, обозначенные термином “физический гидрогель”. Он обратимо распадается под действием возмущений и восстанавливается в течение периодов покоя. Эти два типа ассоциаций выделяют как базовые формы агрегатов в иерархической самоорганизующейся супрамолекулярной архитектуре природного ОВ. Образование метахимического гидрогеля – первичных плотных супрамолекул внутри больших рыхлых супрамолекул, по мнению авторов, и есть механизм, ответственный за присущую природному ОВ биохимическую устойчивость.
Следует отметить, что в супрамолекулярных теориях на второй план отодвинуты модели надмолекулярных структур, образующихся из органических молекул-лигандов и ионов металлов в качестве мостиков, существование которых не вызывает сомнений [158]. Кроме того, эти модели разработаны в основном для водных растворов. Поэтому перенесение такого подхода на нерастворенное ОВ является актуальной задачей. Начало этому подходу положено работами Вершау [176], где ОВ почв представляется в виде мембран на минеральной поверхности, зональной моделью самоорганизации ОВ на минеральных поверхностях [98], а также работами о гумусовой матрице почвенных гелей [33, 34].
ЩЕЛОЧНАЯ ЭКСТРАКЦИЯ
Одним из главных пунктов для критики гуминовой концепции [3, 97, 108] является способ выделения ГВ с помощью щелочной экстракции (0.1–0.5 М NaOН), которая применяется для извлечения ГВ из почв, торфов и бурых углей, а также для десорбции фракций ГК и ФК с XAD8/DAX8 смол при изучении ОВ природных вод [121]. Основные дискуссионные аспекты, это: 1) селективность экстракции по отношению к продуктам гумификации, так как в раствор могут переходить компоненты свежих микробных и растительных остатков, компоненты биомассы и метаболиты, то есть вещества известного строения, 2) химические изменения нативного ОВ, приводящие к возникновению артефактов, 3) релевантность щелочной экстракции для изучения свойств и функций ОВ природных объектов [3, 108, 121, 122, 144].
Селективность экстракции. Количественный вклад веществ известного строения в состав щелочных экстрактов точно неизвестен. Он определяется степенью окислительной трансформации отмерших остатков и считается незначительным (10–15% от Сорг [15]) в объектах с высокой степенью гумификации ОВ (рассчитываемой как Сгк/Собщ × 100%) [26]. Индивидуальные низкомолекулярные вещества частично отделяются на стадии осаждения фракции ГК или при очистке фракции ФК на ионообменных смолах [121]. Однако, полностью отделить биомолекулы от продуктов гумификации в экстрактах невозможно. Например, белковые компоненты микробных тканей, внеклеточные ферменты, связанные с ОВ почв и минералами, грибные меланины переходят в экстракты и соосаждаются с фракцией ГК. Трансформация растительных остатков в почвах сопровождается окислительными реакциями, приводящими к карбоксилированию и гидроксилированию биополимеров, таких, как лигнин, что приводит к увеличению растворимости исходных биомолекул в водных растворителях. Поэтому щелочная экстракция является в определенном смысле селективной по отношению к полярным продуктам трансформации растительных остатков. Интактная лигноцеллюлоза слабо растворима в щелочах, как и липиды, элементы кутикул и другие гидрофобные вещества, остающиеся во фракции “гумина” [80]. Как уже обосновано выше, не стоит трактовать щелочную экстракцию как способ селективного извлечения особой группы специфических веществ (ГВ). Следует относится к этой процедуре как к способу растворения количественно значимой (30–80% от Сорг почв [158]) части ОВ, включающей интактные и измененные (гумифицированные) его компоненты, как способу, позволяющему изучить компонентный состав этой части, молекулярно-массовые свойства веществ [121], в том числе методами масс-спектрометрии ионного циклотронного резонанса c преобразованием Фурье (FT ICR-MS). Осаждение фракции ГК кислотой – это эффективный способ концентрирования растворимого в щелочных условиях ОВ.
Артефакты экстракции. Этот вопрос обсуждается с самого начала изучения веществ щелочных экстрактов и изложен в ряде недавних обзоров [81, 97, 121, 140]. Наиболее радикальные критики считают ГК и ФК лабораторным артефактом [3, 108].
Экстракция компонентов гумуса в щелочной среде сопровождается потреблением кислорода. Величины могут составлять 700–800 мм3 O2/0.2 г почвы в 0.5 M NaOH против 7–37 мм3 O2/0.2 г в 0.1 M Na4P2O7 (рН 7.0) (Бремнер, 1950 цит. по [158]). В присутствии кислорода в щелочной среде может происходить автоокисление фенольных соединений [168], являющихся ключевыми ароматическими компонентами ГК и ФК наземного происхождения. Окислению способствуют активные формы кислорода и ионизированное состояние ОН-групп [54]. В результате возможны реакции как конденсации, так и деградации. Например, продувка кислородом в течение 30 дней ГК торфа, растворенной в 1 М NaOH, привела к конвертации почти половины ГК в ФК и другие низкомолекулярные соединения, а также к потере азота аминокислот в ГК (Свифт и Познер, 1972, цит. по [121]). При продувке щелочных растворов галловой и протокатеховой кислот кислородом показана окислительная полимеризация этих полифенолов с образованием темноокрашенных поликонденсатов, сходных с ГК по pKa карбоксильных и фенольных групп, и спектрам в УФ-, видимой и ИК-областях [59, 66 ]. Возможным изменением ОВ при щелочной экстракции может быть гидролиз сложноэфирных групп, приводящий к увеличению количества карбоксильных групп [121].
Для того, чтобы минимизировать химические изменения ОВ в щелочной среде, обществом IHSS рекомендовано выделение ГК и ФК в атмосфере инертного газа, например азота [160]. Было показано (Свифт и Познер, 1972 цит. по [121]), что при экстракции 1 М NaOH в течение 30 дней в атмосфере N2 конверсия ГК в более низкомолекулярные соединения составляет менее 15%. Условия выделения ГК и ФК из почв, рекомендованные IHSS – 0.1 М NaOH и 24 ч экстракции в атмосфере N2, предполагают еще меньшую трансформацию ОВ. На примере смесей фенольных кислот и их анализа до и после щелочной обработки методом обратнофазовой жидкостной хроматографии, нами показано, что трансформации полифенолов (галловой и кофейной кислот) не происходит в течение 1 ч постоянной продувки. Без продувки азотом галловая и кофейная кислоты “исчезают” c хроматограмм практически сразу после добавления щелочи (рис. 3). Постоянную продувку и отсутствие доступа кислорода при выделении препаратов ГК в больших объемах обеспечить крайне трудно. В практике отечественных исследований гумуса использование N2 не принято [26]. Однако сравнение физико-химических свойств ГК, выделенных 0.1 M NaOH с продувкой азотом и без его использования из гумусовых горизонтов двух контрастных по условиям гумусообразования типов почв: дерново-подзолистой и чернозема, показало отсутствие значимых деградационных и конденсационных процессов при экстракции ГК без N2. В почве с глубокой переработкой ОВ, сопровождающей процессы гумификации (черноземе), присутствие O2 и щелочная среда не вызывали окисления компонентов ОВ. В дерново-подзолистой почве продувка азотом снижала окислительные процессы при экстракции [7].
Рис. 3.
