

Поверхность. Рентгеновские, синхротронные и нейтронные исследования, 2019, № 11, стр. 20-26
Локальная атомная и электронная структура нанолистов β-СoOOH для реакции выделения водорода
М. А. Солдатов 1, *, П. В. Медведев 1, **, Ш. Вей 2, А. В. Солдатов 1
1 Международный исследовательский институт интеллектуальных материалов,
Южный федеральный университет
344090 Ростов-на-Дону, Россия
2 National Synchrotron Radiation Laboratory, University of Science and Technology of China
230029 Anhui, Hefei, People’s Republic of China
* E-mail: mikhail.soldatov@gmail.com
** E-mail: pavelmedvedev1994@gmail.com
Поступила в редакцию 18.12.2018
После доработки 18.03.2019
Принята к публикации 04.04.2019
Аннотация
Проведены расчеты геометрической и зонной структуры нанолистов метагидроксида кобальта, которые относятся к новому классу двумерных нанокатализаторов реакции выделения водорода. В результате отладки методики были отобраны оптимальные обменно-корреляционные функционалы, базисные наборы и размеры “замороженного” остова для расчетов атомной и электронной структур, плотности состояний и ширины запрещенной зоны. Результаты расчетов показали уменьшение запрещенной зоны у нанолистов метагидроксида кобальта в сравнении с кристаллической фазой. Спектры рентгеновского поглощения показали высокую чувствительность метода к изменению локального атомного окружения, особенно в первой координационной сфере атомов кобальта. Рассчитанные модельные структуры позволяют определить особенности строения локальной атомной и электронной структуры на поверхности нанолистов метагидроксида кобальта в ходе реакции выделения водорода.
ВВЕДЕНИЕ
В настоящее время одной из наиболее актуальных проблем в области энергетики является увеличение эффективности технологий возобновляемых источников энергии. В частности, к таким источникам относится солнечная энергия. Важнейшая задача – хранение полученной энергии. Ее можно решить, превращая солнечную энергию в химическую для дальнейшего использования химического топлива в топливных элементах. Водород является одним из наиболее перспективных видов химического топлива в силу высочайшей удельной эффективности для хранения энергии. Улучшение существующих методов каталитического получения водорода в ходе реакции расщепления воды упирается в разработку коммерчески выгодных материалов для производства фотохимических ячеек [1, 2]. Такие установки состоят, как правило, из трех ключевых компонентов: катализатора, на котором выделяется кислород, катализатора, на котором выделяется водород, и полупроводниковых электродов.
Ввиду высокой стоимости существующих катализаторов на основе платины [3] ведутся многочисленные исследования, цель которых – поиск новых материалов, обладающих более низкой стоимостью при столь же высокой эффективности [4]. В последнее время наибольший интерес вызывают материалы на основе недорогих переходных металлов: чистые металлы, их сплавы, смеси и соединения [3]. Ширина их запрещенной зоны может варьироваться в интервале 1–3 эВ [5], что позволяет поглощать основную часть спектра видимого света для эффективной генерации электронно-дырочных пар. Среди оксидов переходных металлов особое место занимают наноматериалы с уникальными структурами: полые, пористые частицы, а также частицы с иерархическими структурами [6]. Благодаря большой площади поверхности, упорядоченности пор, которые обеспечивают высокую скорость переноса и возможности сольватированных ионов взаимодействовать с электрохимически активными атомами, наноматериалы обладают более выгодными характеристиками для применения в области накоплении энергии [7]. В реакции каталитического выделения водорода ключевым этапом является генерация фотоэлектрона и дырки, а также их последующее перемещение к каталитически активной поверхности для восстановления с образованием H2. Однако слабая электрическая проводимость оксидов и короткая диффузионная длина материала приводит к неэффективному разделению электронно-дырочных пар. Поэтому для повышения эффективности таких катализаторов целесообразно уменьшить их толщину до величины, сравнимой с дистанцией переноса электронно-дырочных пар в материале.
