

Поверхность. Рентгеновские, синхротронные и нейтронные исследования, 2021, № 10, стр. 5-13
Локальное атомное строение и магнитные свойства комплексов Cu(II), Сo(II) и Zn(II) 1-(2-гидроксибензилиденамино)бензимидазолинона-2
И. С. Васильченко a, В. Г. Власенко b, *, Т. А. Кузьменко a, Л. Н. Диваева a, Г. С. Бородкин a, С. И. Левченков c, С. Б. Зайченко a, Е. В. Коршунова a, Б. В. Чальцев a, А. С. Бурлов a, **
a Научно-исследовательский институт физической и органической химии
Южного федерального университета
344090 Ростов-на-Дону, Россия
b Научно-исследовательский институт физики Южного федерального университета
344090 Ростов-на-Дону, Россия
c Южный научный центр РАН
344006 Ростов-на-Дону, Россия
* E-mail: vgvlasenko@sfedu.ru
** E-mail: anatoly.burlov@yandex.ru
Поступила в редакцию 26.01.2021
После доработки 25.02.2021
Принята к публикации 28.02.2021
Аннотация
Синтезированы и исследованы новые азометин 1-(2-гидроксибензилиденамино)бензимидазолинон-2 и комплексы Cu(II), Сo(II) и Zn(II) на его основе. Строение азометина и комплексов металлов установлено по данным элементного анализа, ИК-спектроскопии, спектроскопии ЯМР 1H, рентгеновской спектроскопии поглощения и магнетохимии. Показано, что все комплексы имеют моноядерное строение c общей формулой M(HL)2. Для комплекса меди наблюдается слабый обмен антиферромагнитного типа между ионами меди за счет образования межмолекулярных водородных связей N–H···O=C.
ВВЕДЕНИЕ
Гетероциклические соединения, содержащие бензимидазольный фрагмент, вызывают постоянный научный и практический интерес из-за их важной роли в каталитических и медицинских областях [1–3]. Азометиновые соединения N-аминоазолов и их комплексы металлов обладают широким спектром биологической и хемосенсорной активности [4–8]. Изучение координационной способности оснований Шиффа по отношению к ионам металлов, исследование их спектральных характеристик в последние годы является очень важной областью в разработке зондов молекул ДНК и химиотерапевтических препаратов [9–11], в качестве сенсоров для мониторинга клеточных процессов в живых организмах и флуоресцентных датчиков для отслеживания изменений концентраций ионов металлов [12]. Также производные пиразолов и бензимидазолов играют важную роль в разработке противораковых лекарств, эффективно подавляющих развитие новообразований [13, 14]. Особенно большое значение приобрели производные бензимидазолона при создании различных фармакологических препаратов, являющихся ненуклеозидными ингибиторами обратной транскриптазы ВИЧ-1 [15]. Исследования этих соединений показали, что природа заместителей бензольного кольца бензимидазолонового фрагмента значительно влияет на активность против ВИЧ этого класса сильнодействующих антиретровирусных агентов [16–19].
Наличие в основаниях Шиффа N-аминоазолов нескольких донорных центров делает их перспективными моделями для изучения конкурентной координации металлов и позволяет получать моноядерные и полиядерные комплексы с различным лигандным окружением и геометрией хелатного узла, обладающих интересными магнитными характеристиками [20–22].
Ранее [23, 24] были получены альдимины 1-аминобензимидазолтиона-2 (1) и металлокомплексы 2 (M 2+ = Co, Ni, Cu, Cd) на их основе. Было показано, что в твердом виде и в растворе хлористого метилена исследованные альдимины существуют в тионном виде 1б и образуют со всеми исследованными двухвалентными металлами биядерные комплексы типа 2.
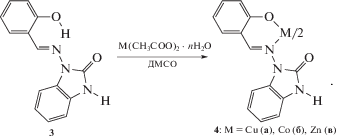
Представляет интерес исследование строения и комплексообразования кислородного аналога азометина 1. В настоящей работе приведены результаты синтеза и исследования строения и свойств новых соединений: 1-(2-гидроксибензилиденамино)бензимидазолинона-2 (3) и его комплексов Cu(II), Сo(II) и Zn(II) (4а–4в).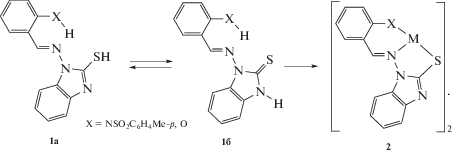
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Исходными компонентами для получения азометина 3 служили 2-гидроксибензальдегид (CAS 90-02-8) промышленного производства и синтезированный по описанным методикам [25, 26] 1-аминобензимидазолон-2.
