

Почвоведение, 2021, № 2, стр. 183-195
Исследование кислотности сильнокислых почв рНKCl < 3.3, содержащих обменные ионы железа (III)
Е. В. Ванчикова a, Е. В. Шамрикова a, *, М. А. Королев a, Е. В. Кызъюрова a, В. И. Михайлов b
a Институт биологии Коми научного центра Российской академии наук
167982 Сыктывкар, ул. Коммунистическая, 28, Россия
b Институт химии Коми научного центра Российской академии наук
167000 Сыктывкар, ул. Первомайская, 48, Россия
* E-mail: shamrik@ib.komisc.ru
Поступила в редакцию 28.03.2020
После доработки 25.06.2020
Принята к публикации 20.07.2020
Аннотация
Сильнокислым почвам (рНKCl ≤ 3.3), широко распространенным на европейском северо-востоке, свойственна низкая прецизионность потенциометрического измерения компонентов обменной кислотности. Цель работы – выявить причины, снижающие точность измерения обменной кислотности почв с использованием одно- и двухкомпонентных модельных систем ионов алюминия и железа (III) в концентрациях, аналогичных вытяжкам из почв. С помощью модельных растворов показано, что высокая кислотность солевых вытяжек из почв вызвана присутствием в них ионов железа (III). Точность измерения ионов металлов определяется порядком перемешивания анализируемого раствора (непрерывное или дискретное), а также временем взаимодействия титранта с компонентами кислотности. Это связано с наличием в исходных модельных растворах дисперсных систем. Параметры частиц (гидродинамический диаметр 70–90 нм, дзета-потенциал 19–30 мВ) определены методами динамического рассеяния света и лазерного доплеровского электрофореза. Присутствие в солевых вытяжках из почв иных компонентов: органических соединений, тонкодисперсных частиц силикатов и гидроксидов Fe, способных проходить через поры фильтра “синяя лента”, еще более снижает точность измерения суммарного содержания Al3+ и Fe3+ (обменную кислотность) потенциометрическим методом и ставит под сомнение возможность его использования. При работе с образцами сильнокислых почв рекомендовано применять атомно-эмиссионный метод.
ВВЕДЕНИЕ
Кислотность почвы – способность почвы проявлять свойства кислот. Под кислотами понимают все компоненты почвы, способные присоединять ионы ОН–. Эти компоненты могут находиться в жидкой фазе почв, на поверхности частиц, в составе комплексных соединений, а также представлять самостоятельные твердые фазы [10, 12]. Обменная кислотность почв определяется компонентами, которые вытесняются катионами в раствор хлорида калия. При обработке почвы раствором KCl (с = = 1 моль/дм3) ионы калия замещают протоны кислотных группировок (рKа < 8) и катионы металлов почвенного поглощающего комплекса, способные к гидролизу: алюминия(III), железа(III), марганца(II) и др., переводя их в жидкую фазу. Отделяют раствор от осадка фильтрованием. Первую аликвотную часть фильтрата титруют раствором гидроксида натрия до рН 8.2, контролируя рН раствора с помощью рН-метра:
При титровании раствором основания происходит депротонирование карбоксильных групп органических кислот, а также нейтрализация протонов, образовавшихся за счет диссоциации кислот и гидролиза вытесненных в раствор обменных Al3+, Fe3+, Mn2+ и их гидроксокомплексов разной основности с образованием гидроксидов.
Обменную кислотность представляют как сумму количеств эквивалентов ионов H+, Al3+, Fe3+ и Mn2+, способных извлекаться раствором хлорида калия из 1000 г почвы. Число эквивалентности алюминия(III) и железа(III) принимают равным трем, марганца(II) – двум. Кислотность почв, обусловленную всеми ионами, рассчитывают по формуле:
Как правило, в почвах содержание алюминия(III), способного обмениваться на ионы калия, многократно больше, чем железа(III) и марганца(II). Поэтому, кислотность, обусловленную подвижными ионами металлов, относят только к ионам алюминия. Число эквивалентности (z) алюминия в стандартном растворе, рассчитанное по формуле: $z = \frac{{c({1 \mathord{\left/ {\vphantom {1 3}} \right. \kern-0em} 3}{\text{AlC}}{{{\text{l}}}_{{\text{3}}}})}}{{с{\text{(AlC}}{{{\text{l}}}_{{\text{3}}}})}},$ z = 2.998, где c(1/3AlCl3) – измеренная молярная концентрация эквивалентов хлорида алюминия в приготовленном растворе, моль/дм3; c(AlCl3) – заданная молярная концентрация хлорида алюминия в растворе, моль/дм3 [14].
Таким образом, в подобных случаях в системе протекают реакции:
Отдельные стадии образования осадка гидроксида алюминия не проявляются на кривой титрования. При добавлении раствора гидроксида натрия в системе существует равновесие всех форм алюминия(III), которое постепенно смещается в сторону образования осадка. Осаждение осадка заканчивается при рН 8.2. Избыток гидроксида натрия первоначально приводит к частичному растворению осадка с образованием тетрагидроксоалюминат-ионов
Однако из-за малой растворимости осадка гидроксида алюминия и разбавленного раствора гидроксида натрия, используемого в качестве титранта, этот процесс обратим. Стабильным в системе является гидроксид алюминия.
В солевом растворе (с(KCl) = 1 моль/дм3) коэффициент активности трехвалентных катионов не превышает γ = 3 × 10–5, двухвалентных – 10–2, одновалентных – 0.3.
В сильнокислых почвах (${\text{р}}{{{\text{Н}}}_{{{{{\text{Н}}}_{{\text{2}}}}{\text{О}}}}}$ < 4.4 и рНКСl < < 3.2) содержание ионов железа может быть существенным [16]. Такие почвы широко распространены в районах Крайнего Севера. В столь кислой среде начинают растворяться многие соединения Al, Fe и Mn, которые не являются обменными [12].
В литературе есть сведения, что вытяжки из почв, приготовленные согласно ГОСТ26423-85-2009, не всегда являются истинными растворами. Ранее доказанная нами низкая прецизионность измерения обменной кислотности сильнокислых почв, содержащих ионы железа(III) [1, 15], может быть связана с образованием дисперсных систем. В этой связи целесообразно изучение состава дисперсных частиц и учет факта их присутствия. При проведении титрования систем, содержащих коллоидные частицы, время взаимодействия почвы с титрантом с момента добавления очередной порции раствора основания до определения рН может быть существенным. Данное обстоятельство не отражено в прописях по оценке кислотности почв (ГОСТ 26423-85-2009).
