

Поверхность. Рентгеновские, синхротронные и нейтронные исследования, 2020, № 11, стр. 36-44
Изменения формы спектров ИК поглощения в интервале 2000–2300 см–1 при старении химически карбонизированной пленки поливинилиденфторида
В. Е. Живулин a, b, Р. Х. Хайранов a, Н. А. Злобина a, Л. А. Песин a, *
a Южно-Уральский государственный гуманитарно-педагогический университет
454080 Челябинск, Россия
b Южно-Уральский государственный университет
(Национальный исследовательский университет)
454080 Челябинск, Россия
* E-mail: pesinla@mail.ru
Поступила в редакцию 30.03.2020
После доработки 28.04.2020
Принята к публикации 30.04.2020
Аннотация
Для практического использования новых материалов ключевое значение имеет стабильность их атомной структуры и физико-химических свойств. Методом ИК-спектроскопии проведен долговременный (около 120 000 мин) и регулярный мониторинг состояния молекулярной структуры двух образцов пленки поливинилиденфторида после ее пятичасового химического дегидрофторирования и промывки этанолом. Образцы отличались продолжительностью выдержки при пониженном давлении перед началом измерений. Анализируются изменения спектров поглощения валентных колебаний тройных углерод-углеродных связей в интервале волновых чисел 2000–2300 см–1. Исходная пленка поливинилиденфторида в этой области прозрачна, но после дегидрофторирования в ней наблюдается поглощение в виде полосы сложной формы. Визуально полоса состоит из трех широких особенностей с центрами около 2050, 2100 и 2160 см–1. Первая и вторая сразу после синтеза отсутствуют, но при старении образцов появляются и растут пропорционально друг другу, причем наблюдаются временны́е интервалы быстрого и медленного роста, а также стабилизации.
ВВЕДЕНИЕ
В последние десятилетия появляется все большее количество работ, посвященных синтезу и изучению новых материалов на основе углерода (графен, углеродные нанотрубки, фуллерены). Интерес к углеродным материалам обусловлен их уникальными физико-химическими свойствами, перспективными для практического применения. Синтез и исследование низкоразмерных углеродных структур, содержащих цепочечные фрагменты [1–10], до сих пор остаются актуальной научной проблемой. В этом плане привлекает внимание гипотеза о возможности существования одномерной модификации химически чистого углерода (карбина) [1]. Предполагается, что идеальный карбин состоит из линейных углеродных цепей, в которых соседние атомы соединены либо двойными, либо чередующимися тройными и одиночными связями (соответственно кумуленовые или полииновые типы упорядочения). В ряде работ рассчитана ширина запрещенной зоны карбина, причем различные методы расчета приводят к большому разнообразию значений в диапазоне от 0.2 до 8.5 эВ [11]. В то же время большинство экспериментальных исследований показывают, что карбиноподобные структуры обладают полупроводниковыми свойствами [1, 12, 13]. Существуют различные и часто противоречивые структурные модели карбиновых цепей и их взаимного упорядочения [1, 2, 4]. Открытие кристаллического карбина в природных минералах [3] показывает принципиальную возможность его лабораторного синтеза и (как следствие) перспективу практического применения.
Тем не менее, до сих пор идеальные кристаллы карбина не синтезированы. В продуктах синтеза, получивших название “карбиноиды”, присутствуют не только наноразмерные фрагменты линейно полимеризованного углерода, но и большое количество дефектов (неуглеродные включения, межцепочечные сшивки и др.) [1, 2, 4]. Этот тип углерода впервые был синтезирован в СССР в 60-х годах прошлого века [1].
Одним из путей синтеза карбиноподобных углеродных наноструктур является карбонизация цепных полимеров, имеющих углеродную основу, например поливинилиденфторида (ПВДФ). Сам ПВДФ обладает рядом полезных свойств, благодаря которым он широко используется в мембранных технологиях [14], электронике, медицине, акустике и др. [15, 16]. Его молекулы представляют собой углеродные цепи, к каждому атому которых попеременно присоединены два атома фтора и водорода. Существует три основных типа конформации цепей: α, β и γ [17]. В зависимости от преобладающего конформационного типа полимерный материал может обладать различными свойствами. Например, когда преобладают цепи β-типа (плоский зигзаг), полимер обладает пьезоэлектрическими свойствами. Как правило, в ПВДФ сосуществуют кристаллические и аморфные области.