Не так давно были продемонстрированы кристаллиты CoO размером 10 нм, которые обладают более высокой эффективностью (5%) по сравнению с InTaO4, допированным никелем, с квантовой эффективностью ~0.66% или же гетерогенным композитом Ga1 –xZnxN1 –xOx с квантовой эффективностью ~2.5%. Однако стоит отметить, что уже через 1 ч этот катализатор деактивируется, что связано с “отравлением” каталитических центров в ходе окислительно-восстановительных реакций, проходящих вследствие агрегации дырок на поверхности наночастиц. Для предотвращения этого явления можно использовать тонкие наноразмерные пленки на основе соединений переходных металлов [5, 8].
Как и наночастицы, такие нанопленки обладают большой площадью поверхности, но одновременно более высокой эффективностью. В своей экспериментальной работе Huang с соавторами [5] показали, что эффективность разделения электронно-дырочных пар для нанолистов β-CoOOH достигает 60–90% при облучении светом в интервале длин волн λ = 350–350 нм. Также стоит отметить, что β-CoOOH показывает более высокую структурную и каталитическую стабильность и достаточно узкую запрещенную зону. Пленки на основе β-CoOOH имеют слоистую гексагональную структуру, в которой атомы кобальта координированы шестью гидроксид-анионами, формирующими октаэдр [5, 9]. Это обеспечивает восстановление катионов H+ на их поверхности с последующей десорбцией H2 [9]. Благодаря этим свойствам, а также низкой стоимости β-CoOOH является одним из наиболее перспективных материалов для создания топливных элементов [10] и конденсаторов [11].
Для детального понимания механизма действия и повышения квантовой эффективности сверхтонких двумерных нанолистов метагидроксида кобальта (β-CoOOH) необходимо глубокое понимание фундаментальных взаимосвязей между параметрами его структуры и физико-химическими свойствами. Информацию о зонной структуре, плотности состояний, эффективной массе носителей заряда и периодической структуре могут дать расчеты на основе теории функционала плотности. В свою очередь, теоретический анализ XANES-спектров рентгеновского поглощения (X-ray Absorption Near Edge Structure – ближняя тонкая структура спектров рентгеновского поглощения) может дать важную информацию об особенностях локальной атомной и электронной структуры материала, в том числе в ходе реакции, если спектры получены в режиме operando, т.е. в реальных условиях фотокаталитической реакции.
В настоящей работе были рассчитаны зонная структура, плотность состояний и спектры рентгеновского поглощения для модельных структур кристаллического и наноразмерного метагидроксида кобальта β-CoOOH.
МЕТОДЫ РАСЧЕТА
Структура была рассчитана в рамках теории функционала плотности с использованием программного комплекса BAND [12–17]. Аналитические градиенты энергии для систем с трансляционной инвариантностью сформулированы в рамках теории функционала плотности Кона–Шэма. Энергетические градиенты реализованы в программе BAND, в которой непосредственно используется базисный набор функций Блоха, состоящий из численно рассчитанных атомных орбиталей и орбиталей слейтеровского типа [18].
При разработке методики расчетов были проведены вычисления с использованием обменно-корреляционных функционалов различных типов. В частности, использовалось приближение локальной электронной плотности (LDA) [19]. В данном подходе однодетерминантная электронная волновая функция была представлена в виде линейной комбинации центрированных на атомах базисных функций. Другими классами протестированных обменно-корреляционных функционалов были: приближение обобщенных градиентов (GGA:PBE [20], BLYP [21, 22]), эмпирические поправки на дисперсионное взаимодействие (PBE-D [23], PBE-D3 [24], PBE-D3-BJ [25]), приближение функционалов группы meta-GGA (SCAN и TPSS [26]). Если в приближении LDA используется только локальная плотность, то в приближении обобщенных градиентов – поправка на градиенты плотности, а в случае примененных в настоящей работе функционалов группы meta-GGA – зависимость от кинетической энергии и отсутствие эмпирических параметров [26].
Базисные наборы состояли из атомных орбиталей, полученных в ходе численного решения уравнений Кона–Шэма для изолированных сферических атомов, дополненных набором слейтеровских орбиталей. Были протестированы различные базисные наборы слейтеровских функций: типа DZ (двухэкспонентные атомные орбитали), DZP (двухэкспонентные атомные орбитали с добавлением поляризационных функций) и TZP (трехэкспонентные атомные орбитали с добавлением поляризационных функций).