Влияние рН на состав смеси фенольных кислот по данным обратнофазовой жидкостной хроматографии: А – исходная смесь кислот в 20 мМ ацетатном буфере (рН 4.5), Б – смесь кислот после 1 ч выдерживания в щелочных условиях (NaOH, рН 12) при постоянной продувке азотом, В – то же, без продувки азотом: 1 – галловая, 2 – протокатеховая, 3 – гидроксибензойная, 4 – ванилиновая, 5 – кофейная, 6 – сиреневая, 7 – ванилиновая, 8 – феруловая кислоты. Колонка SunergiHydro-RP (150 × 4.6 мм, 4 мкм, Phenomenex, США). Кислоты определяли как описано ранее [5].
Еще одним предметом дискуссии является темная окраска растворов ГВ. Высказаны предположения, что это результат щелочной обработки [108]. Однако деструкция свежих растительных остатков сопровождается их потемнением со временем (например, в компостах, [93]), гумусовые горизонты почв имеют природную темную окраску [38], и темноокрашенные вещества выделяются из почв не только щелочами [55, 64, 79, 121]. Установлено, что применение разбавленной щелочи не приводит к существенному изменению компонентного состава экстрагируемого природного ОВ. С помощью пиролитической масс-спектрометрии (Pi-FIMS) в почве были идентифицированы те же 10 классов соединений, что и во фракциях ГК, ФК и гумин, причем относительная распространенность 8 из 10 классов была идентична для всех 3 фракций ГВ и почвы в целом [143]. Масс-спектрометрией ионно-циклотронного резонанса (MS ICR) установлено, что десорбция ОВ щелочью с XAD-8 смол не оказывала существенного влияния на молекулярный состав растворенного ОВ (РОВ) природных вод – из около 13 000 молекулярных брутто-формул, найденных в спектрах исходного РОВ почти 90% присутствуют также в трех его фракциях (ГК, ФК и гидрофильное РОВ), что является очень высоким коэффициентом подобия [121]. Следовательно, фракции ГК, ФК, гумин удовлетворительно аппроксимируют основные тенденции в структурно-композиционном составе природного ОВ.
В качестве аргументов против “искусственного” действия щелочи на природное ОВ, де Нобили с соавт. [55] отмечают, что свежие растительные остатки и вещества гумуса могут испытывать щелочное воздействие (рН 8–10) и до экстракции в результате деятельности педофауны – при прохождении через пищеварительный тракт беспозвоночных (см. ссылки в [55]). В гумусовых горизонтах почв с типом гумуса модер или амфимулль такие зоогенно трансформированные материалы могут составлять от 70 до 100% от объема почвенной массы [180]. Таким образом, даже в кислых почвах природное ОВ испытывает локальное воздействие щелочных условий. Несмотря на модификацию нативного ОВ, фракции ГК и ФК, по мнению [121] состоят из реально существующих природных веществ и могут быть использованы для изучения компонентного состава и функций ОВ почв и природных вод. Однако структура нативного коллоидного гумуса имеет мало общего с состоянием веществ в щелочных экстрактах.
Применение щелочной экстракции. Экстракция щелочью (0.1–0.5 М NaOH) наиболее эффективна в количественном плане, позволяет выделить до 80% от Сорг гумуса по сравнению с около 30% от Сорг при использовании растворов нейтральных солей – Na4P2O7, NaF [158], поэтому разбавленным растворам щелочей отдается предпочтение перед другими экстрагентами. Тем не менее, щелочная экстракция приводит к селективному выделению веществ, обогащенных полярными группами и обладающих повышенной реакционной способностью относительно ОВ в целом [97, 108]. Поэтому использование щелочной экстракции требует обоснованного выделения областей ее применения [121]. Щелочная экстракция является общепринятым методом получения гуминовых препаратов [139]. Прикладное значение таких препаратов для сельского хозяйства, экологии и медицины не вызывает сомнений даже у противников гуминовой концепции [96]. Содержание фракции ГК служит индикатором зрелости компостов [53], степени гумификации ОВ почв [21, 23]. Величина Cгк/Cфк может служить мерой гидролизуемости ОВ почв, если не придавать фульвокислотам значения реально и самостоятельно существующей группы почвенного гумуса [23]. Щелочная экстракция позволяет определить молекулярные массы и брутто-формулы десятков тысяч индивидуальных компонентов ОВ методом FT-ICR MS, а также приблизительно оценить их относительную распространенность [89, 130]. Ни почва в целом, ни денсиметрические фракции не подходят для таких исследований. Эксперименты с ГК и ФК позволили сделать хорошие прогнозные модели по миграции и распределению ионов металлов и пестицидов в почве [73, 106, 165], что свидетельствует в пользу ГК и ФК как представительных фракций ОВ почв при изучении и моделировании таких процессов [121]. С другой стороны, возникает вопрос о возможности оценки реакционной способности ОВ в почвах на основании реакционной способности веществ щелочных изолятов. В почве часть функциональных групп ОВ “занята” образованием связей с минеральной матрицей, что хорошо отражено в модели [98]. Например, связывание фенольных кислот минералами происходит в первую очередь за счет карбоксильных групп, которые являются основными реакционными центрами в диапазоне рН 4–7, характерном для большинства почв [5, 6]. Следовательно, величины комплексообразующей способности по отношению к ионам металлов, полученные для растворов гумусовых кислот, завышены по сравнению с таковыми для связанного с минеральной матрицей состояния этих веществ.
В заключение следует сказать, что разработано множество альтернативных подходов к процедуре выделения ОВ из твердых объектов, позволяющих уменьшить гетерогенность и расширить круг доступных структурному анализу компонентов. В числе таких подходов использование экстрагентов на водной основе: буферных и комплексообразующих растворов [158], полярных и неполярных органических растворителей, предшествующих щелочной экстракции (“гумеомика” Пикколо [117]), и, наконец, системы растворителей, включая ДМСО, позволяющей выделить компоненты “гумина” [78, 80, 153, 154]. В результате последовательных экстракций выделяют спектр различающихся по гидрофильности фракций ОВ, в сумме составляющих бóльшую долю от Сорг почвы, чем классический щелочной изолят, и более полно отражающих многообразие свойств различных компонентов ОВ почв. В русле снижения гетерогенности ОВ находятся и работы, в которых сочетаются методы денсиметрического фракционирования и экстракции [52]. Развитие в последнее время спектральных методов анализа (Pi-FIMS, nano-SIMS, твердофазной 13С-ЯМР) является перспективным подходом к изучению природного ОВ in situ [140], но не может полностью заменить экстракционные методы. Экстракция и исследования in situ должны быть взаимодополняющими.
ЗАКЛЮЧЕНИЕ
В составе неживого ОВ гумус и гумифицированное ОВ четко отличаются морфологически, химически и функционально от детрита (слаборазложившихся органических остатков). По своему составу гумус представляет собой совокупность исходных биомолекул растительных и животных тканей, продуктов их деградации и трансформации, и продуктов внеклеточного синтеза. Все эти вещества составляют единый динамический ансамбль коллоидной природы, стабилизированный ковалентными, ионными, и межмолекулярными взаимодействиями. Поэтому следует признать условность принятого в гуминовой номенклатуре разделения этого ансамбля на вещества плохо определенной структуры (ГВ) и вещества известного строения (не-ГВ). Такое разделение вызывает противоречия и с операционным подходом к выделению ГВ. Возникает проблема индивидуальных соединений, входящих в состав операционных фракций, нет консенсуса, что считать ГВ – только полярные карбоксилированные вещества щелочных экстрактов (ГК и ФК) или же и гумин, содержащий значительную долю идентифицируемых неполярных соединений (липидов, кутина, и др). Трактовка ГВ как темноокрашенных веществ, образующихся в результате реакций биодеградации и свободнорадикальной конденсации и отличных по структуре от биополимеров растительных и микробных тканей, на практике создает проблемы с идентификацией этих веществ в сформировавшемся гумусе. Возможность протекания свободнорадикальных реакций конденсации и сополимеризации в почвах не вызывает сомнений, однако их количественный вклад в формирование состава природного ОВ требует дальнейшего изучения.