Известны два основных пути карбонизации ПВДФ: радиационный (облучение фотонами и бомбардировка микрочастицами различной массы и энергии) [18–27] и химический [1, 28–34]. ПВДФ содержит одинаковое количество атомов фтора и водорода, которые в результате радиационного или химического воздействия удаляются из полимера в виде фтористого водорода. Это позволяет глубоко, хотя и не исчерпывающим образом [26], карбонизировать ПВДФ без разрушения углеродной цепочечной структуры. Соседние атомы углерода, освободившиеся от фтора и водорода, могут соединяться друг с другом одинарными и кратными связями, образуя кумуленовые или полииновые структуры [1]. При этом становятся возможными также межцепочечные сшивки. Однако неполнота карбонизации приводит к сохранению некоторого количества молекулярных комплексов CF2 (CH2) или/и CF (CH) внутри частично “голой” цепи. Эти комплексы могут играть стабилизирующую роль, удерживая карбонизированные фрагменты соседних цепей на расстоянии, достаточном для предотвращения немедленного коллапса линейного углерода в структуры более высокой размерности.
Наиболее простым и продуктивным методом глубокой карбонизации ПВДФ, позволяющим модифицировать достаточно большое количество полимера, является химическая обработка. Согласно известной модели химической карбонизации, под воздействием жидкой дегидрофторирующей смеси фтор и водород в равных количествах отделяются от углеродного скелета [1]. К настоящему времени выявлены некоторые характерные особенности химически синтезированных производных ПВДФ [34]. Сделаны оценки глубины проникновения дегидрофторирующей смеси в частично кристаллическую пленку ПВДФ при различной продолжительности дегидрофторирования. Проводимость карбонизированного слоя оказалась выше, чем у его полимерного предшественника [35], что имеет перспективу синтеза проводящих и/или полупроводниковых нанопленок на упругой и прозрачной диэлектрической подложке для опто- и акустоэлектронных наноустройств.
Изучение продуктов химической карбонизации ПВДФ методом ЯМР и ИК-спектроскопии показало, что увеличение продолжительности обработки в дегидрофторирующей смеси повышает отношение числа протонов к числу ядер фтора, несмотря на синхронное и монотонное уменьшение числа СН2- и CF2-групп [34]. Это связано с интенсивной реакцией нуклеофильного замещения атомов фтора гидроксильными группами, содержащимися в компонентах смеси и атмосферном воздухе. Этот эффект препятствует образованию кратных углерод-углеродных связей, присущих карбиноидным структурам.
В работе [36] нам удалось уменьшить присоединение ОН-групп к углеродной основе за счет замены дистиллированной воды этанолом при финальной промывке образца, химически карбонизированного в течение 30 минут. Мониторинг эволюции его молекулярной структуры при старении регулярно проводился в течение почти целого календарного года методом ИК-спектроскопии. Обнаружено долговременное немонотонное изменение формы ИК-спектров в области частот, соответствующих полосе поглощения тройных углерод-углеродных связей. Именно эта полоса поглощения представляет особый интерес, так как существование тройных углерод-углеродных связей возможно только в цепочечной (карбиноидной) форме углерода.
В настоящей статье описаны результаты модификации этой полосы ИК-поглощения при старении карбиноидной пленки, синтезированной химическим дегидрофторированием ПВДФ в течение пяти часов. Десятикратное по сравнению с [36] увеличение продолжительности реакции преследовало две цели: увеличить концентрацию тройных углерод-углеродных связей за счет большего количества прореагировавшего полимерного вещества и (возможно) уменьшить время, прошедшее после синтеза образца, по истечении которого его молекулярная структура перестанет изменяться. Очевидно, что стабильность атомной структуры и физико-химических свойств новых материалов, а также выявление необходимых условий их хранения и эксплуатации имеют ключевое значение для их практического использования. В работе [37] показано, что на кинетику дезактивации парамагнитных центров в химически карбонизированных образцах ПВДФ значительное влияние оказывает взаимодействие с воздухом. В этой связи мы дополнительно провели изучение влияния атмосферного воздуха на характер и кинетику модификации молекулярной структуры химически карбонизированного материала и получили совершенно неожиданный результат.
