

Поверхность. Рентгеновские, синхротронные и нейтронные исследования, 2021, № 6, стр. 33-38
Локальная атомная и электронная структура нанолистов сульфида железа
М. А. Солдатов a, *, П. В. Медведев a, И. Е. Горбань a, К. Лю b, Ш. Вей b, А. В. Солдатов a
a Международный исследовательский институт интеллектуальных материалов,
Южный федеральный университет
344090 Ростов-на-Дону, Россия
b National Synchrotron Radiation Laboratory, University of Science and Technology of China
230029 Anhui, Hefei, China
* E-mail: mikhail.soldatov@gmail.com
Поступила в редакцию 26.09.2020
После доработки 22.12.2020
Принята к публикации 27.12.2020
Аннотация
Проведены расчеты геометрической и зонной структуры нанолистов сульфида железа (макинавита). Данный материал является одним из представителей нового класса двумерных нанокатализаторов для реакции выделения водорода. Расчеты показывают отсутствие запрещенной зоны для модельных структур кристаллического и наноразмерного макинавита. В модели окисленного наноразмерного макинавита половина атомов серы была замещена атомами кислорода, ширина запрещенной зоны составила 0.37 эВ. Уширение запрещенной зоны может объяснить появление каталитической активности у окисленного наноразмерного макинавита. Спектры рентгеновского поглощения показали высокую чувствительность метода к изменению локального атомного окружения, особенно в первой координационной сфере атомов железа. Рассчитанные модельные спектры позволяют определить особенности локальной атомной и электронной структуры поверхности наноразмерного макинавита в ходе реакции выделения водорода.
ВВЕДЕНИЕ
Глобальное изменение климата какое-то время назад перешло из разряда предостережений ученых [1, 2] в область того, с чем сталкиваются отрасли, не занимающиеся климатическими исследованиями [3]. Климатические изменения связывают с систематическим повышением уровня парниковых газов – углекислого газа, метана, оксидов азота и других. Значительный вклад в выделение парниковых газов вносит энергетика, построенная на базе таких сжигаемых источников энергии, как уголь, нефть и газ. Стоит отметить, что парниковые газы активно выделяются не только в результате работы электростанций, но и при сжигании топлива в двигателях транспортных средств. Таким образом, развитие экологически чистой энергетики является одной из важнейших задач замедления климатических изменений и сохранения окружающей среды. Важной задачей является не только получение энергии из возобновляемых источников, но также ее эффективное преобразование и хранение. Одно из давно развивающихся направлений возобновляемой энергетики – использование солнечной энергии. Комплексное решение этого вопроса предполагает превращение солнечной энергии в химическое топливо для дальнейшего использования в топливных элементах. Благодаря высокой удельной эффективности хранения энергии молекулярный водород является одним из перспективных видов химического топлива. Ячейки для прямого преобразования солнечной энергии в химическую путем реакции расщепления воды существуют на рынке уже долгое время. Однако существующие катализаторы на основе платины обладают высокой стоимостью [4], которая может подняться еще выше при полномасштабном распространении технологии. Для широкого применения технологии требуется решить важные материаловедческие задачи – перейти от платиновых катализаторов к катализаторам на основе распространенных элементов, сохранив стабильность и эффективность платиновых катализаторов.
Заменой платиновых катализаторов могут стать нанообъекты на основе соединений недорогих и распространенных переходных металлов [4]. Ширина их запрещенной зоны может варьироваться в интервале 1–3 эВ [5], что позволяет поглощать основную часть спектра видимого света для эффективной генерации электронно-дырочных пар. Наноструктурированные материалы обладают большой площадью поверхности и могут содержать упорядоченные поры. Такое строение может обеспечить высокую скорость переноса заряда и доступность активных каталитических центров для ионов электролита. Именно это делает наноматериалы на основе соединений переходных металлов отличными кандидатами для накопления энергии [6]. В реакции фотокаталитического выделения водорода ключевым этапом является генерация фотоэлектронов и “дырок”, а также их последующее перемещение к каталитически активной поверхности для восстановления с последующим образованием молекулярного водорода. В силу низкой электрической проводимости и короткой диффузионной длины материала эффективность разделения электронно-дырочных пар может снижаться. Для повышения эффективности катализаторов целесообразно уменьшить их толщину до величины, сравнимой с расстоянием переноса электронно-дырочных пар в материале.