Дополнительно были протестированы различные размеры “замороженного” остова или количество остовных орбиталей. В данном подходе остовные орбитали каждого элемента рассчитываются один раз для одиночного атома и фиксируются при дальнейших расчетах [27]. Основные “тренды”, полученные в результате отладки методики для оптимизации геометрической структуры, приведены в табл. 1. Для расчета зонной структуры, плотности состояний и ширины запрещенной зоны также был протестирован ряд обменно-корреляционных функционалов (табл. 2).
Таблица 1.
Результаты геометрической оптимизации периодической структуры метагидроксида кобальта
| Метод | Остовные орбитали | Параметр решетки a, Å | Параметр решетки c, Å | Ширина запрещенной зоны, эВ |
|---|---|---|---|---|
| Рентгеновская дифракция [28] | – | 2.851 | 13.150 | – |
| Оптический спектр [5] | – | – | – | ≈ 2.0 |
| LDA DZ | Co3p, O1s | 2.837 | 12.954 | 0.92 |
| LDA DZ | Co2p, O1s | 2.835 | 12.934 | 0.92 |
| LDA DZ | – | 2.836 | 12.942 | 0.88 |
| LDA DZP | Co3p, O1s | 2.825 | 12.856 | 0.87 |
| LDA TZP | Co3p, O1s | 2.825 | 12.875 | 0.84 |
| BLYP DZ | Co3p, O1s | 2.905 | 13.182 | 0.96 |
| BLYP DZ | Co2p, O1s | 2.892 | 13.187 | 0.99 |
| BLYP DZ | – | 2.899 | 13.142 | 0.94 |
| BLYP TZP | Co3p, O1s | 2.899 | 13.062 | 0.92 |
| BLYP TZP | Co2p, O1s | 2.896 | 13.081 | 0.93 |
| BLYP TZP | – | 2.899 | 13.097 | 0.93 |
| PBE DZP | Co3p, O1s | 2.855 | 13.148 | 0.99 |
| PBE-D DZ | Co3p, O1s | 2.898 | 13.096 | 0.98 |
| PBE-D3 DZ | Co2p, O1s | 2.873 | 13.148 | 1.03 |
| PBE-D3-BJ DZ | Co2p, O1s | 2.889 | 13.140 | 0.99 |
| TPSS DZ | – | 2.884 | 13.163 | 1.29 |
| TPSS DZP | – | 2.853 | 13.145 | 1.32 |
| TPSS TZP | – | 2.853 | 13.144 | 1.31 |
| SCAN DZP | – | 2.849 | 12.994 | 2.31 |
| SCAN TZP | – | 2.847 | 13.021 | 2.30 |
Таблица 2.
Параметры и результаты расчетов ширины запрещенной зоны объемной фазы метагидроксида кобальта
| Метод | Остов | Базис | Ширина запрещенной зоны, эВ |
|---|---|---|---|
| Эксперимент | – | – | ≈2.0 |
| LDA | Co3p, O1s | DZ | 0.8872 |
| LDA | Co2p, O1s | TZP | 0.8142 |
| BLYP | Co3p, O1s | DZ | 1.2535 |
| BLYP-D | Co3p, O1s | DZ | 1.2535 |
| BLYP-D3 | Co3p, O1s | DZ | 1.2535 |
| BLYP-D3 | Co2p, O1s | TZP | 1.2059 |
| PBE | Co3p, O1s | DZ | 1.0965 |
| PBE | Co2p, O1s | TZP | 1.0445 |
| SCAN | Co2p, O1s | TZP | 2.3576 |
В силу таких особенностей атомной структуры на границе раздела между жидкой и твердой фазами в системах жидкость–2D наноструктурированный фотокатализатор, как полости и отсутствие периодичности, для расчета спектров XANES был выбран метод конечных разностей, реализованный в программе fdmnes [29–33]. Метод конечных разностей использовался при расчете волновой функции фотоэлектрона для энергии 100 эВ выше края поглощения. Этот подход позволил избежать приближения muffin-tin [19], которое используется в большинстве программ для расчетов спектров рентгеновского поглощения, основанных на теории многократного рассеяния. При использовании приближения muffin-tin потенциал считают сферическим внутри соприкасающихся или перекрывающихся атомных сфер и постоянным между ними. Однако во многих случаях потенциал muffin-tin может не полностью отражать реальный потенциал, особенно в случае, когда исследуемое вещество пористое, с выраженными ковалентными связями или содержит большие границы раздела. В рамках вычислений методом конечных разностей удается избежать такого упрощения реального потенциала, и для моделирования спектра XANES можно использовать “полный” потенциал системы. Данный подход хорошо зарекомендовал себя при исследовании пористых материалов на основе металлорганических каркасных структур [31, 32]. Поверхность данных материалов, по сути, представляет собой границу раздела между твердой и жидкой/газовой фазами, аналогичную границе в наноструктурированной двумерной модификации метагидроксида кобальта β-CoOOH в водных растворах.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