К компонентам вытяжек из почв, способным образовывать устойчивые дисперсные системы, относятся гидроксиды алюминия и железа(III), частицы глинистых минералов, силикат-ион, органические соединения. Гидроксиды в кислой среде имеют положительный заряд поверхности, силикат-ион и глинистые минералы – отрицательный [9, 18]. Состав дисперсной фазы почвенных вытяжек, вероятно, определяется элементным составом почвы, который в свою очередь зависит от типа почвы и горизонта, гранулометрического состава, специфических свойств химического элемента. Количество органических и органо-минеральных соединений в минеральных горизонтах может быть существенным за счет миграции в условиях промывного водного режима [11]. Полярные органические соединения в водных растворах преимущественно являются поверхностно активными веществами. Заряд их поверхности зависит от сочетания значений констант диссоциации органических кислот и рН вытяжек.
Удобным инструментом для понимания физико-химических процессов, протекающих в вытяжках из почв в процессе их титровании, являются модельные солевые (KCl) растворы. При этом авторы отдают себе отчет в том, что прямой перенос данных, полученных в модельных системах, на почвенные экстракты не всегда возможен из-за сложности состава последних.
Экспрессным методом изучения водных дисперсных систем является метод динамического рассеяния света (ДРС, DynamicLightScattering, DLS). Метод позволяет исследовать системы, содержащие частицы диаметром (D) от 0.3 нм до 10 мкм. Принцип измерения размеров частиц основан на фиксации и анализе временной автокорреляционной функции флуктуации интенсивности рассеянного света при прохождении лазерного луча через систему. Флуктуации интенсивности возникают благодаря случайному броуновскому движению частиц, ведущему к колебаниям локальной концентрации частиц, и, следовательно, к флуктуациям показателя преломления. Рассеянный свет принимается фотоприемником. Сигнал с выхода фотоприемника обрабатывается цифровым коррелятором. Из анализа полученной автокорреляционной функции можно определить коэффициент диффузии, средний гидродинамический диаметр частиц (по уравнению Стокса–Эйнштейна) и их распределение по размерам. В случае, если частицы плавают в виде одного агрегата, прибор детектирует их как одну крупную частицу, так как агрегат двигается с соответствующей для крупной частицы скоростью [7].
Помимо определения размерных характеристик дисперсной фазы, ряд лазерных анализаторов позволяет измерять электрокинетический потенциал (ζ-потенциал) – потенциал на плоскости скольжения, который не равен поверхностному потенциалу, но его измерение часто является единственным доступным способом для оценки свойств двойного электрического слоя (ДЭС). ДЭС представляет собой слой ионов, образующийся на поверхности в результате адсорбции ионов из раствора и диссоциации поверхностных функциональных групп. ζ-потенциал является важным экспериментально определяемым параметром двойного электрического слоя, который во многих случаях характеризует агрегативную устойчивость коллоидных систем. В лазерных анализаторах определение ζ-потенциала частиц основано на измерении электрофоретической подвижности частиц с использованием эффекта Доплера (метод лазерного доплеровского электрофореза).
Ранее метод ДРС был использован для исследования как синтетических наноразмерных систем неорганического [17] и органического [21] состава, так и для природных объектов, в том числе наночастиц почв [22]. Последняя работа посвящена исследованию агрегативной устойчивости наночастиц почвы в присутствии электролитов и природных органических веществ.
Цель работы – исследование влияния ионов железа(III) и дисперсной фазы, присутствующих в солевых (KCl) вытяжках из почв, на оценку компонентов обменной кислотности почв потенциометрическим методом.
ОБЪЕКТЫ ИМЕТОДЫ
В качестве объектов исследований выбрали сильнокислые (${\text{р}}{{{\text{Н}}}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ ≤ 4.8, рНKCl ≤ 3.3) образцы минеральных горизонтов почв разного генезиса (табл. 1). Названия почв даны в соответствии с [6, 8].
Таблица 1.
Характеристика образцов почв
| Горизонт | Глубина, см | рН | Массовая доля компонента | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Н2О | KCl | СВОС, мг/кг | Сорг | SiO2 | Fe2O3 | Al2O3 | СаO | MgO | K2O | Na2O | ИФ | ||
| % | |||||||||||||
| Разрез 21-Ж. Подзол иллювиально-гумусово-железистый (на элювии алевролитовых сланцев), Albic Podzols, 61°41′ N, 54°53′ E [3–5] | |||||||||||||
| Е | 4–16 | 4.4 | 3.3 | 4– | – | 86.2 | 1.93 | 6.26 | 0.26 | 0.33 | 1.45 | 1.08 | 6 |
| Разрез 3. Подзол иллювиально-гумусово-железистый (на флювиогляциальных отложениях, подстилаемых моренными суглинками), Albic Podzols, 65°22′ N, 49°23′ E | |||||||||||||
| Е | 7–13 | 4.1 | 3.2 | 0.50 | 0.8 | 88.4 | 1.52 | 5.24 | 0.35 | 0.86 | 1.07 | – | 7 |
| Разрез 57. Подзол иллювиально-гумусово-железистый (на флювиогляциальных отложениях, подстилаемых моренными суглинками), Albic Podzols, 65°23′ N, 50°47′ E | |||||||||||||
| Е | 3–14 | 4.2 | 3.2 | 0.91 | 1.3 | 90.3 | 1.30 | 4.09 | 0.24 | 0.47 | 0.80 | – | 6 |
| Разрез 25. Торфяно-подзол глееватый (на флювиогляциальных отложениях), Gleyic Histic Podzols, 65°38′ N, 49°22′ E | |||||||||||||
| Eg | 15–29 | 4.1 | 3.2 | 0.14 | 0.3 | 94.2 | 0.50 | 3.26 | 0.20 | 0.59 | 0.54 | – | 2 |
| Разрез 27-Ж. Светлозем иллювиально-железистый (на пылеватых суглинках), Albic Luvisols [5] | |||||||||||||