Спектр ЯМР 1Н раствора лиганда 3 и комплекса цинка 4в в ДМСО-d6 записан на приборе UNITY-300 (Varian) в режиме внутренней стабилизации полярно-резонансной линии 2Н в ДМСО-d6.
Магнетохимические измерения проведены на SQUID-магнетометре MPMS-5S Quantum Design в интервале температур 2–300 К и магнитном поле 5 кЭ. ИК-спектры регистрировали на приборе Varian-Excalibur 3100 FT-IR методом нарушенного полного внутреннего отражения в порошке.
Рентгеновские спектры K-краев поглощения Cu, Co и Zn получены в режиме прохождения на EXAFS-спектрометре в Сибирском синхротронном центре (г. Новосибирск). Энергия электронного пучка, который использовался в качестве источника рентгеновского синхротронного излучения, была 2 ГэВ при среднем токе 80 мА. Рентгеновские спектры поглощения обрабатывали путем стандартных процедур выделения фона, нормирования на величину скачка K-края и выделения атомного поглощения μ0, после чего проводилось фурье-преобразование выделенного EXAFS (χ)-спектра в интервале волновых векторов фотоэлектронов k от 2.5 до 12–13 Å–1 с весовой функцией k3. Полученный модуль фурье-трансформанты EXAFS с точностью до фазового сдвига соответствовал радиальной функции распределения атомов вокруг поглощающего иона металла. Точные значения параметров ближайшего окружения иона металла в исследованных соединениях определяли путем нелинейной подгонки параметров соответствующих координационных сфер при сопоставлении рассчитанного EXAFS и выделенного из полного спектра поглощения методом фурье-фильтрации. Указанную нелинейную подгонку проводили с использованием пакета программ IFFEFIT [27]. Необходимые для построения модельного спектра фазы и амплитуды рассеяния фотоэлектронной волны рассчитывали по программе FEFF7 [28]. В качестве исходных атомных координат, необходимых для расчета фаз и амплитуд рассеяния и дальнейшей подгонки, использовали рентгеноструктурные данные для монокристаллов комплексов металлов с близкой молекулярной структурой из Кембриджской базы данных.
Квантово-химические расчеты проводили в рамках теории функционала плотности с использованием гибридного обменно-корреляционного функционала B3LYP [29, 30] и валентно-расщепленного базиса гауссовых функций, расширенного поляризационными d-функциями на тяжелых атомах 6-311++G(d,p) [31]. Использовалась программа Gaussian’09 [32]. Геометрию молекул оптимизировали без ограничения по симметрии, минимумы поверхности потенциальной энергии характеризовали отсутствием мнимых частот рассчитанных нормальных колебаний. Влияние среды учитывали в рамках модели непрерывной поляризуемой среды [33] с использованием параметров для растворителя (ДМСО), принятых в программе Gaussian’09 по умолчанию.
1-(2-Гидроксибензилиденамино)бенз- имидазолинон-2 (3)
Раствор 1.04 г (7 ммоль) 1-аминобензимидазолона-2 [25, 26] и 0.75 мл (7 ммоль) 2-гидроксибензальдегида в 15 мл ледяной уксусной кислоты кипятили в течение 3 ч. Выделяющийся после охлаждения осадок отфильтровывали, промывали водой и высушивали в вакуумном шкафу. Выход 1.350 г (76%). Получены бесцветные волокнистые кристаллы, температура плавления 261°С (из бутанола). Найдено: С 66.39; H 4.16; N 16.49 мас. %. Для С14Н11N3О2 вычислено: С 66.40; H 4.38; N 16.59 мас. %. Спектр ЯМР 1Н (ДМСО-d6), химический сдвиг δ (м – мультиплет, с – синглет): 6.80–7.80 (8H, м, CAr–H), 9.92 (1Н, c, HC=N), 10.33 (1H, с, NH), 11.21 м.д. (1H, с, ОH). ИК-спектр, ν: 1601 (C=N), 1701 (C=O), 3144 (NH), 3430 см–1 (OH).