Традиционную щелочную экстракцию следует рассматривать как способ растворения полярных веществ гумуса, а осаждение фракции ГК кислотой – как способ концентрирования и грубой очистки ряда компонентов для их дальнейшего изучения. Это также общепринятый способ получения препаратов, имеющих большое прикладное значение. Термины ГК, ФК, гумин следует закрепить за операционными фракциями (препаратами), не придавая этим понятиям значения специфических и химически уникальных веществ. Соотношение Сгк/Сорг является простым и удобным показателем типов гумуса, отражающим биоклиматические условия его образования. Выяснение механизмов формирования, структурной организации, функций веществ, составляющих гумифицированное ОВ природных объектов, требует дальнейших исследований.
Список литературы
Александрова Л.Н. Органическое вещество почвы и процессы его трансформации. Л.: Наука, 1980. 288 с.
Богдан В.И., Сергеева Я.Э., Лунин В.В., Перминова И.В., Константинов А.И., Зинченко Г.Е., Богдан К.В. Биоконверсия фенольных мономеров лигнина и лигнинсодержащих субстратов базидиальным грибом Lentinus tigrinus // Прикл. Биохим. Микробиол. 2018. 54(2). С. 186–194.
Ваксман С.А. Гумус. Происхождение, химический состав и значение его в природе. М.: Огиз-Сельхозгиз, 1937. 471 с.
Дергачева М.И. Система гумусовых веществ почв (пространственные и временные аспекты). М.; Новосибирск: Наука, Сиб. отд-ние, 1989. 113 с.
Заварзина А.Г., Ермолин М.С., Демин В.В., Федотов П.С. Взаимодействие смеси фенольных кислот с модифицированным каолинитом в статических и динамических условиях // Почвоведение. 2018. № 8. С. 1004–1013. https://doi.org/10.1134/S0032180X18080129
Заварзина А.Г., Ермолин М.С., Демин В.В., Федотов П.С. Влияние уксусной кислоты и ацетат-ионов на сорбцию–десорбцию смеси фенольных кислот модифицированным каолинитом // Почвоведение. 2020. № 8. С. 948–958. https://doi.org/10.31857/S0032180X20080171
Заварзина А.Г., Кравченко Е.Г., Константинов А.И., Перминова И.В., Чуков С.Н., Демин В.В. Сравнение свойств препаратов гуминовых кислот, выделенных из почв щелочной экстракцией в отсутствие и присутствие кислорода // Почвоведение. 2019. № 8. С. 910–922.
Заварзина А.Г, Семенова Т.А., Белова О.В., Лисов А.В., Леонтьевский А.А., Иванова А.Е. Продукция лакказы и деструкция гуминовых кислот почвенными микроскопическими грибами // Микробиология. 2018. Т. 87. № 3. С. 233–241.
Загоскина Н.В., Николаева Т.Н., Лапшин П.В., Заварзин А.А., Заварзина А.Г. Водорастворимые фенольные соединения у лишайников // Микробиология. 2013. Т. 82(3). С. 1–8.
Запрометов М.Н. Фенольные соединения: Распространение, метаболизм и функции в растениях. М.: Наука, 1993. 271 с.
Звягинцев Д.Г., Мирчинк Т.Г. О природе гуминовых кислот почв // Почвоведение. 1986. № 5. С. 68–75.
Иванов А.Л., Когут Б.М., Семенов В.М., Тюрина Оберландер М., Ваксман Шанбахер Н. Развитие учения о гумусе и почвенном органическом веществе: от Тюрина и Ваксмана до наших дней // Бюл. Почв. ин-та им. В.В. Докучаева. 2017. Вып. 90. С. 3–38.
Ковалева Н.О., Ковалев И.В. Биотрансформация лигнина в дневных и погребенных почвах разных экосистем // Почвоведение. 2009. № 11. С. 84–96.
Когут Б.М., Семенов В.М., Артемьева З.С., Данченко Н.Н. Дегумусирование и почвенная секвестрация углерода // Агрохимия. 2021. № 5. С. 3–13.
Кононова М.М. Органическое вещество почвы. М.: Изд-во АН СССР, 1963. 315 с.
Кононова М.М. Проблема почвенного гумуса и современные задачи его изучения. М.: Изд-во АН СССР, 1951. 390 с.
Красильников П.В. Устойчивые соединения углерода в почвах: происхождение и функции // Почвоведение. 2015. № 9. С. 1131–1144.
Лен Ж.-М. Супрамолекулярная химия: концепции и перспективы. Новосибирск: Наука, 1998. 334 с.
Лисов А.В., Заварзина А.Г., Белова О.В., Леонтьевский А.А. Трансформация гуминовой кислоты грибом cerrena unicolor в условиях роста на целлюлозе и глюкозе // Микробиология. 2020. № 3. С. 300–307.
Орлов Д.С. Гуминовые вещества в биосфере // Соросовский образовательный журнал. 1997. № 2. С. 56–63.
Орлов Д.С. Гумусовые кислоты почв и общая теория гумификации. М.: Изд-во Моск. ун-та, 1990. 325 с.
Орлов Д.С. Гумусовые кислоты почв. М.: Изд-во Моск. ун-та, 1974. 334 с.
Орлов Д.С. Почвенные фульвокислоты: история их изучения, значение и реальность // Почвоведение. 1999. № 9. С. 1165–1171.
Орлов Д.С. Химия почв. М.: Изд-во Моск. ун-та, 1992. 401 с.
Орлов Д.С., Бирюкова О.Н., Суханова Н.И. Органическое вещество почв Российской Федерации. М.: Наука, 1996. 258 с.
Орлов Д.С., Гришина Л.А. Практикум по химии гумуса. М.: Изд-во Моск. ун-та, 1981. 273 с.
Перминова И.В. Гуминовые вещества – вызов химикам XXI века // Химия и жизнь – XXI век (до 1997 г. Химия и жизнь). 2008. № 1. С. 50–55.
Рабинович М.Л., Болобова А.В., Кондращенко В.И. Теоретические основы биотехнологии древесных композитов. Кн. 1. Древесина и разрушающие ее грибы. М.: Наука, 2001. 264 с.
Семенов В.М., Когут Б.М. Почвенное органическое вещество. М.: ГЕОС, 2015. 233 с.
Семенов В.М., Тулина А.С., Семенова Н.А., Иванникова Л.А. Гумификационные и негумификационные пути стабилизации органического вещества в почве (обзор) // Почвоведение. 2013. № 4. С. 393–407.
Тюрин И.В. Органическое вещество почв и его роль в почвообразовании и плодородии. Учение о почвенном гумусе. М.–Л.: Сельхозгиз, 1937. 287 с.
Тюрин И.В. Органическое вещество почвы и его роль в плодородии. М.: Наука, 1965. 322 с.
Федотов Г.Н., Шоба С.А. Влияние структурного перехода в гумусовой матрице почвенных гелей на некоторые свойства почв // Докл. РАН. 2014. Т. 457. № 1. С. 57–60.