| E | 10–18 | 4.3 | 3.1 | – | – | 74.8 | 2.86 | 10.54 | 0.75 | 0.58 | 1.98 | 1.57 | 14 |
| Разрез 1. Глееподзолистая почва с микропрофилем подзола (сильноопесчаненные моренные суглинки), Stagnic Cutanic Albeluvisols (Siltic), 65°24′ N, 49°25′ E | |||||||||||||
| EL[e-hf]g | 5–20 | 3.8 | 2.9 | – | 0.8 | – | – | – | – | – | – | – | 8 |
| Разрез 44. Торфяно-подзолисто-глеевая почва (на флювиогляциальных отложениях), Stagnic Histic Albeluvisols, 65°25′ N, 50°51′ E | |||||||||||||
| ELg | 17–24 | 4.0 | 3.1 | 1.56 | 2.4 | 74.7 | 3.77 | 11.89 | 0.27 | 1.22 | 1.42 | – | 16 |
| Разрез 37-Ж. Подзол иллювиально-гумусово-железистый (на элювии алевролитовых сланцев), Albic Podzols, 63°57′ N, 52°12′ E | |||||||||||||
| E | 4–10 | 3.9 | 2.8 | 0.36 | 0.82 | 86.6 | 1.94 | 5.92 | 0.27 | 0.96 | 1.44 | 0.81 | 5 |
| E | 10–15 | 4.2 | 3.1 | – | 0.54 | 85.0 | 2.24 | 6.97 | 0.30 | 1.05 | 1.73 | 0.09 | 5 |
| Разрез 3-Ж. Подзол иллювиально-гумусово-железистый (на элювии алевролитовых сланцев), Albic Podzols, 63°57′ N, 52°12′ E [4] | |||||||||||||
| Е | 8–16 | 4.1 | 2.8 | – | 1.63 | 81.8 | 1.81 | 8.60 | 0.12 | 0.21 | 1.68 | 0.75 | 8 |
| Разрез 29-Ж. Светлозем иллювиально-железистый (на пылеватых суглинках), Albic Luvisols [4] | |||||||||||||
| E | 4–11 | 4.3 | 3.1 | – | – | 80.5 | 2.17 | 8.92 | 0.65 | 0.37 | 1.9 | 1.8 | 11 |
| Разрез 4. Глееподзолистая (на сильноопесчаненных моренных суглинках), Stagnic Albeluvisols (Siltic), 65°28′ N, 49°32′ E | |||||||||||||
| ELg | 6–16 | 4.2 | 3.2 | 0.47 | 1.0 | 85.2 | 2.56 | 6.78 | 0.304 | 0.87 | 1.19 | – | 13 |
| Разрез 56. Торфяно-карболитозем потечно-гумусовый (на элювии известняков), Histic Calcaric Leptosols Humic, 65°23′ N, 50°48′ E | |||||||||||||
| ELg | 14–25 | 4.1 | 3.1 | 0.87 | 0.6 | 87.4 | 1.30 | 6.55 | 0.48 | 0.58 | 1.39 | – | 4 |
| Разрез 17. Серогумусовая глееватая мерзлотная почва (морские отложения), Reductaquic Umbric Cryosols, 68°21′ N, 59°28′ E | |||||||||||||
| AY | 6–22 | 4.8 | 3.1 | 0.40 | – | 75.3 | 3.85 | 10.54 | 0.80 | 1.25 | 1.78 | – | 18 |
| Разрез 20. Маршевая дерново-глеевая (морские отложения), Tidalic Fluvisol (Arenic, Ochric, Epiprotosalic), 68°19′ N, 59°31′ E [16] | |||||||||||||
| ACao,g | 19–30 | 3.6 | 3.3 | 0.53 | 0.5 | 82.0 | 1.75 | 9.65 | 0.68 | 1.10 | 1.46 | 0.57 | 8 |
Рассматриваемые почвы сформированы в широком диапазоне природно-климатических условий: в подзоне средней тайги (разрезы 27-Ж, 29-Ж), северной тайги (разрезы 3-Ж, Р 21-Ж, 37-Ж, 3, 57, 25, 1, 4, 44, 56), в маршевой зоне побережья Баренцева моря (разрезы 17, 20). Существенные различия в биоклиматических, литолого-геоморфологических и геокриологических условиях определили специфические черты почвообразования исследуемого ряда почв и отличия в физико-химических свойствах. При этом ведущая роль в пространственном разнообразии сравниваемых объектов принадлежит минералогическому и гранулометрическому составам, а также литологической неоднородности почвообразующих пород. Различия климатических условий сказываются на гидротермических режимах почв и их отдельных свойствах.
Выбранные объекты характеризуются следующими показателями: массовая доля илистой фракции составляет 2–13%, валовое содержание оксида кремния 75–87%, оксида алюминия – 3–12%, оксида железа(III) – 0.5–4%, углерода органических соединений – <2.5%. Детальная аналитическая полнопрофильная характеристика почв, включающих эти объекты, приведена ранее [3–5, 16].
Водные и солевые вытяжки из почв готовили в соответствии с ГОСТ 26423-85-2009. Содержание ионов Fe(III), Al(III) и Si(IV) в солевых и водных вытяжках из образцов почв определяли методом атомно-эмиссионной спектроскопии (табл. 2). Относительная погрешность измерения содержания ионов железа и алюминия, приписанная аттестованной методике, равна δ = 28%. Массовая доля углерода водорастворимых органических соединений почв измерена методом высокотемпературного каталитического окисления. Относительная погрешность измерения составляет δ(Сорг) = 12%.
Таблица 2.
Массовая доля ионов в солевых вытяжках из образцов почв, измеренная атомно-эмиссионным методом
| Объект | Массовая доля элемента, мг/кг | ||
|---|---|---|---|
| Si(IV) | Al(III) | Fe(III) | |
| Е(4–16), разрез 21-Ж | – | 181.8 | 57.4 |
| Е(7–13), разрез 3 | – | 178.8 | 22.3 |
| Е(3–14), разрез 57 | 0.6 | 238.1 | 47.1 |
| Еg(15–29), разрез 25 | 0.7 | 46.5 | 3.2 |
| Е(10–18), разрез 27-Ж | 2.8 | 277.7 | 151.3 |
| ELg(17–24), разрез 44 | 1.6 | 429.0 | 59.8 |
| Е(4–10), разрез 37-Ж | – | 214.9 | 103.9 |
| Е(10–15), разрез 37-Ж | – | 362.2 | 180.9 |
| Е(8–16), разрез 3-Ж | – | 186.3 | 6.9 |
| ELg(6–16), разрез 4 | 1.8 | 370.7 | 51.9 |
| Е(4–11), разрез 29-Ж | 1.5 | 286.6 | 162.2 |
| ELg(16–25), разрез 56 | 0.9 | 79.5 | 13.6 |
| АY(6–22), разрез 17 | 2.6 | 70.0 | 1.5 |
Для понимания проблем, снижающих точность потенциометрического измерения обменной кислотности, исследованы модельные системы ионов алюминия и железа(III). Выбор двух указанных компонентов вызван многократным (до 400 раз) превышением их содержания в солевых вытяжках над кремнием(IV). Для приготовления модельных растворов использовали гексагидрат хлорида алюминия и гексагидрат хлорида железа(III). Растворы из кристаллогидратов готовили на основе хлорида калия (с = 1 моль/дм3). Исследовали изомолярную серию растворов, содержащих Al3+ и Fe3+, в количествах, сопоставимых с их содержанием в исследуемых солевых вытяжках из почв (табл. 3, 4).