ОБЪЕКТ И МЕТОДИКА ИССЛЕДОВАНИЯ
Исследуемый материал получен пятичасовым дегидрофторированием при комнатной температуре пленки ПВДФ Ф-2м толщиной 20 мкм производства фирмы “Пластполимер” (Санкт-Петербург). Дегидрофторирующая смесь состояла из 1 объемной части насыщенного раствора едкого кали в этаноле и девяти объемных частей химически чистого ацетона. Процедура синтеза практически аналогична подробно описанной в работе [34], за исключением того, что после предварительной промывки ацетоном финальную промывку образца проводили не водой, а этанолом [36]. В результате химического воздействия первоначально прозрачная полимерная пленка стала полностью матово-черной. Для изучения влияния атмосферного воздуха дегидрофторированная пленка сразу после окончания синтеза и промывки была разделена на две части. Обе части прочно закреплялись в отдельные держатели с прямоугольными отверстиями. Это позволяло обеспечивать при последующих измерениях единообразное прохождение ИК-излучения через одни и те же участки образцов. Перед измерениями оба образца выдерживались в форвакуумной камере при комнатной температуре и давлении остаточных газов около 5 × 10–2 мм рт. ст. в течение 1 и 13 суток (соответственно, образцы 1 и 2). Мы полагали, что пониженное давление сможет ингибировать на начальном этапе процессы старения образца 2, и предполагали наблюдать соответствующее отставание хода ожидаемой модификации его молекулярной структуры по сравнению с образцом 1. В этой же камере образцы хранились в промежутках времени между измерениями, в частности, в ночное время.
Как правило, применение лишь одного экспериментального метода не может дать исчерпывающей информации об исследуемом объекте. Тем не менее, метод ИК-спектроскопии обладает высокой чувствительностью к самой незначительной модификации молекулярной структуры вещества [38]. Поэтому именно этот метод был выбран нами для мониторинга процесса старения синтезированного материала. ИК-спектры измерялись в интервале частот 400–4000 см–1 с помощью Фурье-спектрометра Shimadzu IRAffinity-1 в геометрии на пропускание. Измерения начались с 1500 и 18406 минуты, а закончились на 116 528 и 116 530 минутах (соответственно, образцы 1 и 2) после окончания химической обработки и промывки. Получено всего 230 и 171 спектров образцов 1 и 2. Это позволилодостаточно подробно наблюдать изменение формы ИК-спектров при старении. Экспериментальные данные по пропусканию пересчитывались на величину оптической плотности.
ЭКСПЕРИМЕНТАЛЬНЫЕ РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Наиболее существенная необратимая модификация формы ИК-спектров при увеличении продолжительности старения обоих образцов наблюдается в частотном интервале 1800–2300 см–1. Исходная пленка ПВДФ в этой области прозрачна, но после дегидрофторирования в ней наблюдается поглощение в виде полосы сложной формы. Известно [36, 38], что полоса поглощения 2000–2300 см–1 возникает в результате валентных колебаний тройных углерод-углеродных связей. На рис. 1 и 2 представлены соответствующие фрагменты ИК-спектров образцов 1 и 2, измеренных в различные моменты времени, прошедшего после окончания синтеза. Форма спектров при старении образцов изменяется исключительно закономерным образом. В основном модификация происходит вследствие появления и роста довольно широких пиков поглощения вблизи 2050 и 2100 см–1 по мере увеличения продолжительности старения.
Рис. 1.
Модификация формы полосы поглощения тройных углерод-углеродных связей при старении образца 1. Регистрация представленных в качестве примера спектров проведена спустя 2942, 22 893, 34 646, 50 342, 50 488, 61 892, 114 997 и 116 725 минут после окончания синтеза. Рост пиков поглощения вблизи 2050 и 2100 см–1 происходит по мере увеличения продолжительности старения, поэтому кривые на рисунке легко идентифицируются.
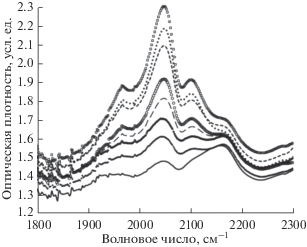
Рис. 2.