Федотов Г.Н., Шоба С.А. Структурный переход в гумусовой матрице почвенных гелей и электросопротивление почв // Почвоведение. 2015. № 11. С. 1346–1353.
Штаудингер Г. Высокомолекулярные органические соединения. Каучук и целлюлоза. М., 1935. 548 с.
Aiken G.R. Isolation and concentration techniques for aquatic humic substances // Humic substances in soil, sediment, and water: Geochemistry and isolation / Eds. G.R. Aiken et al. N.Y.: John Wiley & Sons, 1985. P. 363–385.
Alexandrova L.N., Arshavskay T.Th., Dorfman F.M., Lyuzin M.F., Yurlova O.V. Humus acids and their organo-mineral derivatives in soil // Int. Soil Sci. Congr. Trans. 1968. V. 3(9). P. 143–152.
Allison S.D. Brown ground: a soil carbon analogue for the green world hypothesis? // The American Naturalist. 2006. V. 167(5). P. 619–27. https://doi.org/10.1086/503443
Amelung W., Brodowski S., Sandhage-Hofmann A., Bol R. Combining biomarker with stable isotope analysis for assessing the transformation and turnover of soil organic matter // Adv. Agron. 2008. V. 100. P. 155–250. https://doi.org/10.1016/S0065-2113(08)00606-8
Angst G., Mueller K.E., Nierop K.G.J., Simpson M.J. Plant- or microbial-derived? A review on the molecular composition of stabilized soil organic matter // Soil Biol. Biochem. 2021. V. 156. 108189. https://doi.org/10.1016/j.soilbio.2021.108189
Arantes V., Goodell B. Current Understanding of Brown-Rot Fungal Biodegradation Mechanisms: A Review // Deterioration and Protection of Sustainable Biomaterials. Oxford University Press, 2014. ACS Symp. Ser. 1158. Peer Reviewed Book Chapter. P. 3–22. https://doi.org/10.1021/bk-2014-1158.ch001
Baigorri R., Fuentes M., Gonzfilez-Gaitano G., Garcia-Mina J.M. Simultaneous presence of diverse molecular patterns in humic substances in solution // J. Phys. Chem B. 2007. V. 111. P. 10577–10582.
Baldock J.A., Broos K. Soil organic matter // Handbook of soil sciences: Properties and processes.: Boca Raton: CRC Press/Taylor Francis Group, 2011. P. 11.11–11.52.
Batjes N.H. Total carbon and nitrogen in the soils of the world // Eur. J. Soil Sci. 2014. V. 65. P. 4-21. https://doi.org/10.1111/ejss.12114_2
Baveye Ph.C., Wander M. The (bio)chemistry of soil humus and humic substances: why is the “New view” still considered novel after more than 80 years? // Front. Environ. Sci. 2019. V. 7. P. 27. https://doi.org/10.3389/fenvs.2019.00027
Beckett R.P., Zavarzina A.G., Liers C. Oxidoreductases and cellulases in lichens: Possible roles in lichen biology and soil organic matter turnover // Fungal Biol. 2013. V. 117. P. 431–438. https://doi.org/10.1016/j.funbio.2013.04.007
Bollag J.M., Liu S.Y., Minard R.D. Enzymatic oligomerization of vanillic acid // Soil Biol. Biochem. 1982. V. 14. P. 157–163.
Brady N.C., Weil R.R. The nature and properties of soils. New Jersey: Prentice Hall, Upper Saddle River, 2008.
Burdon J. Are the traditional concepts of the structures of humic substances realistic? // Soil Sci. 2001. V. 166. P. 752–769.
Burns R.G., DeForest J.I., Marxsen J., Sinsabaugh R.I., Stromberger M.E., Wallenstein M.D., Weintraub M.N., Zoppini A. Soil enzymes in a changing environment: current knowledge and future directions // Soil Biol. Biochem. 2013. V. 58. P. 216–234.
Cao X., Schmidt-Rohr K. Abundant nonprotonated aromatic and oxygen-bonded carbons make humic substances distinct from biopolymers // Environ. Sci. Technol. Lett. 2018. V. 5. P. 476−480. https://doi.org/10.1021/acs.estlett.8b00107
Cao X., Olk D.C., Chappell M., Cambardella C.A., Miller L.F., Mao J. Solid-state NMR analysis of soil organic matter fractions from integrated physical-chemical extraction // Soil Sci. Soc. Am. J. 2011. V. 75. P. 1374–1384. https://doi.org/10.2136/sssaj2010.0382
Chefetz B., Hatcher P.G., Hadar Y., Chen Y. Chemical and Biological Characterization of Organic Matter during Composting of Municipal Solid Waste // J. Environ. Qual. 1996. V. 25(4). P. 776–785. https://doi.org/10.2134/jeq1996.00472425002500040018x
Chiorcea-Paquim A.-M., Enache T.A., De Souza Gil E., Oliveira-Brett A.M. Natural phenolic antioxidants electrochemistry: Towards a new food science methodology // Compr. Rev. Food Sci. Food Saf. 2020. V. 19. P. 1680–1726. https://doi.org/10.1111/1541-4337.12566
De Nobili M., Bravo C., Chen Y. The spontaneous secondary synthesis of soil organic matter components: A critical examination of the soil continuum model theory // Appl. Soil Ecol. 2020. V. 154. P. 103655. https://doi.org/10.1016/j.apsoil.2020.103655
DiDonato N., Chen H.M., Waggoner D., Hatcher P.G. Potential origin and formation for molecular components of humic acids in soils // Geochim. Cosmochim. Acta. 2016. V. 178. P. 210–222. https://doi.org/10.1016/j.gca.2016.01.013
Doerr S.H., Santín C., Merino A., Belcher C.M., Baxter G. Fire as a removal mechanism of pyrogenic carbon from the environment: effects of fire and pyrogenic carbon characteristics // Front. Earth Sci. 2018. V. 6. 127. https://doi.org/10.3389/feart.2018.00127
Dou S., Shan J., Song X., Cao R., Wu M., Li Ch., Guan S. Are humic substances soil microbial residues or unique synthesized compounds? A perspective on their distinctiveness // Pedosphere. 2020. V. 30(2). P. 159–167. https://doi.org/10.1016/S1002-0160(20)60001-7
Drosos M., Jerzykiewiczc M., Louloudi M., Deligiannakis Y. Progress towards synthetic modelling of humic acid: Peering into the physicochemical polymerization mechanism // Colloids Surf., A. 2011. V. 389. P. 254–265. https://doi.org/10.1016/j.colsurfa.2011.08.016
Ertel J.R., Hedges J.I. The lignin component of humic substances: Distribution among soil and sedimentary humic, fulvic, and base-insoluble fractions // Geochim. Cosmochim. Acta. 1984. V. 48(10). P. 2065–2074. https://doi.org/10.1016/0016-7037(84)90387-9
Esfahani M.R., Stretz H.A., Wells M.J.M. Abiotic reversible self-assembly of fulvic and humic acid aggregates in low electrolytic conductivity solutions by dynamic light scattering and zeta potential investigation // Sci. Total Environ. 2015. V. 537. P. 81–92.
Flaig W., Beutelspacher H., Rietz E. Chemical composition and physical properties of humic substances // Soil Components. V. 1. N.Y.: Springer-Verlag, 1975. P. 1–211.