Таблица 3.
Состав однокомпонентных солевых (KCl) растворов хлоридов алюминия и железа(III)
| № раствора | рН | с1(1/3Х3+) | Va(Х3+) | Va(KCl) | V(NaOH) | с2(1/3Х3+) | $\bar {c}$(1/3Х3+)/S |
|---|---|---|---|---|---|---|---|
| моль/дм3 | см3 | моль/дм3 | |||||
| Al(III) | |||||||
| 1 | 4.15 | 0.0181 | 15.00 | 10.00 | 13.4 | 0.01787 | 0.01778/0.00015 |
| 2 | 4.13 | 20.00 | 5.00 | 17.6 | 0.01760 | ||
| 3 | 4.12 | 22.50 | 2.50 | 20.1 | 0.01787 | ||
| Fe(III) | |||||||
| 1 | 2.76 | 0.0183 | 10.00 | 15.00 | 13.4 | 0.01720 | 0.01680/0.00040 |
| 2 | 3.02 | 5.00 | 20.00 | 17.6 | 0.01640 | ||
| 3 | 3.20 | 2.50 | 22.50 | 20.1 | 0.01680 | ||
Примечание. Здесь и далее: с1 – расчетная молярная концентрация эквивалентов компонента в исходном растворе, Va(Х3+) – объем аликвоты исходного раствора, V(NaOH) – объем гидроксида натрия, пошедший на титрование, с2(1/3Х3+) – измеренная молярная концентрация эквивалентов компонента в исходном растворе, $\bar {с}$ – среднее значение измеренной молярной концентрации эквивалентов компонента в исходном растворе, S – стандартное отклонение единичного результата измерений молярной концентрации эквивалентов компонента в исходном растворе.
Таблица 4.
Состав двухкомпонентных солевых (KCl) растворов хлоридов алюминия и железа(III)
| № раствора | рН | Va(Al3+) | Va(Fe3+) | Расчетные значения | Измеренные значения | δ, % | |||
|---|---|---|---|---|---|---|---|---|---|
| с1 (1/3Al3+) | с1 (1/3Fe3+) | с1(1/3M3+) | V(NaOH), см3 |
с2(1/3M3+), моль/дм3 |
|||||
| см3 | моль/дм3 | ||||||||
| 1 | 2.72 | 15.00 | 10.00 | 0.01068 | 0.00672 | 0.01740 | 22.3 | 0.01784 | 2.5 |
| 2 | 2.95 | 20.00 | 5.00 | 0.01424 | 0.00336 | 0.01760 | 21.8 | 0.01744 | –0.9 |
| 3 | 3.08 | 22.50 | 2.50 | 0.01602 | 0.00168 | 0.01770 | 22.3 | 0.01784 | 0.8 |
При титровании применяли различные порядки перемешивания анализируемых растворов: непрерывное перемешивание в течение всего периода титрования и дискретное перемешивание (однократное перемешивание после добавления очередной порции титранта). При непрерывном перемешивании модельных растворов использовали разные режимы, изменяя скорость титрования (время t между добавлением очередных порций титранта):
а) t = 20 мин (время, достаточное для установления равновесия в системах при добавлении раствора щелочи),
б) фиксированный объем титранта V(NaOH) = = 0.5 см3, t = 5 мин, (V(NaOH)/t) = 0.1 см3/мин),
в) фиксированный объем титранта V(NaOH) = = 0.5 см3, t = 1 мин, (V(NaOH)/t) = 0.5 см3/мин).
Титрование солевых вытяжек из почв выполняли в условиях непрерывного перемешивания в режиме t = 20 мин.
Измерения проводили в Экоаналитической лаборатории Института биологии ФИЦ Коми НЦ УрО РАН, аккредитованной в Системе аккредитации аналитических лабораторий Росстандарта России (аттестат РОСС RU.0001.511257 от 16.04.14).
Характеристики коллоидных частиц модельных коллоидных растворов, такие как средний диаметр частиц и ζ-потенциал, измеряли в Институте химии ФИЦ Коми НЦ УрО РАН с помощью методов ДРС и лазерного доплеровского электрофореза (табл. 5). Данные получали с использованием лазерного анализатора ZetaSizerNanoZS (MalvernInstruments). Источник излучения – He–Ne-газовый лазер с длиной волны излучения 633 нм и мощностью 4 мВт. Измерения выполняли при фиксированном значении угла рассеяния света – 173°. Данная оптическая конфигурация, использующая в своей основе детектирование обратно рассеянного излучения, является более чувствительной и позволяет проводить измерения образцов с более низкими концентрациями и меньшими размерами частиц по сравнению со стандартной оптической схемой с углом рассеяния 90°. Для контроля повторяемости результатов на каждом образце выполняли не менее трех повторных измерений. Для измерения размеров и ζ-потенциала частиц использовали U-образные капиллярные поликарбонатные кюветы с позолоченными электродами DTS1070 (MalvernInstruments).
Таблица 5.