Бис[1-(2-гидроксибензилиденамино)бенз- имидазолинонато-2]медь (4а)
К горячему раствору 0.25 г (1 ммоль) имина 3 в 10 мл бутанола добавляли горячий раствор 0.1 г (0.5 ммоль) моногидрата ацетата меди(II) в 10 мл метанола. После кипячения реакционной смеси в течение 2 ч выпавший осадок отфильтровывали, промывали горячим метанолом и сушили в вакуумном шкафу при 100°С. Получен темно-зеленый порошок, температура плавления выше 300°C. Выход 0.322 г (51%). Найдено: С 59.23; H 3.53; N 14.77 мас. %. Для C28H20N6O4Сu вычислено: С 59.20; H 3.55; N 14.79 мас. %. ИК-спектр, ν: 1618 (C=N), 1634 (C=O), 3171 см–1 (NH).
Бис[1-(2-гидроксибензилиденамино)бенз- имидазолинонато-2]кобальт (4б)
Синтезировали по аналогичной методике с использованием тетрагидрата ацетата кобальта(II). Получен оранжевый порошок, температура плавления выше 300°C. Выход 0.240 г (43%). Найдено: С 59.64; H 3.56; N 14.85 мас. %. Для C28H20N6O4Со вычислено: С 59.69; H 3.58; N 14.92 мас. %. ИК-спектр, ν: 1620 (C=N), 1700 (С=О), 3177 см–1 (NH), μэф = 4.85 μВ (294 К).
Бис[1-(2-гидроксибензилиденамино)бенз- имидазолинонато-2]цинк (4в)
Синтезировали по аналогичной методике с использованием дигидрата ацетата цинка(II). Получен бледно-желтый порошок, температура плавления выше 300°C. Выход 0.296 г (52%). Найдено: С 59.67; H 3.56; N 14.89 мас. %. Для C28H20N6O4Zn вычислено: С 59.69; H 3.58; N 14.92 мас. %. Спектр ЯМР 1Н (ДМСО-d6), химический сдвиг δ: 6.62–7.52 (8H, м, CAr–H), 9.62 (1Н, c, HC=N), 10.28 м.д. (1H, с, NH). ИК-спектр, ν: 1620 (C=N), 1662 (ν С=О), 3164 см–1 (ν NH).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В настоящей работе получен 1-салицилидениминобензимидазолон-2 (3), исследованы его строение в растворе ДМСО и твердой фазе, а также комплексообразование с ионами Cu2+, Co2+, Zn2+.
Для азометина 3, так же, как и для его аналога 1, можно ожидать образование нескольких таутомерных форм.
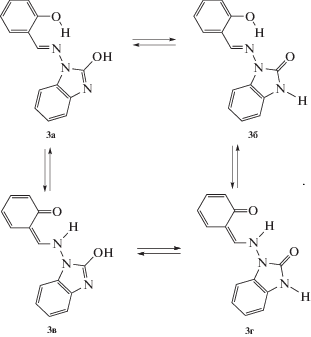
Для определения относительной устойчивости таутомерных форм соединения 3 был проведен квантово-химический расчет методом функционала плотности (табл. 1). На основании полученных расчетных данных о прототропной таутомерии в азометинах 3 установлено, что равновесие осуществляется в основном между енолиминной (3б) и кетоаминной (3г) формами (в растворе ДМСО энергия составляет 4.1 ккал/моль), при котором осуществляется обратимый внутримолекулярный перенос протона, приводящий к перераспределению π-электронов в ароматической части молекулярной структуры соединения. Такой обратимый внутримолекулярный перенос протона, обусловленный электростатическими различиями между атомами кислорода и азота салицилидениминового фрагмента, может осуществляться через таутомерную цвиттерионную форму основания Шиффа. Существование таутомерного равновесия в растворах для салицилальдегидных оснований Шиффа и их производных многократно подтверждено различными экспериментальными методами [34–37]. Также установлено, в том числе и методом рентгеноструктурного анализа, что большинство этих соединений кристаллизуются преимущественно в виде енолиминовых таутомеров [38, 39], хотя существует ряд примеров, подтверждающих наличие кетоаминовой формы в твердом состоянии, сосуществование как енол/кето-таутомеров в кристалле, так и некоторых десмотропных систем [40–43].
Таблица 1.
Рассчитанные значения энергии Е и относительная устойчивость Δ таутомеров 3a–3г в газовой фазе (ГФ) и растворе ДМСО
| Таутомер | E, ат. ед. (ГФ) | Δ, ккал/моль (ГФ) | E, ат. ед. (ДМСО) | Δ, ккал/моль (ДМСО) |
|---|---|---|---|---|
| 3б | –855.0476883 | 0 | –855.0764083 | 0 |
| 3г | –855.0358246 | 7.4 | –855.0699515 | 4.1 |
| 3а | –855.0231294 | 15.4 | –855.0490305 | 17.2 |
| 3в | –855.0123382 | 22.2 | –855.0459433 | 19.1 |
Наличие в ИК-спектре азометина 3 характерной полосы поглощения группы С=О при 1701 см–1, полосы поглощения группы NH бензимидазольного цикла при 3144 см–1, а также полосы поглощения гидроксильной группы при 3430 см–1 позволяет приписать указанному соединению в твердом состоянии фенолиминную бензимидазолоновую таутомерную форму 3б.