Fujitake N., Asakawa D., Yanagi Y. Characterization of soil humic acids by 13C NMR spectroscopy and high performance size exclusion chromatography (HPSEC). Bunseki Kagaku, 2012. V. 61. P. 287–298. (Japanese with English summary)
Gerke J. Concepts and misconceptions of humic substances as the stable part of soil organic matter: A review // Agronomy. 2018. V. 8(5). P. 76. https://doi.org/10.3390/agronomy8050076
Gessler N.N., Egorova A.S., Belozerskaya T.A. Melanin Pigments of Fungi under Extreme Environmental Conditions (Review) // Appl. Biochem. Microbiol. 2014. V. 50(2). P. 105–113.
Giannakopoulos E., Drosos M., Deligiannakis Y. A humic-acid-like polycondensate produced with no use of catalyst // J. Colloid Interface Sci. 2009. V. 336. P. 59–66. https://doi.org/10.1016/j.jcis.2009.03.037
Gilli G., Gilli P. The nature of the hydrogen bond: Outline of a comprehensive hydrogen bond theory. Oxford: Oxford Univ. Press, 2013.
Glaser B., Haumeier L., Guggenberger G., Zech W. The Terra preta phenomenon: A model for sustainable agriculture in the humid tropics // Naturwissenschaften. 2001. V. 88. P. 37–41.
Gomez-Toribio V., Garcia-Martin A.B., Martinez M.J., Martinez A.T., Guillen F. Enhancing the production of hydroxyl radicals by Pleurotus eryngii via quinone redox cycling for pollutant removal // Appl. Environ. Microbiol. 2009. V. 75. V. 3954–3962.
Grinhut T., Hadar Y., Chen Y. Degradation and transformation of humic substances by saprotrophic fungi: processes and mechanisms // Fungal Biol. Rev. 2007. V. 21(4). P. 179–189. https://doi.org/10.1016/j.fbr.2007.09.003
Grinhut T., Salame T.M., Chen Y., Hadar Y. Involvement of ligninolytic enzymes and Fenton-like reaction in humic acid degradation by Trametes sp. // Appl. Microbiol. Biotechnol. 2011. V. 91. P. 1131–1140.
Guggenberger G., Zech W. Dissolved organic carbon in forest floor leachates: simple degradation products or humic substances? // Sci. Total Environ. 1994. V. 152. P. 37–47. https://doi.org/10.1016/0048-9697(94)90549-5
Gustafsson J.P. Modeling the acid-base properties and metal complexation of humic substances with the Stockholm humic model // J. Colloid Interface Sci. 2001. V. 244. P. 102–112. https://doi.org/10.1006/jcis.2001.7871
Haider K., Frederick L.R., Flaig W. Reactions between amino acid compounds and phenols during oxidation // Plant Soil. 1965. V. 22. P. 49–64.
Harvey G.R., Boran D.A., Chesal L.A., Tokar J.M. The structure of marine fulvic and humic acids // Marine Chem. 1983. V. 12. P. 119–132.
Harvey G.R., Boran D.A., Piotrowicz S.R., Weisel C.P. Synthesis of marine humic substances from unsaturated lipids // Nature. 1984. V. 309. P. 244–246.
Hatcher P.G., Schnitzer M., Dennis L.W., Maciel G.E. Aromaticity of humic substances in soils // Soil Sci. Soc. Am. J. 1981. V. 45. P. 1089–1094.
Hayes M.H.B. Solvent systems for the isolation of organic components from soils // Soil Sci. Soc. Am. J. 2006. V. 70. P. 986–994.
Hayes M.H.B., Clapp C.E. Humic substances: considerations of compositions, aspects of structure, and environmental influences // Soil Sci. 2001. V. 166. P. 723–737.
Hayes M.H.B., Mylotte R., Swift R.S. Humin: its composition and importance in soil organic matter // Adv. Agron. 2017. V. 143. P. 47–138. https://doi.org/10.1016/bs.agron.2017.01.001
Hayes M.H.B., Swift R.S. An appreciation of the contribution of Frank Stevenson to the advancement of studies of soil organic matter and humic substances // J. Soils Sedim. 2018. V. 18(4). P. 1212–1231. https://doi.org/10.1007/s11368-016-1636-6
Hayes M.H.B., Swift R.S. The chemistry of soil organic colloids // The Chemistry of Soil Constituents. Chichester: Wiley, 1978. P. 179–320.
Hayes M.H.B., Swift R.S. Vindication of humic substances as a key component of organic matter in soil and water // Adv. Agronomy. 2020. V. 163. Ch. 1. https://doi.org/10.1016/bs.agron.2020.05.001
Hayes M.H.B., Swift R.S., Byrne C.M., Song G., Simpson A.J. Humin: The simplest of the humic substances? // Advances in natural organic matter and humic substances research. Proceedings Book of the Communications presented to the 15th Meeting of the IHSS. Tenerife – Canary Islands. June 27–July 2, 2010. V. 1. P. 64–68.
Hedges J.I., Eglinton G., Hatcher P.G., Kirchman D.L., Arnosti C., Derenne S., Evershed R.P. et al. The molecularly-uncharacterized component of nonliving organic matter in natural environments // Org. Geochem. 2000. V. 31. P. 945–958.
Hedges J.I., Mann D.C. The characterization of plant tissues by their lignin oxidation products // Geochim. Cosmochim. Acta. 1979. V. 43. P. 1803–1807.
Hofrichter M. Review: Lignin conversion by manganese peroxidase (MnP) // Enzyme Microb. Technol. 2002. V. 30. P. 454–466. https://doi.org/10.1016/S0141-0229(01)00528-2
Huang P.M., Hardie A.G. Formation Mechanisms of Humic Substances in the Environment // Biophysico-Chemical Processes Involving Natural Nonliving Organic Matter in Environmental Systems. Ch. 2. Hoboken: John Wiley & Sons, 2009. P. 84–98. https://doi.org/10.1002/9780470494950.ch2
Ikeya K., Sleighter R.L., Hatcher P.G., Watanabe A. Characterization of the chemical composition of soil humic acids using Fourier transform ion cyclotron resonance mass spectrometry // Geochim. Cosmochim. Acta. 2015. V. 153. P. 169–182. https://doi.org/10.1016/j.gca.2015.01.002
International Humic Substances Society (IHSS). 2007. What are humic substances? Available online at http://humic-substances.org/what-arehumic-substances-2/
Jenkinson D.S. The fate of plant and animal residues in soil // The chemistry of soil processes. Wiley, Chichester, 1981. P. 505–561.
Kelleher B.P., Simpson A.J. Humic substances in soils: are they really chemically distinct? // Environ. Sci. Technol. 2006. V. 40. P. 4605–4611.
Khan K.S., Mack R., Castillo X., Rainer M.K., Joergensen G. Microbial biomass, fungal and bacterial residues, and their relationships to the soil organic matter C/N/P/S ratios // Geoderma. 2016. V. 271. P. 115-123. https://doi.org/10.1016/j.geoderma.2016.02.019
Kim S., Kramer R.W., Hatcher P.G. Graphical method for analysis of ultrahigh-resolution broadband mass spectra of natural organic matter, the van Krevelen diagram // Anal. Chem. 2003. V. 75. № 20. P. 5336–5344.
Kirk T.K. Degradation and conversion of lignocelluloses // The filamentous fungi. V. 4. Fungal technology. London: Edward Arnold, 1983. P. 266–295.