Параметры коллоидных частиц модельных систем
| Молярная концентрация эквивалентов с(1/3Х3+), моль/дм3 |
Средний размер частиц (D), нм |
ζ-потенциал, мВ | pH |
|---|---|---|---|
| Солевой (KCl) раствор | |||
| с(1/3Al3+) = 0.01072 | Частицы однозначно не диагностируются | 4.08 | |
| с(1/3Al3+) = 0.01480 | 52.0 (3.0)* | 20.4 (1.9) | 4.00 |
| с(1/3Fe3+) = 0.00688 | 67.6 (1.4) | 24.3 (6.7) | 2.86 |
| с(1/3Fe3+) = 0.00328 | 83.1 (7.1) | 18.9 (9.0) | 3.06 |
| с(1/3Fe3+) = 0.00168 | 89.7 (2.2) | 29.5 (2.8) | 3.20 |
| с(1/3Fe3+) = 0.00672 | |||
| с(1/3Al3+) = 0.01068 | 77.5 (6.9) | 25.5 (2.8) | 2.82 |
| Σс(1/3M3+) = 0.01740 | |||
| с(1/3Fe3+) = 0.00336 | |||
| с(1/3Al3+) = 0.01424 | 76.6 (5.8) | – | 3.00 |
| Σс(1/3M3+) = 0.01740 | |||
| с(1/3Fe3+) = 0.00168 | |||
| с(1/3Al3+) = 0.01602 | 84 (22) | 23 (15) | 3.08 |
| Σс(1/3M3+) = 0.01770 | |||
| Водный раствор | |||
| с(1/3Fe3+) = 0.00328 | 56 (18) | 18.8 (3.8) | 3.06 |
| с(1/3Fe3+) = 0.00168 | 31.2 (2.0) | 7.5 (3.1) | 3.18 |
| с(1/3Fe3+) = 0.00672 | |||
| с(1/3Al3+) = 0.00107 | 69 (35) | 21.8 (4.4) | 2.82 |
| Σс(1/3M3+) = 0.00779 | |||
| с(1/3Fe3+) = 0.00336 | |||
| с(1/3Al3+) = 0.00142 | 72 (15) | 17.1 (5.3) | 3.05 |
| Σс(1/3M3+) = 0.00478 | |||
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Исследование модельных солевых (KCl) растворов алюминия и железа(III)
Оценка молярной концентрации хлоридов алюминия или железа(III), их смеси в солевых растворах (KCl) по результатам титрования. При титровании солевых растворов Al3+ раствором NaOH в режиме непрерывного перемешивания и времени взаимодействия с титрантом, достаточном для установления равновесия при добавлении раствора щелочи (t = 20 мин – время от добавления очередной порции титранта до регистрации каждого шага титрования), на кривых титрования наблюдается три участка. Увеличение рН до 4.3, выход на плато (связывание добавляемых ионов ОН–), образование гидроксоионов и гидроксида алюминия, и резкое увеличение до рН 8.2, свидетельствующее об осаждении гидроксида алюминия. Результат измерений – молярная концентрация эквивалентов ионов алюминия равна $\bar {с}$(Al3+) = 0.0178 ± 0.0003 моль/дм3 (рис. 1, А).
Рис. 1.
Кривые потенциометрического титрования хлорида алюминия (А):Va(Al3+) = 15 см3, Va(KCl) = 10 см3 (1), Va(Al3+) = 20 см3, Va(KCl) = 5 см3 (2), Va(Al3+) = 22.5 см3, Va(KCl) = 2.5 см3 (3) и хлорида железа(III) (Б): Va(Fe3+) = = 2.5 см3, Va(KCl) = 22.5 см3(1), Va(Fe3+) = 5 см3, Va(KCl) = 20 см3 (2), Va(Fe3+) = 10 см3, Va(KCl) = 15 см3(3).
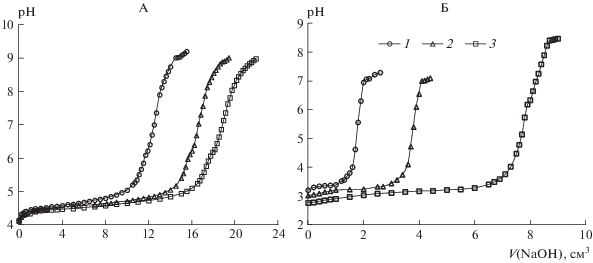
При титровании солевых растворов железа(III) раствором NaOH в режиме непрерывного перемешивания и времени взаимодействия с титрантом, достаточном для установления равновесия при добавлении раствора щелочи, формирование гидроксоионов и гидроксида железа(III) начинается сразу при добавлении раствора гидроксида натрия и заканчивается при рН 7.0 (резкое увеличение рН). Результат измерений $\bar {с}$ (1/3Fe3+) = 0.0168 ± 2S (рис. 1, Б). Поскольку раствор хлорида железа(III) готовили из неустойчивого кристаллогидрата, молярная концентрация эквивалентов железа(III) в растворе меньше рассчитанного значения.
Значения рН модельных солевых систем ионов алюминия равны 4.1–4.2, ионов железа(III), а также двухкомпонентных систем (Al3+ и Fe3+) – 2.7–3.2 (табл. 3, 4). Последние совпадают со значениями рН солевых (KCl) вытяжек из почв, содержащих сопоставимые количества названных обменных ионов. Следовательно, высокая кислотность солевых экстрактов изучаемых почв может обусловливаться обменными формами ионов железа(III).
В двухкомпонентных системах относительная погрешность измерения молярных концентраций эквивалентов хлоридов алюминия и железа(III) незначительна |δ| ≤ 2.5. Таким образом, совместное присутствие ионов и высокая ионная сила раствора не влияют на измерение суммарной молярной концентрации эквивалентов ионов алюминия и железа(III) методом потенциометрического титрования (рис. 2) в случае, если выдержано время установления равновесия между исследуемым раствором и электродной системой. Однако столь длительный промежуток времени, необходимый для достижения равновесия в системе при добавлении каждой порции титранта, ставит под сомнение возможность использования данного режима титрования при выполнении массовых исследований.
Рис. 2.
Кривые потенциометрического титрования суммы хлоридов алюминия и железа(III) в солевых растворах: Va(Al3+) = 15 см3, Va(Fe3+) = 10 см3 (1), Va(Al3+) = 20 см3, Va(Fe3+) = 5 см3 (2), Va(Al3+) = 22.5 см3, Va(Fe3+) = 2.5 см3 (3).

В двухкомпонентных системах происходят следующие реакции взаимодействия с водой
На кривых титрования двухкомпонентных модельных растворов четко прослеживаются только два скачка – в точке эквивалентности для ионов железа(III) и алюминия. Следовательно, стадии гидролиза ионов Al3+ и Fe3+ не отражаются на кривых титрования. Кроме того, ход кривой титрования ионов железа(III) при совместном присутствии ионов Al3+ и Fe3+ в солевых вытяжках искажается. Это, вероятно, связано с тем, что гидролиз ионов железа(III) препятствует взаимодействию ионов алюминия с водой. В то же время ионы Н+ расходуются на взаимодействие с гидроксоионами алюминия, что усиливает гидролиз ионов железа(III). Поэтому нет “вертикального” скачка на кривой титрования для образования гидроксида железа(III), и невозможно четко выявить значение рН конца образования гидроксида железа(III). Причем, чем меньше молярная концентрация ионов железа(III) в растворе, тем меньшее значение рН соответствует эквивалентному количеству добавленных гидроксид-ионов, необходимых для образования гидроксида железа(III). Если молярная концентрация ионов железа(III) в растворе с(1/3Fe3+) > 0.005 моль/дм3, можно оценить ее значение с погрешностью <10%, используя для расчета объем титранта, израсходованный до рН 4.3.