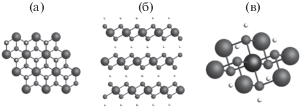
Метагидроксид β-CoOOH представляет собой слоистое, нерастворимое в воде соединение. Он образует черные кристаллы тригональной сингонии, пространственная группа R$\overline 3 $m, параметры ячейки a = 0.2852 нм, c = 1.3150 нм (база данных COD, индекс 9009884 [28]). Слоистость структуры наглядна при рассмотрении ее перпендикулярно оси с (рис. 1б). Расстояние между атомами кислорода двух соседних слоев составляет величину порядка RO–O ≈ 2.5 Å, а межатомные расстояния RO–H ≈ 1.25 Å. При рассмотрении процессов катализа, в первую очередь, интересна локальная структура вблизи атомов кобальта, которые являются активными каталитическими центрами (рис. 1в). Первая координационная сфера атома кобальта состоит из шести атомов кислорода, образующих октаэдр, межатомные расстояния RCo–O = 1.89(7) Å. Если учесть и атомы водорода, то они составляют вторую координационную сферу на расстоянии RCo–H = 2.74(1) Å и находятся над атомами кислорода, по три в каждую сторону от листа β-CoOOH. Третья координационная сфера представляет собой шесть атомов кобальта, лежащих в одной плоскости с центральным атомом кобальта и образующих шестигранник с межатомными расстояниями RCo–Co = 2.85(1) Å. В случае рассмотрения двумерных нанолистов метагидроксида кобальта β-CoOOH практически весь материал представляет собой границу раздела между жидкостью и твердым телом. В данном случае активными центрами 2D наноструктурированных фотокатализаторов выступают отдельные атомы кобальта, их окружение может существенно меняться в ходе реакции.
Рис. 1.
Изображение структуры β-CoOOH: а – вид вдоль оси с; б – вид перпендикулярно оси с; в – локальное окружение кобальта. Атомы кобальта изображены крупными темно-серыми шарами, атомы кислорода – серыми, атомы водорода – светло-серыми.
В ходе проведенных вычислений было показано, что приближение локальной плотности недооценивает параметры кристаллической решетки (отклонение параметра c на величину порядка 0.3 Å). Использование приближения обобщенных градиентов хотя и более затратно по времени, тем не менее, улучшает точность определения расположения атомов и параметров решетки (до 0.01 Å для GGA:PBE). В ходе анализа полученных результатов не было отмечено определенных закономерностей улучшения точности определения параметров решетки и расположения атомов в зависимости от выбора базисного набора, остовных орбиталей или типа использованной эмпирической поправки дисперсии. При рассмотрении результатов геометрической оптимизации с применением функционалов meta-GGA стоит отметить, что TPSS показал более высокую точность по сравнению с формализмом SCAN. Тем не менее, именно расчеты зонной структуры с использованием формализма SCAN дают значения ширины запрещенной зоны (2.3 эВ), близкие к экспериментальным данным (2.0 эВ). Приближения LDA и GGA существенно недооценивают ширину запрещенной зоны (в среднем ≈1.0 эВ).
Так как параметры кристаллической решетки, полученные с помощью более широких базисных наборов не улучшили согласие результатов расчетов с экспериментальными данными, для геометрической оптимизации структуры нанолистов метагидроксида кобальта было выбрано приближение локальной плотности и базис типа DZ (двухэкспонентные атомные орбитали), а в качестве остовных орбиталей – 3p-орбитали атомов кобальта и 1s-орбитали атомов кислорода.