Kleber M., Johnson M.G. Advances in understanding the molecular structure of soil organic matter: Implications for interactions in the environment // Adv. Agron. 2010. V. 106. P. 77–142. https://doi.org/10.1016/S0065-2113(10)06003-7
Kleber M., Lehmann J. Humic substances extracted by alkali are invalid proxies for the dynamics and functions of organic matter in terrestrial and aquatic ecosystems // J. Environ. Qual. 2019. V. 48. P. 207–216. https://doi.org/10.2134/jeq2019.01.0036
Kleber M., Sollins S., Sutton R.A. A conceptual model of organo-mineral interactions in soils: self-assembly of organic molecular fragments into multilayered structures on mineral surfaces // Biogeochem. 2007. V. 85. P. 9–24.
Klucakova M., Veznikova K. Micro-organization of humic acids in aqueous solutions // J. Mol. Struct. 2017. V. 1144. P. 33–40. https://doi.org/10.1016/j.molstruc.2017.05.012
Knicker H. Solid state CPMAS 13C and 15N NMR spectroscopy in organic geochemistry and how spin dynamics can either aggravate or improve spectra interpretation // Org. Geochem. 2011. V. 42. P. 867–890.
Knicker H., Rosario-Ortiz F.L., Zaccone C. Preface—special issue in memory of Frank J. Stevenson // J. Soils Sedim. 2018. V. 18. P. 1209–1211. https://doi.org/10.1007/s11368-018-1955-x
Kögel-Knabner I. The macromolecular organic composition of plant and microbial residues as inputs to soil organic matter: Fourteen years ago // Soil Biol. Biochem. 2017. V. 105. P. A3–A8.
Kögel-Knabner I., Amelung W. Soil organic matter in major pedogenic soil groups // Geoderma. 2021. V. 384. P. 114785. https://doi.org/10.1016/j.geoderma.2020.114785
Kögel-Knabner I., Guggenberger G., Kleber M., Kandeler E., Kalbitz K., Scheu S., Eusterhues K., Leinweber P. Organo-mineral associations in temperate soils: Integrating biology, mineralogy, and organic matter chemistry // J. Plant Nutr. Soil Sci. 2008. V. 171. P. 61–82.
Kopittke P.M., Dalal R.C., Hoeschen C., Lia C., Menziesa N.W., Mueller C.W. Soil organic matter is stabilized by organo-mineral associations through two key processes: The role of the carbon to nitrogen ratio // Geoderma. 2020. V. 357. P. 113974. https://doi.org/10.1016/j.geoderma.2019.113974
Kulikova N., Perminova I. Binding of atrazine to humic substances from soil, peat, and coal related to their structure // Environ. Sci. Technol. 2002. V. 36. P. 3720–3724. https://doi.org/10.1021/es015778e
Lehmann J., Solomon D., Kinyang J., Dathe L., Wirick S., Jacobsen C. Spatial complexity of soil organic matter forms at nanometre scales // Nat. Geosci. 2008. V. 1. P. 238–242. https://doi.org/10.1038/ngeo155
Lehmann J., Kleber M. The contentious nature of soil organic matter // Nature. 2015. V. 528. P. 60–68.
Lisov A.V., Trubitsina L.I., Lisova Z.A., Trubitsin I.V., Zavarzina A.G., Leontievsky A.A. Transformation of humic acids by two-domain laccase from streptomyces anulatus // Process Biochem. 2018. V. 76. P. 128–135.
MacCarthy P. The Principles of Humic Substances // Soil Sci. 2001. V. 166(11). P. 738–751. https://doi.org/10.1097/00010694-200111000-00003
Mahieu N., Randall E.W., Powlson D.S. Statistical Analysis of Published Carbon-13 CPMAS NMR Spectra of Soil Organic Matter // Soil Sci. Soc. Amer. J. 1999. V. 63(2). P. 307–319. https://doi.org/10.2136/sssaj1999.03615995006300020008x
Mao J.-D., Cao X., Olk D.C., Chu W., Schmidt-Rohr K. Advanced solid-state NMR spectroscopy of natural organic matter // Prog. Nucl. Magn. Reson. Spectrosc. 2017. V. 100. P. 17–51.
Mao J.-D., Hu W.-G., Schmidt-Rohr K., Davies G., Ghabbour E.A., Xing B. Quantitative Characterization of Humic Substances by Solid-State Carbon-13 Nuclear Magnetic Resonance // Soil Sci. Soc. Am. J. 2000. V. 64. P. 873–884. https://doi.org/10.2136/sssaj2000.643873x
Merino C., Matus F., Kuzyakov Ya., Dyckmanse J., Stock S., Dippold M.A. Contribution of the Fenton reaction and ligninolytic enzymes to soil organic matter mineralisation under anoxic conditions // Sci. Total Environ. 2021. V. 760. P. 143397. https://doi.org/10.1016/j.scitotenv.2020.143397
Monreal C., Schnitzer M. The chemistry and biochemistry of organic components in the soil solutions of wheat rhizospheres // Adv. Agron. 2013. V. 121. P. 179–251. https://doi.org/10.1016/B978-0-12-407685-3.00004-9
Naidja A., Huang P.M., Dec J., Bollag J.M. Comparison of the reaction products from the transformation of catechol catalyzed by birnessite or tyrosinase // Soil Sci. Soc. Am. J. 1998. V. 62. P. 188–195.
Nebbioso A., Piccolo A. Advances in humeomics: Enhanced structural identification of humic molecules after size fractionation of a soil humic acid // Anal. Chim. Acta. 2012a. V. 720. P. 77–90. https://doi.org/10.1016/j.aca.2012.01.027
Nebbioso A., Piccolo A. Molecular characterization of dissolved organic matter (DOM): a critical review // Anal. Bioanal. Chem. 2012b. V. 405. P. 109–124. https://doi.org/10.1007/s00216-012-6363-2
Nelson P.N., Baldock J.A. Estimating the molecular composition of a diverse range of natural organic materials from solid-state 13C NMR and elemental analyses // Biogeochemistry. 2005. V. 72. P. 1–34.
Nosanchuk J.D., Stark R.E., Casadevall A. Fungal melanin: what do we know about structure? // Front. Microbiol. 2015. V. 6. P. 1463.
Olk D.C., Bloom P.R., Perdue E.M., McKnight D.M., Chen Y., Farenhorst A., Senesi N. et al. Environmental and agricultural relevance of humic fractions extracted by alkali from soils and natural waters // J. Environ. Qual. 2019. V. 48. P. 217–232. https://doi.org/10.2134/jeq2019.02.0041
Orlov D.S., Ammosova Y.M., Glebova G.I. Molecular parameters of humic acids // Geoderma. 1975. V. 13. P. 211–229. https://doi.org/10.1016/0016-7061(75)90019-1
Ortner A., Huber D., Haske-Cornelius O., Weber H.K., Hofer K., Bauer W., Nyanhongo G.S., Guebitz G.M. Laccase mediated oxidation of industrial lignins: is oxygen limiting? // Process Biochem. 2015. V. 50. P. 1277–1283.
Osono T. Ecology of ligninolytic fungi associated with leaf litter decomposition // Ecol. Res. 2007. V. 22. P. 955–974. https://doi.org/10.1007/s11284-007-0390-z
Perdue E.M., Ritchie J.D. Dissolved organic matter in freshwaters // Treatise Geochem. 2014. V. 7. P. 237–272.
Perminova I.V. Size-exclusion chromatography of humic substances: complexities of data interpretation attributable to non-size exclusion effects // Soil Sci. 1999. V. 164(11). P. 834–840.