По данным спектроскопии ЯМР 1Н сделан вывод о том, что азометиновое производное 1-аминобензимидазолин-2-она в растворе ДМСО также существует исключительно в фенолиминной бензимидазолоновой таутомерной форме 3б. В пользу данного вывода свидетельствует синглетный характер сигнала протона группы НС=N при δH = 9.92 м.д. и наличие сигнала протона группы NH бензимидазольного цикла при δH = = 10.33 м.д. Согласно данным элементного анализа комплексы 4 имеют состав M(HL)2, где H2L – альдимин 1-аминобензимидазолинона-2.
В ИК-спектрах комплексов 4, полученных на основе имина 3, исчезает широкая полоса поглощения ν O–H (3430 см–1) и проявляются полосы поглощения, соответствующие колебаниям групп ν C=O (1634, 1700 и 1662 см–1), ν N–H (3171, 3177 и 3164 см–1) для комплексов меди, кобальта и цинка соответственно.
Величина эффективного магнитного момента для комплекса кобальта 4б составляет μэф = 4.85 μВ (294 К), она характерна для высокоспиновых тетраэдрических комплексов Co(II) и не меняется с понижением температуры, что в совокупности с данными элементного анализа позволяет приписать ему моноядерное строение с составом Co(HL)2.
Согласно данным элементного анализа и ИК-спектроскопии комплекс цинка 4в также имеет аналогичное с Zn(HL)2 моноядерное строение. В спектре ЯМР 1Н этого комплекса исчезает сигнал протона OH-группы лиганда, синглетный сигнал протонов CH=N-групп смещается в сильное поле и проявляется при 9.62 м.д. Сигнал протонов NH бензимидазольного фрагмента лиганда незначительно смещается до 10.28 м.д. В ИК-спектре комплекса 4в исчезают полосы поглощения около 3430 см–1, соответствующие OH-группе лиганда, полоса поглощения νCH=N смещается по сравнению с 3 в высокочастотную область на 19 см–1, а полоса поглощения νС=О смещается в низкочастотную область на 39 см–1. Такое поведение спектроскопических (ИК и ЯМР 1Н) характеристик свидетельствует о депротонировании лиганда 3 и образовании хелатной структуры 4в. Понижение частоты валентных колебаний связи ν C=O в комплексе меди 4а по сравнению с положением этой полосы в спектрах азометина 3 и данные элементного анализа позволяют предположить реализацию в данном соединении моноядерного строения состава Cu(HL)2, аналогичного комплексам кобальта и цинка 4б, 4в.
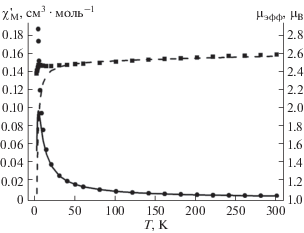
Подтверждением этого вывода является магнетохимическое исследование комплекса 4а в интервале температур 300–30 К. Температурная зависимость магнитной восприимчивости комплекса 4а (рис. 1) хорошо описывается в рамках изотропной модели Гейзенберга–Дирака–Ван Флека [44, 45]: 2J = –4.6 см–1, g = 2.06, среднеквадратичная ошибка R = 0.84%. Близкие значения параметра магнитного обмена были ранее зафиксированы в моноядерных комплексах меди, молекулы которых объединены в димеры, связанные водородными связями, посредством молекул ассоциированного метанола [46, 47]. Ниже 30 К наблюдается существенное отклонение экспериментальных значений от теоретической зависимости; можно предположить, что имеет место фазовый переход второго рода и комплекс является слабым антиферромагнетиком. На основании ИК-спектров и данных магнетохимических измерений можно утверждать, что молекулы комплекса Cu(II) 4а образуют димеры за счет межмолекулярных водородных связей N–H···O=C.
Рис. 1.
Температурная зависимость $\chi _{{\text{M}}}^{{\text{'}}}$ (⚫) и μэф (◼) комплекса 4a; линии соответствуют теоретической зависимости.