Модификация формы полосы поглощения тройных углерод-углеродных связей при старении образца 2. Регистрация представленных в качестве примера спектров проведена спустя 24 520, 30 009, 34 645, 50 489, 61 856, 83 466, 102 221 и 116 727 минут после окончания синтеза. Рост пиков поглощения вблизи 2050 и 2100 см–1 происходит по мере увеличения продолжительности старения, поэтому кривые на рисунке легко идентифицируются.
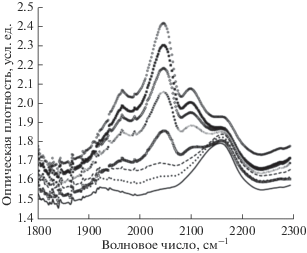
Из сравнения рис. 1 и 2 хорошо видно качественное подобие модификации формы спектров обоих образцов. При внимательном рассмотрении рисунков можно заметить и отличия. Видно, что поглощение вблизи 2050 и 2100 см–1 в спектрах образца 2 меньше по сравнению с образцом 1, если спектры обоих образцов регистрируются в близкие моменты времени. Для этого достаточно сравнить на обоих рисунках, например, третьи спектры снизу, которые регистрировались практически одновременно (34 646 и 34 645 минут), или первые сверху (116 725 и 116 727 минут).
Для параметризации формы спектров из них сначала единообразно вычиталась фоновая составляющая, которая аппроксимировалась отрезком прямой в интервале волновых чисел 1830–2240 см–1, а затем измерялись величины оптической плотности в точках 2050, 2102 и 2162 см–1. Эти точки соответствуют частотным положениям максимумов трех явно наблюдаемых пиков, которые, как мы полагаем, связаны с валентными колебаниями С≡С-связей. В результате изменения формы спектров некоторые пики превращаются в наплывы и наоборот, поэтому максимумы всех трeх пиков могут не наблюдаться в явном виде в одни и те же моменты времени. В дальнейшем условимся называть эти три пика или наплыва спектральными особенностями 1–3 с центрами вблизи 2050, 2102 и 2162 см–1. Другая широкая спектральная особенность в области 1850–2000 см–1 (скорее всего) возникает вследствие валентных колебаний, кумулированных С=С-связей различной протяженности [36], а изменение ее формы происходит в результате частичного наложения на низкочастотное плечо особенности 1.
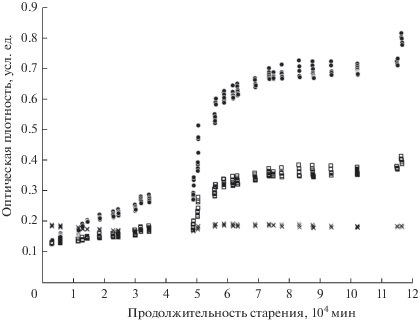
На рис. 3 показаны зависимости величин оптической плотности в центрах особенностей 1–3 от продолжительности старения образца 1. Для образца 2 эта зависимость качественно очень похожа, поэтому мы не стали ее приводить на отдельном рисунке. В спектрах обоих образцов особенность 3 изменяется слабо. Наиболее заметны немонотонные синхронные изменения особенностей 1 и 2. Эта связь явно прослеживается при рассмотрении рис. 1–3. Мы построили в оболочке Excel соответствующие линии тренда и убедились, что зависимости между величинами оптической плотности D1 и D2 в центрах особенностей 1 и 2 для обоих образцов очень хорошо описываются линейными уравнениями с достаточно близкими коэффициентами:
Рис. 3.
Зависимость оптической плотности особенностей 1 (⚫), 2 (◻) и 3 (×) от продолжительности старения образца 1.
D1 = 2.34D2 – 0.15 (коэффициент корреляции R2 = 0.997, образец 1) и
D1 = 2.37D2 – 0.17 (коэффициент корреляции R2 = 0.997, образец 2).
Такая очевидная связь свидетельствует о влиянии на рост обеих особенностей одного и того же молекулярного процесса, происходящего при старении образцов.