Perminova I.V., Frimmel F.H., Kudryavtsev A.V., Kulikova N.A., Abbt-Braun G., Hesse S. et al. Molecular weight characteristics of humic substances from different environments as determined by size exclusion chromatography and their statistical evaluation // Environ. Sci. Technol. 2003. V. 37. P. 2477–2485.
Peuravuori J., Koivikko R., Pihlaja K. Characterization, differentiation and classification of aquatic humic matter separated with different sorbents: synchronous scanning fluorescence spectroscopy // Water Res. 2002. V. 36(18). P. 4552–4562. https://doi.org/10.1016/S0043-1354(02)00172-0
Piccolo A. Humus and Soil Conservation // Humic substances in terrestrial ecosystems. Elsevier, 1996. P. 225–264.
Piccolo A. The supramolecular structure of humic substances: A novel understanding of humus chemistry and implications in soil sciences // Adv. Agron. 2002. V. 75. P. 57–134.
Piccolo A., Nardi S., Concheri G. Macromolecular changes of humic substances induced by interaction with organic acid // Eur. J. Soil Sci. 1996. V. 47(3). P. 319–328. https://doi.org/10.1111/j.1365-2389.1996.tb01405.x
Piccolo A., Nardi S., Concheri G. Micelle-1ike conformation of humic substances as revealed by size exclusion chromatography // Chemosphere. 1996. V. 33. № 4. P. 595–602.
Piccolo A., Spaccini R., Nieder R., Richter J. Sequestration of a biologically labile organic carbon in soils by humified organic matter // Clim. Change. 2004. V. 67. № 2. P. 329–343.
Preston C.M. Carbon-13 solid-state NMR of soil organic matter – Using the technique effectively // Can. J. Soil Sci. 2001. V. 81(3). P. 255–270.
Preston C.M., Nault J.R., Trofymow J.A. Chemical changes during 6 years of decomposition of 11 litters in some Canadian forest sites. P. 2. 13C Abundance, solid-state 13C NMR spectroscopy and the meaning of “lignin” // Ecosystems. 2009. V. 12. P. 1078–1102.
Rice J.A. Humin // Soil Sci. 2001. V. 166(1). P. 848–857.
Rodionov A., Pätzold S., Welp G., Pallares R.C., Damerow L., Amelung W. Sensing of soil organic carbon using visible and near-infrared spectroscopy at variable moisture and surface roughness // Soil Sci. Soc. Am. J. 2014. V. 78(3). P. 949–957. https://doi.org/10.2136/sssaj2013.07.0264
Ruiz-Dueñas F.J., Martínez Á.T. Microbial degradation of lignin: how a bulky recalcitrant polymer is efficiently recycled in nature and how we can take advantage of this // Microb. Biotechnol. 2009. V. 2(2). P. 164–177.
Saito B., Seckler M.M. Alkaline extraction of humic substances from peat applied to organic-mineral fertilizer production // Braz. J. Chem. Engineer. 2014. V. 31(3). P. 675–682. https://doi.org/10.1590/0104-6632.20140313s00002512
Schaeffer A., Nannipieri P., Kästner M., Schmidt B., Botterweck J. From humic substances to soil organic matter–microbial contributions. In honor of Konrad Haider and James P. Martin for their outstanding research contribution to soil science // J. Soils Sediments. 2015. V. 15. P. 1865–1881. https://doi.org/10.1007/s11368-015-1177-4
Schmidt M.W.I., Skjemstad J.O., Gehrt E., Kögel-Knabner I. Charred organic carbon in German chernozemic soils // Eur. J. Soil Sci. 1999. V. 50(2). P. 351–365. https://doi.org/10.1046/j.1365-2389.1999.00236.x
Schmidt M.W.I., Torn M.S., Abiven S., Dittmar T., Guggenberger G., Janssens I.A., Kleber M. et al. Persistence of soil organic matter as an ecosystem property // Nature. 2011. V. 478. P. 49–56. https://doi.org/10.1038/nature10386
Schnitzer M., Monreal C.M. Quo vadis Soil Organic Matter Research?: A Biological Link to the Chemistry of Humification // Adv. Agron. 2011. V. 113. P. 139–213.
Schnitzer M., Khan S.U. Humic Substances in the Environment. N.Y.: Marcel Dekker, Inc., 1972. 327 p.
Shleev S., Shumakovich G., Mazhugo Yu., Yaropolov A., Ruzgas T., Gorton L. Interaction of fungal laccases and laccase-mediator systems with lignin // Enzyme Microb. Technol. 2006. V. 39. P. 841–847. https://doi.org/10.1016/j.enzmictec.2006.01.010
Simpson A.J. Determining the molecular weight, aggregation, structures and interactions of natural organic matter using diffusion ordered spectroscopy // Magn. Reson. Chem. 2002. V. 40. S72–S82.
Simpson A.J., Kingery W.L., Hayes M.H.B., Spraul M., Humpfer E., Dvortsak P., Kerssebaum R., Godejohann M., Hofmann M. Molecular structures and associations of humic substances in the terrestrial environment // Naturwissenschaften. 2002. V. 89. P. 84–88.
Simpson A.J., Kingery W.L., Spraul M., Humpfer E., Dvortsak P., Kerssebaum R. Separation of structural components in soil organic matter by diffusion ordered spectroscopy // Environ. Sci. Technol. 2001. V. 35. P. 4421–4425. https://doi.org/10.1021/es0106218
Simpson A.J., Simpson M.J., Smith E., Kelleher B.P. Microbially derived inputs to soil organic matter: Are current estimates too low? // Environ. Sci. Technol. 2007. V. 41. P. 8070–8076.
Sinsabaugh R.L. Phenol oxidase, peroxidase and organic matter dynamics of soil // Soil Biol. Biochem. 2010. V. 42. P. 391–404.
Sleighter R.L., Hatcher P.G. Fourier transform mass spectrometry for the molecular level characterization of natural organic matter: Instrument capabilities, applications, and limitations. INTECH, 2011. https://doi.org/10.5772/15959
Smernik R.J., Oades J.M. The use of spin counting for determining quantitation in solid state 13C NMR spectra of natural organic matter 2. HF-treated soil fractions // Geoderma. 2000. V. 96(3). P. 159–171. https://doi.org/10.1016/S0016-7061(00)00007-0
Song G., Hayes M.H.B., Novotny E.H., Simpson A.J. Isolation and fractionation of soil humin using alkaline urea and dimethylsulphoxide plus sulphuric acid // Naturwissenschaften. 2011. V. 98. P. 7–13.
Song G., Novotny E.H., Simpson A.J., Clapp C.E., Hayes M.H.B. Sequential exhaustive extraction of a Mollisol soil, and characterizations of the humic components, including humin, by solid and solution state NMR // Eur. J. Soil. Sci. 2008. V. 59. P. 505–516.
Sposito G. The Chemistry of Soils. Oxford: Oxford University Press, 2008. 344 p.
Steffen K.T., Hatakka A., Hofrichter M. Degradation of humic acids by the litter-decomposing basidiomycete Collybia dryophila // Appl. Environ. Microbiol. 2002. V. 68. P. 3442–3448.
Stevenson F.J. Humus Chemistry: Genesis, Composition, Reactions. Wiley: 1982. 443 p.
Stevenson F.J. Humus Chemistry, Genesis, composition, reactions. N.Y.: John Wiley & Sons, 1994. 512 p.