Локальное атомное строение комплексов 4а–4в определено по данным рентгеновской спектроскопии K-краев поглощения Cu, Co и Zn: XANES (X-Ray Absorption Near Edge Structure) и EXAFS (Extended X-Ray Absorption Fine Structure). На рис. 2 приведены нормированные спектры XANES и соответствующие модули фурье-трансформанты спектров EXAFS для комплексов 4а–4в, на вставке к рис. 2а показаны также первые производные dμ/dE, где μ – массовый коэффициент поглощения рентгеновского излучения.
Рис. 2.
Нормированные спектры XANES и их первые производные dμ/dE (на вставке) (а) и соответствующие модули фурье-трансформанты спектров EXAFS K-краев поглощения Cu (1), Co (2) и Zn (3) соединений 4a–4в (б): сплошные линии – экспериментальные данные, пустые кружки – теория.
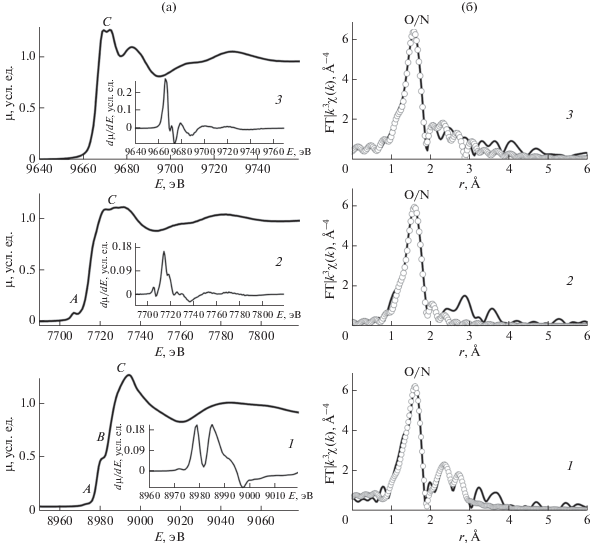
Энергетические и структурные особенности спектров XANES K-края поглощения, включающие пики до ∼10–15 эВ перед краем, непосредственно область края и до ∼50 эВ после края, обусловлены многими факторами: они чувствительны к степени окисления поглощающего атома из-за эффектов экранирования 1s-дырки, химическому составу, количественным характеристикам координационного окружения иона металла и симметрии координационного полиэдра. Предкраевые особенности возникают из-за квадрупольных электронных переходов 1s → 3d атомных орбиталей металла, которые дают очень слабую интенсивность рентгеновских спектров поглощения из-за низкой вероятности перехода. Однако в случае симметрии окружения, отличной от идеально октаэдрической или плоско-квадратной (при отсутствии центра инверсии), эти запрещенные электрические дипольные переходы могут быть интенсивными из-за смешивания атомных 3d–4p-орбиталей металла [48–50].
Спектры XANES комплекса 4а характеризуются наличием малоинтенсивного предкраевого пика А при 8973.6 эВ и интенсивного пика B при 8981.2 эВ непосредственно на CuK-крае поглощения. Наличие такого интенсивного и хорошо разрешимого пика B в спектре XANES характерно для плоско-квадратного координационного центра в комплексах и обусловлено проявлением вакантной атомной орбитали металла pz (ось z перпендикулярна плоскости N2O2). Проявление компоненты B наряду с очень малой интенсивностью предкраевого пика А в рентгеновском CuK-спектре поглощения позволяет заключить, что в комплексе 4а ион меди имеет плоско-квадратное атомное окружение. Этот вывод подтверждает и рассмотрение первой производной dμ/dE спектра XANES этого соединения, которая имеет несколько хорошо разрешимых максимумов, обусловленных расщеплением р-орбиталей меди в поле, образованном плоско-квадратным окружением атомов ближайшей координационной сферы.
Интенсивность предкраевого пика А при 7707.7 эВ в спектре XANES комплекса 4б значительно выше, что указывает на несимметричное окружение иона кобальта, способствующее значительному смешиванию атомных орбиталей 3d–4p металла. Первая производная dμ/dE спектра XANES этого соединения представляет собой широкий максимум, имеющий особенности в виде дополнительного плеча. Такие особенности характерны для тетраэдрических комплексов кобальта(II) [51, 52].
В спектре XANES комплекса 4в отсутствует предкраевой пик в силу заполнения 3d-оболочки Zn(II). Однако положение ZnK-края при ~9667 эВ, определяемого по максимуму dμ/dE, уширение этого максимума и наличие дополнительного высокоэнергетического плеча указывает, что ионы Zn(II) в этом комплексе могут иметь тетраэдрическое окружение.