Скорость увеличения ИК-поглощения в интервале 2000–2250 см–1 существенно отличается на различных этапах старения. Обе обсуждаемые особенности демонстрируют слабый рост приблизительно до 49 000 минут, прошедших после окончания синтеза, затем очень быстро растут до 56 200 минут. В интервале 56 200–74 000 мин снова наблюдается медленный рост, сменяющийся стабилизацией оптической плотности до нового увеличения после 100 000 мин. Описанный немонотонный рост поглощения хорошо иллюстрируется рис. 4, на котором представлены вариации величины оптической плотности D1 в центре доминирующей в спектрах особенности 1 в зависимости от продолжительности старения обоих образцов. На рис. 4 также хорошо видно, что (как отмечалось выше) в близкие моменты времени поглощение около 2050 см–1 в спектрах образца 2 меньше по сравнению с образцом 1. Неожиданными и пока необъяснимыми нам представляются синхронные изменения параметра D1 (как и D2, который на рис. 4 не показан) в спектрах обоих образцов до 100 000 мин, в том числе, во временнóй области бурного роста этих параметров. Выше 100 000 мин эта синхронность нарушается: новый скачок параметров D1 и D2 в спектрах образца 2 происходит на 21758 мин раньше, по сравнению с образцом 1.
Рис. 4.
Немонотонные изменения величины оптической плотности D1 при старении образцов 1 (⚫) и 2 (◻).
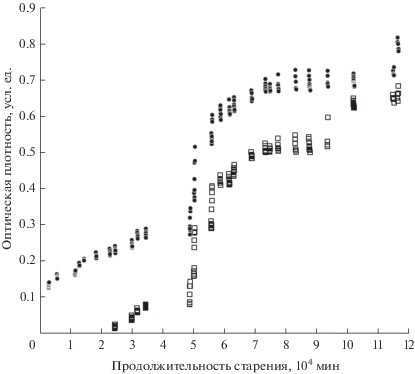
Отметим еще один постоянно возникающий обратимый эффект, причина которого пока неясна. На рис. 3 и 4 видны серии почти вертикально расположенных экспериментальных точек. Это величины параметров D1 и D2 в спектрах, полученных в близкие моменты времени (через 20–30 минут) в одной серии измерений. Как отмечалось выше, между измерениями образцы хранились при пониженном давлении. При регистрации первого спектра только что извлеченного из вакуумной камеры образца величины параметров D1 и D2, как правило, имеют наибольшее значение в данной серии измерений, а затем постепенно уменьшаются.
На рис. 5, 6 этот эффект продемонстрирован на примере двух ИК-спектров образца 1, зарегистрированных на 87 637 и 87 957 минутах старения в начале и в завершение одной серии измерений. Первый получен сразу после извлечения из вакуумной камеры, второй – спустя 320 мин нахождения на воздухе. Предположительно наблюдаемое явление может быть связано с постепенным накоплением влаги на поверхности образцов на протяжении серии измерений. На это указывает увеличение поглощения в спектральной области 2700–3700 см–1, характерной для валентных колебаний ОН-групп (рис. 6). Это означает, что в результате дегидрофторирования поверхность образцов становится гигроскопичной. Вполне вероятно, что увеличение толщины водяной пленки при последовательных измерениях на воздухе приводит к постепенному уменьшению интенсивности проходящего через образец излучения. Не исключено также, что наблюдаемые нами во всех спектрах небольшие пики при 653, 680 и 850 см–1 являются следствием либрационных колебаний кластеров воды различных размеров [39, 40]. Некоторые комбинационные моды либрационных и деформационных колебаний таких кластеров могут давать вклад в полосу поглощения, характерную для валентных колебаний С≡С-связей [40].
Рис. 5.
Фрагменты ИК-спектров образца 1 в интервале 1800–2250 см–1, измеренных сразу после извлечения из вакуумной камеры на 87 637 минуте старения (сплошная линия) и спустя 320 мин пребывания на воздухе (пунктир). Предварительно из спектров вычиталась линейная составляющая фона.
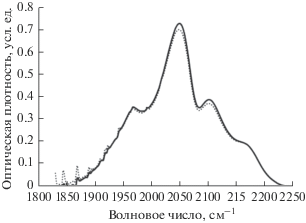
Рис. 6.
Фрагменты ИК-спектров образца 1 в интервале 2600–3800 см–1, измеренных сразу после извлечения из вакуумной камеры на 87 637 минуте старения (сплошная линия) и спустя 320 мин пребывания на воздухе (пунктир).