Sutton R., Sposito G. Molecular structure in humic substances: The new view // Environ. Sci. Technol. 2005. V. 39. P. 9009–9015. https://doi.org/10.1021/es050778q
Swift R. Organic matter characterization // Methods of soil analysis. P. 3. Chemical methods. SSSA, Madison, WI. 1996. P. 1011–1069. https://doi.org/10.2136/sssabookser5.3.c35
Swift R.S. Molecular weight, size, shape, and charge characteristics of humic substances: Some basic considerations // Humic Substances II: In Search of Structure. Chichester: Wiley, 1989. P. 449–466.
Thevenot M., Dignac M.-F., Rumpel C. Fate of lignins in soils: A review // Soil Biol. Biochem. 2010. V. 42. P. 1200–1211.
Thurman E.M., Malcolm R.L. Preparative isolation of aquatic humic substances // Environ. Sci. Technol. 1981. V. 15. P. 463–466. https://doi.org/10.1021/es00086a012
Thurston C.F. The structure and function of fungal laccases // Microbiology. 1994. V. 140. P. 19–26.
Tiberg C., Sjostedt C., Gustafsson J.P. Metal sorption to Spodosol Bs horizons: Organic matter complexes predominate // Chemosphere. 2018. V. 196. P. 556–565. https://doi.org/10.1016/j.chemosphere.2018.01.004
Trubitsina L.I., Lisov A.V., Belova O.V., Trubitsin I.V., Demin V.V., Konstantinov A.I., Zavarzina A.G., Leontievsky A.A. Transformation of low molecular compounds and soil humic acid by two domain laccases of streptomyces puniceus in the presence of ferulic and caffeic acids // PLoS ONE. 2020. V. 15(9). P. 1–17. https://doi.org/10.1371/journal.pone.0239005
Vasilyeva N.A., Abiven S., Milanovskiy E.Y., Hilf M., Rizhkov O.V., Schmidt M.W.I. Pyrogenic carbon quantity and quality unchanged after 55 years of organic matter depletion in a Chernozem // Soil Biol. Biochem. 2011. V. 43. P. 1985–1988. https://doi.org/10.1016/j.soilbio.2011.05.015
Vermerris W., Nicholson R. Biosynthesis of Phenolic Compounds // Phenolic Compound Biochemistry. Springer, Dordrecht. 2008. https://doi.org/10.1007/978-1-4020-5164-7_3
von Lützow M., Kögel-Knabner I., Ekschmitt K., Flessa H., Guggenberger G., Matzner E., Marschner B. Stabilization of organic matter in temperate soils: mechanisms and their relevance under different soil conditions – a review // Eur. J. Soil Sci. 2006. V. 57. P. 426–445.
von Lützow, M., Kögel-Knabner I., Ekschmitt K., Flessa H., Guggenberger G., Matzner E., Marschner B. Stabilization of organic matter in temperate soils: mechanisms and their relevance under different soil conditions – a review // Eur. J. Soil Sci. 2006. V. 57. P. 426–445.
Waggoner D.C., Chen H., Willoughby A.S., Hatcher P.G. Formation of black carbon-like and alicyclic aliphatic compounds by hydroxyl radical initiated degradation of lignin // Org. Geochem. 2015. V. 82 P. 69–76. https://doi.org/10.1016/j.orggeochem.2015.02.007
Wang K., Xing B. Structural and sorption characteristics of adsorbed humic acid on clay minerals // J. Environ. Qual. 2005. V. 34. P. 342–349. https://doi.org/10.2134/jeq2005.0342
Wells M.J.M. Conductivity-dependent flow field-flow fractionation of fulvic and humic acid aggregates // Chromatography. 2015. V. 2. P. 580–593. https://doi.org/10.3390/chromatography2030580
Wells M.J.M. Supramolecular answers to the organic matter controversy // J. Environ. Qual. 2019. V. 48. P. 1644–1651. https://doi.org/10.2134/jeq2019.02.0089
Wells M.J.M., Stretz H.A. Supramolecular architectures of natural organic matter // Sci. Total Environ. 2019. V. 537. P. 81–92. https://doi.org/10.1016/j.scitotenv.2019.03.406
Wershaw R.L. Model for humus in soils and sediments // Environ. Sci. Technol. 1993. V. 27. P. 814–816. https://doi.org/10.1021/es00042a603
Wershaw R.L. The study of humic substances: In search of a paradigm // Humic substances: Versatile components of plants, soil, and water. Cambridge: Royal Society of Chemistry, 2000. P. 1–7. https://doi.org/10.1016/B978-1-85573-807-2.50005-9
Wong D.W.S. Structure and action mechanism of ligninolytic enzymes // Appl. Biochem. Biotechnol. 2009. V. 157(2). P. 174–209.
Yavmetdinov I.S., Stepanova E.V., Gavrilova V.P., Lokshin B.V., Perminova I.V., Koroleva O.V. Isolation and characterization of humin-like substances produced by wood-degrading white-rot fungi // Appl. Biochem. Microbiol. Moscow. 2003. V. 39. P. 257–264.
Zanella A., Ponge J.-F., Matteodo M. Terrestrial humus systems and forms – Field practice and sampling problems // Appl. Soil Ecol. 2018. V. 122. P. 92–102.
Zavarzina A. Heterophase synthesis of humic acids in soils by immobilized phenol oxidases // Soil Enzymology. V. 22 of Soil Biology. Berlin Heidelberg: Springer-Verlag, 2011. P. 207–228.
Zavarzina A., Demin V., Lisov A., Ermolin M., Pogozhev E., Fedotov P. Increased sequestration of aromatic carbon on the mineral phase in the presence of a biocatalyst: the study under batch and dynamic conditions. EUROSOIL, 2021 CONT-2401 (https://b-com. mci-group.com/Abstract/Statistics/AbstractStatisticsViewPage.aspx?AbstractID=486255).
Zavarzina A., Lisov A., Leontievsky A., Zavarzin A. Fungal oxidoreductases and humification in forest soils // Soil Enzymology. V. 22 of Soil Biology. Berlin Heidelberg: Springer-Verlag, 2011. P. 187–205.
Zavarzina A.G. A mineral support and biotic catalyst are essential in the formation of highly polymeric soil humic substances // Eurasian Soil Sci. 2006. V. 39. P. S48–S53.
Zavarzina A.G., Lisov A.V., Leontievsky A.A. The role of ligninolytic enzymes laccase and a versatile peroxidase of the white-rot fungus Lentinus tigrinus in biotransformation of soil humic matter: comparative in vivo study // J. Geophys. Res.: Biogeosciences. 2018. V. 123. P. 1–16.
Zavarzina A.G., Zavarzin G.A. Humic substances in the early biosphere // Paleontol. J. 2013. V. 47(9). P. 984–988.
Zavgorodnyaya Yu.A., Demin V.V., Kurakov A.V. Biochemical degradation of soil humic acids and fungal melanins // Org. Geochem. 2002. V. 33. P. 347–355. https://doi.org/10.1016/S0146-6380(01)00165-6
Zech W., Guggenberger G., Haumaier L., Pohhacker R., Schafer D., Amelung W., Miltner A., Kaiser K., Ziegler F. Organic matter dynamics in forest soils of temperate and tropical ecosystems // Humic substances in terrestrial ecosystems. Amsterdam: Elsevier, 1996. P. 101–170.
Zech W., Senesi N., Guggenberger G., Kaiser K., Lehmann J., Miano T.M., Miltner A., Schroth G. Factors controlling humification and mineralization of soil organic matter in the tropics // Geoderma. 1997. V. 79. P. 117–161.
Дополнительные материалы отсутствуют.