Такое качественное описание локального строения на основе рассмотрения спектров XANES нашло свое подтверждение при анализе спектров EXAFS K-краев поглощения, который позволил получить количественные характеристики структуры ближайшего атомного окружения ионов Cu(II), Сo(II) и Zn(II) в комплексах 4а–4в. На рис. 2б показаны модули фурье-трансформанты спектров EXAFS K-края поглощения этих соединений. На всех кривых наблюдается основной пик при r = 1.61 Å, обусловленный рассеянием фотоэлектронной волны на ближайшей координационной сфере из атомов азота и кислорода лигандов. Остальные пики соответствуют следующим координационным сферам, содержащим различные атомы лигандов, в основном атомы углерода. В результате произведенных расчетов модельных спектров EXAFS установлено, что ближайшее окружение ионов металлов в комплексе 4а–4в состоит из двух атомов азота и двух атомов кислорода со средними расстояниями Cu–O/N около 1.98 Å, Cо–O/N около 2.01 Å и Zn–O/N около 2.03 Å. Полученные значения факторов Дебая–Валлера типичны для координационной сферы такого радиуса и состава – около 0.0038–0.0045 Å2. Среднее расстояние Cu–O/N близко к соответствующему значению 2.005 Å в комплексе меди 2, определенному по данным рентгеноструктурного анализа: Cu–O(1) 1.899(1) Å, Cu–O(2) 1.939(2) Å, Cu–N(1) 1.970(2) Å, Cu–N(2) 2.212(2) Å [24].
ЗАКЛЮЧЕНИЕ
Таким образом, на основании совокупности экспериментальных данных ИК-спектроскопии, элементного анализа, рентгеновской спектроскопии поглощения и магнетохимии установлено, что комплексы Со(II) и Zn(II) 1-(2-гидроксибензилиденамино)бензимидазолинона-2 в отличие от димерных комплексов на основе альдиминов 1-аминобензимидазолтиона-2 имеют моноядерное строение c общей формулой M(HL)2. Молекулы комплекса Cu(II) образуют димеры за счет образования межмолекулярных водородных связей N–H···O=C, между ионами меди в данном соединении наблюдается слабый обмен антиферромагнитного типа.
Список литературы
Wright J.B. // Chem. Rev. 1951. V. 48. P. 397. https://doi.org./10.1021/cr6015a002
Comprehensive Coordination Chemistry II / Eds. McCleverty J.A., Meyer T.J. Amsterdam: Elsevier, 2004. V. 1. P. 125.
Comprehensive Heterocyclic Chemistry III / Eds. Katritsky A.R., Ramsden C.A., Scriven E.F.V., Taylor R.J.K. Amsterdam: Elsevier, 2008. V. 4. P. 143.
Küçükgüzel Ş.G., Çikla-Süzgün P. // Eur. J. Med. Chem. 2015. V. 97. P. 830. https://doi.org./10.1016/j.ejmech.2014.11.033
Singh K., Barwa M.S., Tyagi P. // Eur. J. Med. Chem. 2006. V. 41. № 1. P. 147. https://doi.org./10.1016/j.ejmech.2005.06.006
Bagihalli G.B., Avaji P.G., Patil S.A., Badami P.S. // Eur. J. Med. Chem. 2008. V. 43. № 12. P. 2639. https://doi.org./10.1016/j.ejmech.2008.02.013
Yu J.-W., Wang Y., Wang Y.-B., Wang C.-F. // J. Chem. Res. 2013. V. 37. № 3. P. 164. https://doi.org./10.3184/174751913X13605940678869
Ko K.C., Wu J.-S., Kim H.J., Kwon P.S., Bartsch R.A., Lee J.Y., Kim J.S. // Chem. Commun. 2011. V. 47. № 11. P. 3165. https://doi.org./10.1039/C0CC05421F
Salahuddin, Shaharyar M., Mazumder A. // Arab. J. Chem. 2017. V. 10. P. S157. https://doi.org/10.1016/j.arabjc.2012.07.017
Sankarganesh M., Dhaveethu Raja J., Sakthikumar K., Vijay Soloman R., Rajesh J., Athimoolam S., Vijayakumar V. // Bioorg. Chem. 2018. V. 81. P. 144. https://doi.org/10.1016/j.bioorg.2018.08.006