Следует отметить, что осадки гидроксидов алюминия и железа(III) – аморфные. В отличие от кристаллических аморфные осадки образуются не сразу при прибавлении титранта, а до некоторого предела рН остаются в жидкой фазе из-за образования коллоидных частиц разного состава, которые удерживаются в растворе вплоть до достижения порога коагуляции. При увеличении рН системы их образование сопровождается несколькими процессами, например, гидролизом ионов, смещением равновесия гидроксокомплексных частиц, образованием моно-, поли- и гетероядерных коллоидных частиц и их коагуляцией.
Поэтому еще одна причина, препятствующая образованию осадка гидроксида железа(III) в двухкомпонентных Al–Fe-системах, следующая. В случае, если [Al3+] : [Fe3+] > 1 (именно такое соотношение реализуется в модельных системах и исследуемых вытяжках из почв, табл. 2–4), ионы алюминия стабилизируют частицы золя гидроксида железа(III), а его коагуляция происходит только после полного связывания Al3+ в частицы Al(OH)3 [13].
Кроме того, препятствием к осаждению Fe(OH)3 в подобных системах может выступать окклюзия оксигидроксидами Fe(III), в том числе имеющими в своем составе ионы Al. Известно, что подобные формы железа(III) присутствуют в почвах. Особенно оксигидроксиды Fe(III) распространены в почвах гумидных областей [2, 19, 23].
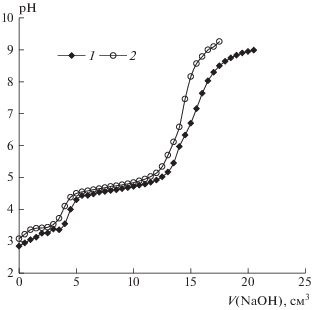
Оценка влияния условий титрования на результат измерений молярной концентрации эквивалентов ионов алюминия и железа(III) и их суммы в солевых растворах. Порядок перемешивания (скорость титрования равна V(NaOH)/t = 0.5 см3/мин). Результат измерения ионов железа(III) в двухкомпонентной системе при обоих способах перемешивания оказывается ниже расчетного. Следует отметить, что в условиях дискретного перемешивания относительная погрешность измерения концентрации эквивалентов ионов железа(III) при их совместном присутствии с ионами алюминия меньше по сравнению с непрерывным перемешиванием. Кривая титрования в диапазоне значений рН > 4.8 при дискретном перемешивании смещена в область бóльших количеств гидроксид-ионов (рис. 3). Следовательно, осаждению гидроксида железа(III) помимо перечисленных выше причин препятствует и непрерывное перемешивание, вероятно, способствующее разрыву связей между дисперсными частицами. Это подтверждается и визуальными наблюдениями – выпадение рыжего осадка в системе при непрерывном перемешивании происходит спустя 10 мин после завершения титрования при рН 9.26, тогда как при дискретном перемешивании при рН 5.91. Результат измерения суммы ионов при обоих порядках перемешивания оказываются выше по сравнению с расчетными, причем относительная погрешность измерения при дискретном перемешивании – больше.
Рис. 3.
Кривые потенциометрического титрования суммы хлоридов алюминия и железа(III) в солевых растворах: Va(Al3+) = 10 см3, Va(Fe3+) = 5 см3, Va(KCl) = 10 см3 в условиях дискретного (1), непрерывного (2) перемешивания. Пунктирными линиями указаны значения рН, начиная с которых визуально отмечено выпадение рыжих хлопьев.

Таким образом, порядок перемешивания анализируемого раствора в процессе титрования влияет на результаты измерения молярной концентрации ионов железа(III) и его суммы с ионами алюминия в солевом растворе.
Скорость титрования. Измеренные значения молярной концентрации эквивалентов ионов железа(III) при разных скоростях титрования отличны от расчетных, что определяется недостаточностью времени взаимодействия и реакциями гидролиза. Результаты показали, что и 1, и 5 мин недостаточно для установления равновесия, удовлетворительной является продолжительность в 20 мин (рис. 4).
Рис. 4.
Кривые потенциометрического титрования суммы хлоридов алюминия и железа(III) в солевых растворах: Va(Al3+) = 10 см3, Va(Fe3+) = 5 см3, Va(KCl) = 10 см3 при скорости титрования V(NaOH)/t = = 0.5 см3/мин (1) и V(NaOH)/t = 0.1 см3/мин (2).
Исследование характеристик коллоидных частиц в солевых и водных растворах, содержащих смеси хлоридов алюминия и железа(III) методом ДРС. Анализ водных и солевых растворов алюминия и железа(III) в концентрациях, аналогичных вытяжкам из почв, выявил наличие коллоидных частиц уже в исходных растворах.
При этом в солевых растворах одно- и двухкомпонентных систем ζ-потенциал частиц колеблется в диапазонах 8–30 мВ и в 1.2–2.4 раз превышает заряд частиц в водных (табл. 5). Наличие положительного заряда частиц в растворах, содержащих только ионы железа и (или алюминия) с рН 2.8–3.2, ожидаемо и соответствует данным литературы, согласно которым в кислой среде ионы алюминия и железа образуют положительно заряженные частицы [9, 18].
Средний размер коллоидных частиц в модельных растворах составляет 30–90 нм. Закономерно, что в солевых растворах размер частиц 1.5–2 раза больше, чем в водных из-за влияния ионов сильного электролита, способствующих объединению и коагуляции коллоидных частиц.
Указанные в таблице размеры частиц существенно (до двух порядков) превышают диаметр мономерных частиц Fe3+ и Al3+, связанных с хлорид-ионами, ОН–-группами и молекулами воды. Следовательно, частицы, присутствующие в исходном растворе, не являются мономерными продуктами гидролиза. Возможно, их образование связано с полимеризацией. Известно, что продукты полимеризации гидроксокомплексов Al имеют общую формулу [Alx(OH)y]3x – y, причем значения x и y могут варьировать в пределах 1 ≤ y ≤ 54 и 1 ≤ x ≤ 144 [20]. Можно предполагать, что обнаруженные методом ДРС коллоидные частицы соизмеримы по размеру с продуктами полимеризации гидроксокомплексов Al.
Кинетику формирования агломератов в данной системе не исследовали, измерение размера частиц осуществляли в свежеприготовленных растворах.