С использованием таких параметров и были проведены описанные ниже расчеты атомной структуры (оптимизация геометрии) нанолистов метагидроксида кобальта. Для учета поверхностных эффектов на границе раздела между жидкостью и твердым телом для 2D наноструктурированных фотокатализатора – нанолистов β-CoOOH – была построена суперъячейка с параметром С = 39.45 Å (рис. 2а). Таким образом, модельная структура представляла собой три слоя H–O–Co–O–H, ближайшие три слоя находились на расстоянии не менее 26.3 Å (рис. 2б). Было показано, что для расчета электронной структуры и корректной оценки ширины запрещенной зоны необходимо использовать приближение обобщенных градиентов типа meta-GGA:SCAN. В ходе оптимизации геометрии параметры атомной структуры значительно не изменились. Далее для данной структуры были проведены расчеты зонной структуры (рис. 3).
Рис. 2.
Моделирование поверхности β-CoOOH: а – суперъячейка с параметром С = 39.45 Å; б – три слоя H–O–Co–O–H.
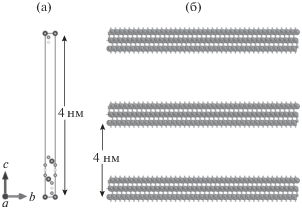
Рис. 3.
Зонная структура, рассчитанная для кристаллического (а) и наноразмерного (б) метагидроксида кобальта.

Для расчета спектров XANES были построены структурные модели поверхности нанолиста β-CoOOH путем проведения сечения по плоскости (001) таким образом, чтобы на поверхности находились атомы водорода (рис. 4), кислорода (рис. 5). Для построенных моделей были вычислены спектры XANES. Рассчитанные спектры модельных структур поверхности метагидроксида кобальта были сравнены с рассчитанными спектрами трехмерной кристаллической структуры. При рассмотрении модели, в которой поверхность катализатора пассивирована атомами водорода, следует отметить уменьшение интенсивности основного максимума и следующего за ним пика (7740–7745 эВ), а также смещение пика в области 7780–7790 эВ в диапазон меньших значений энергии. Также можно заметить небольшое перераспределение интенсивности в предкраевой области (7709–7718 эВ). Интересно, что в случае рассмотрения спектра XANES, рассчитанного для модельной поверхности, не пассивированной атомами водорода (рис. 5), уменьшение высоты основного максимума практически не наблюдается. Также менее выражено и перераспределение интенсивностей в предкраевой области. Однако и для этой модели характерен сдвиг пика в диапазоне 7780–7790 эВ в область меньших значений энергии.
Рис. 4.
Результаты теоретического расчета спектра XANES для кристаллического β-CoOOH (1) и модели поверхности β-CoOOH с атомами водорода на ней (2). На вставке показана структура поверхности.
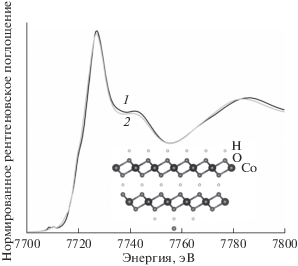
Рис. 5.
Результаты теоретического расчета спектра XANES для кристаллического β-CoOOH (1) и модели поверхности β-CoOOH с атомами кислорода на ней (2). На вставке показана структура поверхности.

Стоит отметить, что аналогичные работы по теоретическому анализу спектров XANES для материалов на границе раздела жидкость–двумерный наноструктурированный материал до сих пор практически отсутствуют. Существует только небольшое число работ, в которых метод теоретического анализа спектров XANES применяется для исследования похожих материалов – пористых металлорганических каркасных структур, в которых вся структура, по сути, является границей раздела жидкость/газ–твердое тело. Следует отметить, что основным преимуществом теоретического анализа данных XANES является возможность провести моделирование структур принципиально новых типов, которые могут реализовываться как статически, так и в ходе динамических процессов, включая процессы, протекающие в ходе реакций в реальных технологических условиях (в режиме operando). Используемый метод теоретического анализа спектров рентгеновского поглощения [34, 35] позволяет определять параметры локальной атомной структуры материалов с высокой точностью и является наиболее быстрым методом среди имеющихся в мировой практике аналогов. Это крайне важно при исследовании преобразования локальной атомной структуры материалов в ходе быстропротекающих процессов (таких как, например, фотокаталитические реакции), когда имеется большой объем экспериментальных данных (в частности, спектров XANES, полученных на различных стадиях исследуемой реакции).