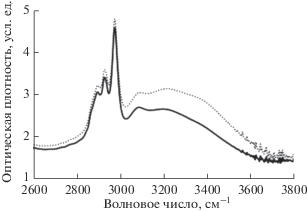
Во всех полученных в данном исследовании спектрах на фоне ОН-полосы наблюдается СН-полоса ИК-поглощения. Она представляет собой суперпозицию линий поглощения, которые по своим частотным положениям очень близки к характерным для четырех типов валентных колебаний молекулярных комплексов в этаноле: симметричных и асимметричных колебаний СН-групп, соответственно, при частотах 2870 (sСН2), 2894 (sСН3), 2927 (aСН2) и 2975 (aСН3) см–1 [41, 42]. Однако в целом форма СН- и ОН-полос отличается от формы этанола. По аналогии с работой [36], можно предположить наличие в карбонизированном материале этоксигрупп, присоединенных к освободившимся от фтора участкам углеродных цепей. Однако, в отличие от результатов [36], в данном исследовании и форма, и интенсивность СН-полосы оказались исключительно стабильны. В частности, для обоих образцов максимальная скорость роста параметров D1 и D2 наблюдается между 48920 и 56200 минутами старения. За это время площадь полосы поглощения в интервале 1830–2240 см–1 увеличивается в 1.7 и 2.1 раза, а площадь СН-полосы уменьшается лишь на 1.6 и 7%, соответственно, для образцов 1 и 2. Это может косвенно свидетельствовать о том, что уменьшение количества этоксигрупп не является причиной роста поглощения за счет вклада колебаний С≡С-связей. Наблюдаемые изменения могут быть следствием либо действительного увеличения в образце концентрации тройных связей, либо изменения симметрии их окружения различными функциональными группами, в частности, кластерами воды.
В отличие от спектра этанола, наблюдается также особенность с центром вблизи 3090–3100 см–1 (рис. 6), которая присутствует в спектрах на любой стадии старении образца. Ее возникновение при дегидрофторировании пленки ПВДФ марки Kynar наблюдалась нами и ранее [34]. Однозначная идентификация происхождения этой особенности в настоящее время также не представляется возможной и требует дополнительного изучения.
ЗАКЛЮЧЕНИЕ
Проведенный нами длительный эксперимент по старению химически дегидрофторированного ПВДФ поставил больше вопросов, чем дал ответов. К настоящему времени в результате проведенных измерений пока удалось лишь выявить некоторые особенности изучаемого явления.
1. После окончания химического синтеза молекулярная структура образца меняется в течение длительного времени. Эти изменения уместно описать термином “старение”.
2. В частотной области, характерной для проявления валентных колебаний тройных связей, происходит сильное изменение формы ИК-спектров поглощения, проявляющееся в появлении и росте двух широких особенностей с центрами около 2050 и 2100 см–1. Их эволюция происходит удивительно синхронно и пропорционально, что является косвенным признаком отражения в ней одного и того же процесса при модификации молекулярной структуры образца. Альтернативное объяснение этой связи могло бы основываться на предположении, что сама по себе особенность 2 не меняется, однако рост особенности 1 вызывает ее пропорциональное увеличение относительно линии фона. Тем не менее, это объяснение противоречит экспериментальным фактам, поскольку особенность 2 не наблюдается в спектрах даже на самых ранних стадиях старения, когда особенность 1 либо отсутствует, либо весьма слаба.
3. Анализ ИК-спектров не позволил однозначно установить причину роста поглощения в частотной области, характерной для валентных колебаний С≡С-связей. Наблюдаемые изменения могут быть следствием либо действительного увеличения в образце концентрации тройных связей, либо изменения симметрии их окружения различными функциональными группами.
4. В явном виде не обнаружена зависимость эволюции молекулярной структуры при старении от продолжительности начальной выдержки дегидрофторированных образцов при пониженном давлении. В спектрах обоих образцов наблюдается синхронный мощный рост вклада валентных колебаний С≡С-связей во временнóй области 49 000–56 200 мин после синтеза.
5. Обнаружена значительная гигроскопичность исследуемого образца, которая проявляется в изменении спектра в области колебаний OH-связей. Когда образец извлекается из вакуумной камеры, то в самом начале очередной серии измерений интенсивность ОН-полосы минимальна, а особенности 1 и 2, напротив, наиболее интенсивны. В каждом последующем измерении сначала наблюдается постепенное увеличение поглощения ОН-связей, а затем достижение некоторого предельного для данной серии измерений значения. При этом особенности 1 и 2 хотя и незначительно, но заметно и синхронно уменьшаются. Возможно, что увеличение толщины водяной пленки при последовательных измерениях на воздухе приводит к постепенному уменьшению интенсивности проходящего через образец излучения. Наличие воды в образцах не исключает ее влияния на форму спектров также и в интервале 2000–2200 см–1.