Таким образом, методом ДРС показано, что исходные модельные растворы помимо ионных форм содержат полимерные коллоидные частицы. При титровании раствором щелочи, последовательное укрупнение коллоидных частиц может препятствовать осаждению осадков гидроксидов Fe3+и Al3+ вплоть до точки эквивалентности. Кроме того, коллоидные частицы вне зависимости от их происхождения (имеют место в исходном растворе или образуются в процессе титрования) могут влиять на результаты титрования и в области значений рН свыше 8 – рН точки нулевого чистого протонного заряда гидроксидов Fe и Al. Это значит, что при более высоких значениях рН может осуществляться депротонирование поверхностных гидроксильных групп этих частиц, и на эту реакцию будут расходоваться ОН–-группы титранта. Данные предположения требуют экспериментальной проверки, для чего нужны дополнительные исследования.
Исследование солевых (KCl) вытяжек из почв. В солевых вытяжках из почв массовая доля ионов Al(III) составляет 46.51–429 мг/кг, Fe(III) – 3–180 мг/кг, Si(IV) – 0.6–2.8 мг/кг (табл. 2). Содержание углерода водорастворимых органических соединений колеблется в диапазоне 0.1–1.56 мг/кг (табл. 1). Таким образом, даже при равенстве молярных концентраций Al3+ и Fe3+ состав солевых экстрактов из почв иной в сравнении с модельными двухкомпонентными системами. Как следствие, состав дисперсных частиц, продуктов гидролиза, а также агрегатов с компонентами фона солевых вытяжек из почв отличен от таковых в модельных растворах. Физико-химические процессы, протекающие при титровании вытяжек почв, еще более не однозначны так как нарушается четкая последовательность взаимодействия гидроксид-ионов с продуктами, образовавшимися в системе.
Некоторые примеры несовпадения результатов измерения содержания ионов железа(III) и алюминия в вытяжках атомно-эмиссионным (табл. 2) и титриметрическим методами представлены на рис. 5. Наблюдаются случаи как не достижения точек эквивалентности, так и их пропуска. Как было указано, образование аморфных осадков, в частности гидроксида алюминия, происходит не сразу при прибавлении титранта. Вплоть до достижения порога коагуляции, до некоторого предела значений рН осадок остается в жидкой фазе из-за образования коллоидных частиц разного состава. Пороги коагуляции коллоидов зависят от концентрации ионов алюминия в растворе и от состава фона – посторонних частиц в растворе. Чем меньше концентрация Al3+ в растворе, тем сложнее получить осадок его гидроксида. Данное явление отчетливо наблюдается на кривых титрования. При высоких концентрациях Al3+ в солевых вытяжках (190–290 мг/кг, табл. 2) на кривой титрования наблюдается достаточно крутой скачок (рис. 5, А–5, В), тогда как при низкой концентрации Al3+ (50 мг/кг) скачок более пологий (рис. 5, Г). Кроме того, отмечено отсутствие скачка титрования, соответствующего эквивалентной точке ионов железа(III). Причины, препятствующие осаждению Fe(OH)3 в подобных системах, приведены выше. Дополнительным фактором, искажающим закономерное осаждение ионов железа в солевых вытяжках, наблюдаемое в однокомпонентной модельной системе Fe3+, может быть присутствие ионов фона: силикат-ионов и органических соединений.
Рис. 5.
Кривые титрования солевых (KCl) вытяжек из почв, полученные в условиях повторяемости (1–3): А – Е(4–10), разрез 37-Ж, Б – Е(4–11), разрез 29-Ж, В – Е(8–16), разрез 3-Ж, Г – Eg(15–29), разрез 25. Вертикальными линиями отмечено значение V(NaOH), при котором в системе достигается значение рН точки эквивалентности при титровании Fe3+ (а), суммы Fe3+ и Al3+ (б).
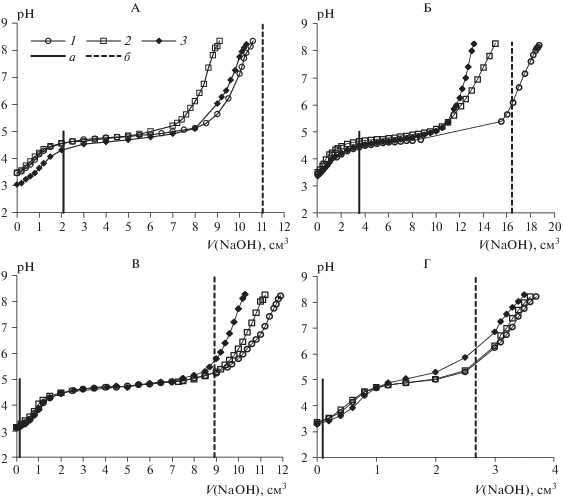
Наложение и выраженность всех указанных факторов, а также старение коллоидных систем (самопроизвольное медленное изменение их свойств, проявляющееся в укрупнении частиц дисперсной фазы), с большой вероятностью имеющее место при выполнении массовых анализов, являются причиной существенного разброса результатов титрования солевых вытяжек из сильнокислых почв, содержащих ионы железа, даже в условиях повторяемости (рис. 5, А–5, В). Следует отметить, что встречаются случаи с высокой воспроизводимостью результатов измерения обменной кислотности почв. Примером является образец горизонта Eg(15–29) торфяно-подзола глееватого (разрез 25, рис. 5, Г). Очевидно, высокая прецизионность результатов связана с тем, что данный объект характеризуется самым низким содержанием в вытяжке Al(III), Fe(III), Si(IV), а также водорастворимых органических соединений (табл. 1).
В солевых вытяжках также возможно присутствие различных химических элементов, в том числе Si, Al и Fe, не только в растворе, но и в составе кристаллических решеток минеральных частиц, способных проходить через поры фильтра квалификации “синяя лента”(3–5 мкм). Факт присутствия минеральных частиц и минералов в вытяжках наблюдается визуально и может привести к завышению значений обменной кислотности в связи с реакциями депротонирования поверхностных ОН–-групп минералов.
Таким образом, в отличие от модельных систем оценка суммарного содержания ионов алюминия и железа(III) (обменная кислотность) в солевых вытяжках из почв с рНКСl < 3.3 может быть не адекватна и при выполнении условий, актуальных для модельных систем: 1) титрование при непрерывном перемешивании, 2) титрование в режиме t = 20 мин. При работе с сильнокислыми образцами почв рекомендовано использовать атомно-эмиссионный метод.