ВЫВОДЫ
В ходе работы были проведены расчеты атомной и электронной структуры нанолистов метагидроксида кобальта, новых перспективных двумерных нанокатализаторов реакции выделения водорода. Было показано, что стандартные обменно-корреляционные функционалы недооценивают ширину запрещенной зоны, и для корректного ее расчета необходимо использовать соответствующие поправки. Было установлено, что оптимальным для расчета геометрической структуры является выбор приближения локальной электронной плотности, расширенного замороженного остова для атома металла (кобальта) (вплоть до 3p) и кислорода (1s) и использование небольшого базисного набора, состоящего всего из двух дзета-функций. Также было показано, что для расчета зонной структуры и корректной оценки ширины запрещенной зоны необходимо использовать приближение обобщенных градиентов формализма meta-GGA:SCAN, расширенный базисный набор, состоящий из трех дзета-функций и одной поляризационной функции, а также следует учитывать в расчете и 3p-орбиталь металла (кобальта). Рассчитанные спектры XANES показывают чувствительность к изменению локального окружения атома кобальта, особенно его первой координационной сферы. Рассчитанные модельные структуры позволят определить особенности строения локальной атомной и электронной структуры на поверхности нанолистов метагидроксида кобальта в ходе реакции выделения водорода.
Список литературы
Gratzel M. // Nature. 2001. V. 414. № 6861. P. 338. https://doi.org/10.1038/35104607
Walter M.G., Warren E.L., McKone J.R. et al. // Chem. Rev. 2010. V. 110. № 11. P. 6446. https://doi.org/10.1021/cr1002326
Kang D., Kim T.W., Kubota S.R. et al. // Chem. Rev. 2015. V. 115. № 23. P. 12839. https://doi.org/10.1021/acs.chemrev.5b00498
Suen N.T., Hung S.F., Quan Q. et al. // Chem. Soc. Rev. 2017. V. 46. № 2. P. 337. https://doi.org/10.1039/c6cs00328a
Huang J.H., Shang Q.C., Huang Y.Y. et al. // Angew. Chem.-Int. Edit. 2016. V. 55. № 6. P. 2137. https://doi.org/10.1002/anie.201510642
Ding S.J., Zhu T., Chen J.S. et al. // J. Mater. Chem. 2011. V. 21. № 18. P. 6602. https://doi.org/10.1039/c1jm00017a
Dhawale D.S., Kim S., Park D.H. et al. // ChemElectroChem. 2015. V. 2. № 4. P. 497. https://doi.org/10.1002/celc.201402365
Huang X., Zeng Z.Y., Zhang H. // Chem. Soc. Rev. 2013. V. 42. № 5. P. 1934. https://doi.org/10.1039/c2cs35387c
Kelpsaite I., Baltrusaitis J., Valatka E. // Mater. Sci.-Medzg. 2011. V. 17. № 3. P. 236. https://doi.org/10.5755/j01.ms.17.3.586
Mansour C., Pauporte T., Ringuede A. et al. // J. Power Sources. 2006. V. 156. № 1. P. 23. https://doi.org/10.1016/j.jpowsour.2005.08.020
Zheng H.J., Tang F.Q., Lim M. et al. // J. Power Sources. 2009. V. 193. № 2. P. 930. https://doi.org/10.1016/j.jpowsour.2009.03.005
Franchini M., Philipsen P.H.T., van Lenthe E. et al. // J. Chem. Theory Comput. 2014. V. 10. № 5. P. 1994. https://doi.org/10.1021/ct500172n