Karale B.K., Rindhe S.S., Rode M.A. // Indian J. Pharm. Sci. 2015. V. 77. № 2. P. 230. https://doi.org/10.4103/0250-474X.156619
Yu C., Fu Q., Zhang J. // Sensors. 2014. V. 14. P. 12560. https://doi.org/10.3390/s140712560
Reddy T.S., Kulhari H., Reddy V.G., Bansal V., Kamal A., Shukla R. // Eur. J. Med. Chem. 2015. V. 101. P. 790. https://doi.org./10.1016/j.ejmech.2015.07.031
Qiao X., Ma Z.-Y., Shao J., Bao W.-G., Xu J.-Y., Qiang Z.-Y., Lou J.-S. // Biometals. 2014. V. 27. P. 155. https://doi.org/10.1007/s10534-013-9696-1
Monforte A.-M., Rao A., Logoteta P., Ferro S., Luca L.D., Barreca M.L., Iraci N., Maga G., Clercq E.D., Pannecouque C., Chimirri A. // Bioorg. Med. Chem. 2008. V. 16. P. 7429. https://doi.org./10.1016/j.bmc.2008.06.012
Theberge C.R., Bednar R.A., Bell I.M., Corcoran H.A., Fay J.F., Hershey J.C., Johnston V.K., Kane S.A., Mosser S., Salvatore C.A., Williams T.M., Zartman C.B., Zhang X.F., Graham S.L., Vacca J.P. // Bioorg. Med. Chem. Lett. 2008. V. 18. P. 6122. https://doi.org./10.1016/j.bmcl. 2008.10.019
Wang W., Cao H.P., Wolf S., Camacho-Horvitz M.S., Holak T.A., Dömling A. // Bioorg. Med. Chem. 2013. V. 21. P. 3982. https://doi.org./10.1016/j.bmc.2012.06.020
Omura H., Kawai M., Shima A., Iwata Y., Ito F., Masuda T., Ohta A., Makita N., Omoto K., Sugimoto H., Kikuchi A., Iwata H., Ando K. // Bioorg. Med. Chem. Lett. 2008. V. 18. P. 3310. https://doi.org./10.1016/j.bmcl.2008.04.032
Berry J.F., Ferraris D.V., Duvall B., Hin N., Rais R., Alt J., Thomas A. G., Rojas C., Hashimoto K., Slusher B.S., Tsukamoto T. // ACS Med. Chem. Lett. 2012. V. 3. P. 839. https://doi.org./10.1021/ml300212a
Zhang R., Wang Q., Li Q., Ma C. // Inorg. Chim. Acta. 2009. V. 362. № 8. P. 2762. https://doi.org./1016/j.jca.2008.12.017
Yu H.-X., Ma J.-F., Xu H.-G., Li S.-L., Yang J., Liu Y.-Y., Cheng Y.-X. // J. Organomet. Chem. 2006. V. 691. № 16. P. 3531. https://doi.org/10.1016/j.jorganchem.2006.05.002
Mamedov V.A., Zhukova N.A., Sinyashin O.G. // Mendeleev Commun. 2017. V. 27. P. 1. https://doi.org./10.1016/j.mencom.2017.01.001
Васильченко И.С., Кузьменко Т.А., Шестакова Т.Е., Борисенко Р.Н., Диваева Л.Н., Бурлов А.С., Борисенко Н.И., Уфлянд И.Е., Гарновский А.Д. // Коорд. химия. 2005. Т. 31. № 10. С. 786.
Vasilchenko I.S., Lyssenko K.A., Kuz’menko T.A., Uraev A.I., Garnovskii D.A., Divaeva L.N., Burlov A.S. // Mendeleev Commun. 2015. V. 25. P. 397. https://doi.org./10.1016/j.mencom.2015.09.030
Kornet M. J., Beaven W., Varia T. // J. Heterocyclic Chem. 1985. V. 22. № 4. P. 1089. https://doi.org/10.1002/jhet.5570220433
Пожарский А.Ф., Нанавян И.М., Кузьменко В.В., Чернышев А.И., Орлов Ю.В., Клюев Н.А. // Химия гетероциклических соединений. 1989. № 11. С. 1486.
Newville M. // J. Synchrotron Rad. 2001. № 8. P. 96. https://doi.org/10.1107/S0909049500016290
Zabinsky S.I., Rehr J.J., Ankudinov A., Albers R.C., Eller M.J. // Phys. Rev. B. 1995. V. 52. P. 2995. http://dx.doi.org./10.1103/PhysRevB.52.2995
Lee C., Yang W., Parr R.G. // Phys. Rev. B. 1988. V. 37. P. 785. http://dx.doi.org./10.1103/PhysRevB.37.785
Becke A.D. // J. Chem. Phys. 1993. V. 98. P. 5648. http://dx.doi.org./10.1063/1.464913
Ditchfield R., Hehre W.J., Pople J.A. // J. Chem. Phys. 1971. V. 54. P. 724. http://dx.doi.org./10.1063/1.1674902
Frisch M.J., Trucks G.W., Schlegel H.B. et al. Gaussian 09, Revision A.02. 2009.