Список литературы
Carbyne and Carbynoid Structures / Ed. Heimann R.B. et al. Dordrecht: Kluwer Academic Publishers, 1999. 446 p.
Luo W., Windl W. // Carbon. 2009. V. 47. P. 367. https://doi.org/10.1016/j.carbon.2008.10.017
Шумилова Т.Г., Данилова Ю.В., Горбунов М.В. и др. // Докл. Акад. наук. 2011. Т. 436. С. 394.
Беленков Е.А., Шахова И.В. // Физика твердого тела. 2011. Т. 53. В. 11. С. 2265.
Freitas A., Azevedo S., Kaschny J.R. // Physica E. 2016. V. 84. P. 444. https://doi.org/10.1016/j.physe.2016.07.018
Prazdnikov Y.// Journal of Modern Physics. 2012. V. 3(9). P. 895. https://doi.org/10.4236/jmp.2012.39117
Buntov E.A., Zatsepin A.F., Guseva M.B. et al. // Carbon. 2017. V. 117. P. 271. https://doi.org/10.1016/j.carbon.2017.03.010
Kang C.-S., Fujisawa K., Ko Y.-I., Muramatsu H. et al. // Carbon. 2016. V. 107. P. 217. https://doi.org/10.1016/j.carbon.2016.05.069
Andrade N.F., Vasconcelos T.L., Gouvea C.P. et al. // Carbon. 2015. V. 90. P. 172. https://doi.org/10.1016/j.carbon.2015.04.001
Ковригин Д.А., Никитенкова С.П. // Физика твердого тела. 2016. Т. 58. В 3. С. 595.
Ravagnan L., Siviero F., Lenardi C. et al. // Phys. Rev. Lett. 2002. V. 89. P. 285506. https://doi.org/10.1103/PhysRevLett.89.285506
Shi L., Rohringer P., Wanko M. et al. // Phys. Rev. Materials. 2017. V. 1(7). P. 075601. https://doi.org/10.1103/Phys.Rev.Materials.1.075601
Korshak V.V., Kudryavtsev Yu.P., Khvostov V.V. et al // Carbon. 1987. V. 25(6). P. 735. https://doi.org/10.1016/0008-6223(87)90143-6
Kudryavtsev Yu.P., Evsyukov S.E., Babaev V.G. et al. // Carbon. 1992. V. 30(2). P. 213. https://doi.org/10.1016/0008-6223(92)90082-8
Zhang S., Shen J., Qiu X. et al. // J. Power Sources. 2006. V. 153. P. 234. https://doi.org/10.1016/j.jpowsour.2005.05.020
Kimoto A., Sugitani N. // Measure. Sci. Technol. 2010. V. 21. P. 075202 / Ed. Anand S.C. et al. Medical and Healthcare Textiles. Manchester. England. Wood Head Publishing, 2010. 558 p. https://doi.org/10.1088/0957-0233/21/7/16
Кочервинский В.В. // Успехи химии. 1996. Т. 65. С. 936.