Franchini M., Philipsen P.H.T., Visscher L. // J. Comput. Chem. 2013. V. 34. № 21. P. 1819. https://doi.org/10.1002/jcc.23323
Velde G.T., Baerends E.J. // Phys. Rev. B. 1991. V. 44. № 15. P. 7888. https://doi.org/10.1103/PhysRevB.44.7888
Wiesenekker G., Baerends E.J. // J. Phys.-Condes. Matter. 1991. V. 3. № 35. P. 6721. https://doi.org/10.1088/0953-8984/3/35/005
Wiesenekker G., Tevelde G., Baerends E.J. // J. Phys. C Solid State. 1988. V. 21. № 23. P. 4263. https://doi.org/10.1088/0022-3719/21/23/012
BAND2018, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, http:// www.scm.com
Kadantsev E.S., Klooster R., De Boeij P.L. et al. // Mol. Phys. 2007. V. 105. № 19–22. P. 2583. https://doi.org/10.1080/00268970701598063
Vosko S.H., Wilk L., Nusair M. // Can. J. Phys. 1980. V. 58. № 8. P. 1200. https://doi.org/10.1139/p80-159
Perdew J.P., Burke K., Ernzerhof M. // Phys. Rev. Lett. 1996. V. 77. № 18. P. 3865. https://doi.org/10.1103/PhysRevLett.77.3865
Becke A.D. // Phys. Rev. A. 1988. V. 38. № 6. P. 3098. https://doi.org/10.1103/PhysRevA.38.3098
Lee C.T., Yang W.T., Parr R.G. // Phys. Rev. B. 1988. V. 37. № 2. P. 785. https://doi.org/10.1103/PhysRevB.37.785
Grimme S. // J. Comput. Chem. 2006. V. 27. № 15. P. 1787. https://doi.org/10.1002/jcc.20495
Grimme S., Antony J., Ehrlich S. et al. // J. Chem. Phys. 2010. V. 132. № 15. P. 19. https://doi.org/10.1063/1.3382344
Grimme S., Ehrlich S., Goerigk L. // J. Comput. Chem. 2011. V. 32. № 7. P. 1456. https://doi.org/10.1002/jcc.21759
Tao J.M., Perdew J.P., Staroverov V.N. et al. // Phys. Rev. Lett. 2003. V. 91. № 14. P. 4. https://doi.org/10.1103/PhysRevLett.91.146401
Te Velde G., Bickelhaupt F.M., Baerends E.J. et al. // J. Comput. Chem. 2001. V. 22. № 9. P. 931. https://doi.org/10.1002/jcc.1056
Delaplane R.G., Ibers J.A., Ferraro J.R. et al. // J. Chem. Phys. 1969. V. 50. № 5. P. 1920. https://doi.org/10.1063/1.1671307
Joly Y. // J. Synchrot. Radiat. 2003. V. 10. P. 58. https://doi.org/10.1107/s0909049502017211
Joly Y. // Phys. Rev. B. 2001. V. 63. № 12. P. 125120. https://doi.org/10.1103/PhysRevB.63.125120
Guda S.A., Guda A.A., Soldatov M.A. et al. // J. Chem. Theory Comput. 2015. V. 11. № 9. P. 4512. https://doi.org/10.1021/acs.jctc.5b00327
Guda A.A., Guda S.A., Soldatov M.A. et al. // J. Phys.: Conf. Ser. 2016. V. 712. P. 012004. https://doi.org/10.1088/1742-6596/712/1/012004
Joly Y., Grenier S., Theory of X-Ray Absorption Near Edge Structure, in X-Ray Absorption and X-Ray Emission Spectroscopy: Theory and Applications. / Ed. van Bokhoven J.A., Lamberti C. Chichester: John Wiley & Sons, 2016.
Smolentsev G., Feiters M.C., Soldatov A.V. // Nucl. Instrum. Methods Phys. Res. A. 2007. V. 575. № 1–2. P. 168. https://doi.org/10.1016/j.nima.2007.01.059
Smolentsev G., Soldatov A.V. // Comput. Mater. Sci. 2007. V. 39. № 3. P. 569. https://doi.org/10.1016/j.commatsci.2006.08.007
Дополнительные материалы отсутствуют.
Инструменты
Поверхность. Рентгеновские, синхротронные и нейтронные исследования