ЗАКЛЮЧЕНИЕ
Исследованием одно- и двухкомпонентных модельных систем Fe(III) и Al(III) с концентрациями, аналогичными солевым вытяжкам из почв, методом динамического рассеяния света доказано, что условия титрования влияют на точность измерения концентрации ионов в растворах. Воспроизводимость измерений повышает непрерывное перемешивание растворов в процессе титрования, а также увеличение времени взаимодействия титранта с компонентами растворов до 20 мин при добавлении каждой порции титранта. Причина низкой прецизионности потенциометрического измерения содержания ионов – присутствие в растворах частиц, средний диаметр которых 70–90 нм, ζ-потенциал 19–30 мВ.
Оценка суммарного содержания ионов алюминия и железа(III) в солевых вытяжках из почв (обменная кислотность почв) с рНKСl < 3.3 может быть не адекватна и при выполнении условий, актуальных для модельных систем из-за наличия в солевых вытяжках компонентов фона (органических соединений и тонкодисперсных частиц силикатов и гидроксидов Fe, способных проходить через поры фильтра “синяя лента”). При работе с сильнокислыми образцами почв (рНKСl < 3.3) рекомендовано использовать атомно-эмиссионный метод.
Список литературы
Ванчикова Е.В., Шамрикова Е.В., Беспятых Н.В., Кызъюрова Е.В., Кондратенок Б.М. Метрологическая оценка методики измерений содержания кислот и ионов металлов, обуславливающих обменную кислотность // Почвоведение. 2015. № 2. С. 188–195. https://doi.org/10.7868/S0032180X14120107
Водяницкий Ю.М. Соединения железа и их роль в охране почв. М.: Почв. ин-т им. В.В. Докучаева, 2010. 154 с.
Жангуров Е.В., Голубева И.И. Морфологическая характеристика и петрографические особенности пород автоморфных почв Северного Тимана // Вестник Ин-та геологии Коми НЦ УрО РАН. 2010. № 2(182). С. 13–17.
Жангуров Е.В., Лебедева (Верба) М.П., Забоева И.В. Микростроение генетических горизонтов автоморфных таежных почв Тимана // Почвоведение. 2011. № 3. С. 288–299.
Жангуров Е.В., Тонконогов В.Д., Забоева И.В. Автоморфные почвы среднего и южного Тимана // Почвоведение. 2008. № 12. С. 1413–1422.
Классификация и диагностика почв России. Смоленск: Ойкумена, 2004. 342 с.
Куликов К.Г., Кошлан Т.В. Определение размеров коллоидных частиц при помощи динамического рассеяния света // Журн. технической физики. 2015. Т. 85. № 12. С. 26–32.
Мировая реферативная база почвенных ресурсов 2014. Международная система почвенной классификации для диагностики почв и создания легенд почвенных карт. Исправленная и дополненная версия 2015 / Научные редакторы перевода: М.И. Герасимова, П.В. Красильников. ФАО и Московский государственный университет им. М.В. Ломоносова, 2017.
Пинский Д.Л. Ионообменные процессы в почвах. Пущино, 1997. 166 с.
Соколов А.В. Агрохимические методы исследования почв. М.: Наука, 1975. 656 с.
Соколова Т.А. Низкомолекулярные органические кислоты в почвах: источники, состав, функции в почвах (обзор) // Почвоведение. 2020. № 5. С. 559–575. https://doi.org/10.31857/S0032180X20050159
Соколова Т.А., Толпешта И.И., Трофимов С.Я. Почвенная кислотность. Кислотно-основная буферность почв. Соединения алюминия в твердой фазе почвы и почвенном растворе. Тула: Гриф и К, 2007. 96 с.
Тарабан Е.А., Криворучко О.П., Плясова Л.М., Оленькова И.П., Буянов Р.А. Соосажденные гидроксиды Fe(III)–Al(III): закономерности формирования и кристаллизации при старении // Известия Сибирского отделения наук СССР. Сер. химических наук. 1990. № 1. С. 10–15.
Шамрикова Е.В., Ванчикова Е.В., Соколова Т.А., Жангуров Е.В., Денева С.В., Боброва Ю.В., Кызъюрова Е.В. Возможные источники обменной кислотности в сильнокислых почвах (рНKCl < 3.3) и оценка правильности ее определения // Почвоведение. 2018. № 12. С. 1431–1445. https://doi.org/10.1134/S0032180X18120110
Шамрикова Е.В., Денева С.В., Кубик О.С., Пунегов В.В., Кызъюрова Е.В., Боброва Ю.И., Зуева О.М. Кислотность органогенных горизонтов арктических почв побережья Баренцева моря // Почвоведение. 2017. № 11. С. 1325–1335. https://doi.org/10.7868/S0032180X17110107
Шамрикова Е.В., Денева С.В., Панюков А.Н., Кубик О.С. Свойства почв и характер растительности побережья Хайпудырской губы Баренцева моря // Почвоведение. 2018. № 4. С. 402–412. https://doi.org/10.7868/S0032180X18040020
Delgado A., González-Caballero F., Bruque J. On the zeta potential and surface charge density of montmorillonite in aqueous electrolyte solutions // Colloid Interface Sci. 1986. V. 113. P. 203–211.
Kosmulski M. Isoelectric points and points of zero charge of metal (hydr)oxides: 50 years after Parks’ review years after Parks’ review // Advances in Colloid and Interface Science. 2016. V. 238. P. 1–61.
Schwertmann U., Comell R.M. Iron Oxides in the Laboratory. Preparation and Characterization. Weinheim: WILEY-VCH Verlag GmbH & Co, 1991. 189 p.
Sposito G. (Ed.). The Environmental Chemistry of Aluminum. London: Lewis Publishers, Boca Raton, 1996. 464 p.
Torlopov M.A., Mikhaylov V.I., Udoratina E.V., Aleshina L.A., Prusskii A.I., Tsvetkov N.V., Krivoshapkin P.V. Cellulose nanocrystals with different length-to-diameter ratios extracted from various plants using novel system acetic acid/phosphotungstic acid/octanol-1 // Cellulose. 2018. V. 25. P. 1031–1046.
Zhu X., Chen H., Li W., He Y., Brookes P.C., White R., Xu J.M. Evaluation of the stability of soil nanoparticles: the effect of natural organic matter in electrolyte solutions // Eur. J. Soil Sci. 2017. V. 68. P. 105–114.
Wells M.A., Fitzpatrick R.W., Gilkes R.J. Thermal and mineral properties of Al-, Cr-, Mn-, Ni- and Ti-substituted goethite // Clays Clay Miner. 2006. V. 54. № 2. P. 176–194.
Дополнительные материалы отсутствуют.