Le Moel A., Duraud J.P., Balanzat E. // Nucl. Instrum. Methods Phys. Res. B. 1986. V. 18(1–6). P. 59. https://doi.org/10.1016/S0168-583X(86)80012-X
Adem E.H., Bean S.J., Demanet C.M. et al. // Nucl. Instrum. Methods Phys. Res. B. 1988. V. 32(1–4). P. 182. https://doi.org/10.1016/0168-583X(88)90206-6
Duca M.D., Plosceanu C.L., Pop T. // J. Appl. Polym.Sci. 1998. V. 67(13). P. 2125. https://doi.org/10.1002/(SICI)1097-4628(19980328)67:13< 2125::AID-APP2>3.0.CO;2-G
Brzhezinskaya M.M., Morilova V.M., Baitinger E.M. et al. // Polym. Degrad. Stab. 2014. V. 99. P. 176. https://doi.org/10.1016/j.polymdegradstab.2013.11
Chebotaryov S.S., Volegov A.A., Pesin L.A. et al // Physica E. 2007. V. 36(2). P. 184. https://doi.org/10.1016/j.physe.2006.10.011
Chebotaryov S.S., Baitinger E.M., Volegov A.A. et al. // Rad. Phys. Chem. 2006. V. 75(11). P. 2024. https://doi.org/10.1016/j.radphyschem.2005.12
Pesin L.A., Morilova V.M., Zherebtsov D.A. et al. // Polym. Degrad. Stab. 2013. V. 98(2). P. 666. https://doi.org/10.1016/j.polymdegradstab.2012.11
Le Moel A., Duraud J.P., Lecomte C. et al. // Nucl. Instrum. Methods Phys. Res. B. 1988. V. 32(1–4). P. 115. https://doi.org/10.1016/0168-583X(88)90192-926
Sidelnikova A.L., Andreichuk V.P., PesinL.A. et al. // Polym. Degrad. Stab. 2014. V. 110. P. 308–311. https://doi.org/10.1016/j.polymdegradstab.2014.09.009
Morikawa E., Choi J., Manohara H.M. et al // J. Appl. Phys. 2000. V. 87(8). P. 4010-6. https://doi.org/10.1063/1.372447
Ross G.J., Watts J.F., Hill M.P. et al. // Polymer. 2000. V. 41. P. 1685. https://doi.org/10.1016/S0032-3861(99)00343-2
Ross G.J., Watts J.F., Hill M.P., Morrissey P. // Polymer. 2001. V. 42. P. 403. https://doi.org/10.1016/S0032-3861(00)00328-1
Кудрявцев Ю.П., Евсюков С.Е., Бабаев В.Г. // Изв. Акад. наук. Сер. хим. 1992. № 5. С. 1223.
Margamov I.G., Pesin L.A., Kudryavtsev Yu.P., Evsyukov S.E. // Appl. Surf. Sci. 1999. V. 148(3–4). P. 183. https://doi.org/10.1016/S0169-4332(99)00154
Mavrinskaya N.A., Pesin L.A., Baumgarten M. et al. // Magnetic Resonance in Solids EJ. 2008. V. 10(1). P. 31.
Живулин В.Е., Москвина Н.А., Грибов И.В. и др. // Поверхность. Рентген., синхротр. и нейтрон. исслед. 2017. № 9. С. 38.
Живулин В.Е., Чернов В.М., Осипов А.А. и др. // Физика твердого тела. 2017. Т. 59. В. 7. С. 1387.
Песин Л.А., Чеботарев С.С., Кувшинов А.М. и др. // Поверхность. Рентген., синхротр. и нейтрон. исслед. 2010. № 3. С. 37.
Zhivulin V.E., Pesin L.A., Belenkov E.A., Greshnyakov V.A., Zlobina N.A, Brzhezinskaya M.M. // Polym. Degrad. Stab. 2020. V. 172. P. 109059. https://doi.org/10.1016/j.polymdegradstab.2019.109059
Живулин В.Е., Злобина Н.А., Песин Л.А. // Известия Томского политехнического университета. Инжиниринг георесурсов. 2015. Т. 326. № 10. С. 150.
Alpert N.L., Keiser W.E., Szymanski H.A. IR: Theory and Practice of Infrared Spectroscopy. Springer Science & Business Media, 2012. 381 p.
Pogorelov V., Doroshenko I., Pitsevich G. et al. // J. Mol. Liquids. 2017. V. 235. P. 7. https://doi.org/10.1016/j.molstruc.2004.03
Vasylieva A., Doroshenko I., Vaskivskyi Ye. et al. // J. Mol. Structure. 2018. V. 1167. P. 232. https://doi.org/10.1016/j.molstruc.2018.05.002
Pitsevich G.A., Doroshenko I.Yu., Pogorelov V.E. et al. // American J. Chemistry. 2012. V. 2(4). P. 218.https://doi.org/10.5923/j.chemistry.20120204
Пицевич Г.А., Дорошенко И.Ю., Погорелов В.Е. и др. // Физика низких температур. 2013. Т. 39. № 4. С. 499.
Дополнительные материалы отсутствуют.
Инструменты
Поверхность. Рентгеновские, синхротронные и нейтронные исследования