Tomasi J., Mennucci B., Cammi R. // Chem. Rev. 2005. V. 105. P. 2999. https://doi.org./10.1021/cr9904009
Минкин В.И., Олехнович Л.П., Жданов Ю.А. Ростов-на-Дону: Изд-во Ростовского ун-та, 1977.
Minkin V.I. // Pure Appl. Chem. 1989. V. 61. P. 661. http://dx.doi.org//10.1351/pac198961040661
Minkin V.I., Garnovskii A.D., Elguero J., Katritzky A.R., Denisko O.V. // Adv. Heterocycl. Chem. 2000. V. 76. P. 157. https://doi.org./10.1016/S0065-2725(00)76005-3
Garnovskii A.D., Nivorozhkin A.L., Minkin V.I. // Coord. Chem. Rev. 1993. V. 126. P. 1. https://doi.org./10.1016/0010-8545(93)85032-Y
Гарновский А.Д., Васильченко И.С. // Успехи химии. 2005. Т. 74. № 3. С. 211.
Власенко В.Г., Бурлов А.С., Кузьменко Т.А., Козаков А.Т., Никольский А.В., Тригуб А.Л., Левченков С.И. // Журн. общей химии. 2018. Т. 88. № 12. С. 2034.
Rubčić M., Užarević K., Halasz I., Bregović N., Mališ M., Đilović I., Kokan Z., Stein R.S., Dinnebier R.E., Tomišić V. // Chem.-Eur. J. 2012. V. 18. P. 5620. https://doi.org./10.1002/chem.201103508
Juribašić M., Bregović N., Stilinović V., Tomišić V., Cindrić M., Šket P., Plavec J., Rubčić M., Užarević K. // Chem.-Eur. J. 2014. V. 20. P. 17333. https://doi.org./10.1002/chem.201403543
Domínguez O., Rodríguez-Molina B., Rodríguez M., Ariza A., Farfán N., Santillan R. // New J. Chem. 2011. V. 35. P. 156. https://doi.org/10.1039/C0NJ00179A
Dominiak P.M., Grech E., Barr G. et al. // Chem.-Eur. J. 2003. V. 9. P. 963. https://doi.org./10.1002/chem.200390118
Kahn O. Molecular Magnetism. N.Y.: VCH Publishers, 1993. 380 p.
Bleaney B., Bowers K.D. // Proc. R. Soc. A. 1952. V. 214. № 1119. P. 451. https://doi.org/10.1098/rspa.1952.0181
Levchenkov S.I., Shcherbakov I.N., Popov L.D., Lukov V.V., Minin V.V., Starikova Z.A., Ivannikova E.V., Tsaturyan A.A., Kogan V.A. // Inorg. Chim. Acta. 2013. V. 405. P. 169. https://doi.org/10.1016/j.ica.2013.05.032
Левченков С.И., Попов Л.Д., Ефимов Н.Н., Минин В.В., Уголкова Е.А., Александров Г.Г., Старикова З.А., Щербаков И.Н., Ионов А.М., Коган В.А. // Журн. неорган. химии. 2015. Т. 60. № 9. С. 1238.
Westre T.E., Kennepohl P., DeWitt J.G., Hedman B., Hodgson K.O., Solomon E.I. // J. Am. Chem. Soc. 1997. V. 119. P. 6297. https://doi.org./10.1021/ja964352a
Leto D.F., Jackson T.A. // Inorg. Chem. 2014. V. 53. P. 6179. https://dx.doi.org./10.1021/ic5006902
Chandrasekaran P., Stieber S.C.E., Collins T.J., Que L., Jr., Neese F., DeBeer S. // Dalton Trans. 2011. V. 40. P. 11070. https://doi.org./10.1039/C1DT11331C
Uchikoshi M., Shinoda K. // Struct. Chem. 2019. V. 30. P. 945. https://doi.org./10.1007/s11224-018-1245-7
Adak S., Hartl M., Daemen L., Fohtung E., Nakotte H. // J. Electr. Spectr. Rel. Phen. 2017. V. 214. P. 8. https://doi.org./10.1016/j.elspec.2016.11.011
Дополнительные материалы отсутствуют.
Инструменты
Поверхность. Рентгеновские, синхротронные и нейтронные исследования