Благодаря низкой цене и распространенности железа сульфид железа обладает высоким потенциалом для применения. Исследования показывают эффективность кристаллического дисульфида железа для катализа реакции выделения водорода [7–11]. Однако по эффективности и стабильности исследованные катализаторы существенно уступают платиновым. Более перспективны наноразмерные катализаторы [12]. Так, наноразмерный катализатор на основе пористого железа и сульфида железа [13] показал стабильность и эффективность, превосходящую платиновые аналоги. Более того, из одного материала на основе пористого железа и сульфида железа можно получать катализаторы, которые показывают высокую эффективность как для реакции выделения водорода, так и для реакции выделения кислорода. Другим подходом, позволяющим получить высокую эффективность, является допирование нанолистов сульфида железа кобальтом [14].
Для детального анализа механизма действия и повышения квантовой эффективности катализаторов на основе наноструктурированного сульфида железа необходимо глубокое понимание фундаментальных взаимосвязей между параметрами его структуры и физико-химическими свойствами. Информацию о зонной структуре, плотности состояний, эффективной массе носителей заряда и периодической структуре могут дать расчеты на основе теории функционала плотности. Часто для исчерпывающего анализа структуры используют методы рентгеновской дифракции, просвечивающей и растровой электронной микроскопии, рентгеновской фотоэлектронной спектроскопии и спектроскопии рентгеновского поглощения. К преимуществам спектроскопии поглощения можно отнести элементную селективность, чувствительность к локальному окружению атомов в структурах без дальнего порядка и возможность получать данные в режиме operando, т.е. в реальных условиях исследуемой каталитической реакции. Данные спектроскопии рентгеновского поглощения, как правило, делятся на две области, которые обрабатывают, используя различные подходы. Анализ спектров EXAFS (Extended X-Ray Absorption Fine Structure – протяженная тонкая структура рентгеновского спектра поглощения) позволяет путем подгонки параметров установить координационные числа и расстояния до нескольких координационных сфер. Теоретический анализ спектров рентгеновского поглощения XANES (X-Ray Absorption Near-Edge Structure – околопороговая структура спектров рентгеновского поглощения) в принципе может дать важную информацию о таких особенностях локальной атомной и электронной структуры материала, как зарядовое состояние, межатомные расстояния и углы между связями. Методы искусственного интеллекта для анализа спектров XANES позволяют получать данные о структуре даже без измерения эталонных соединений [15].
В настоящей работе были рассчитаны зонная структура, плотность состояний и спектры рентгеновского поглощения для модельных структур кристаллического и наноразмерного сульфида железа.
МЕТОДЫ РАСЧЕТА
Геометрическая и зонная структуры макинавита были рассчитаны в рамках теории функционала плотности с использованием программного комплекса BAND [16–20]. Аналитические градиенты энергии для систем с трансляционной инвариантностью сформулированы в рамках теории функционала плотности Кона–Шэма. Энергетические градиенты реализованы в программе BAND, в которой непосредственно используется базисный набор функций Блоха, состоящий из численно рассчитанных атомных орбиталей и орбиталей слейтеровского типа [21].
При разработке методики расчетов были проведены вычисления с использованием обменно-корреляционных функционалов различных типов. В частности, рассматривали приближение обобщенных градиентов (GGA: BLYP [22, 23]) и приближение функционалов группы meta-GGA (SCAN) [24], которые показали хорошее согласие с экспериментальными данным для наноразмерных листов метагидроксида кобальта [25]. В случае приближения обобщенных градиентов использовали поправку на градиенты плотности, а в случае функционалов группы meta-GGA – зависимость от кинетической энергии и отсутствие эмпирических параметров [24]. Базисные наборы состояли из атомных орбиталей DZP (двухэкспонентные атомные орбитали с добавлением поляризационных функций), полученных в ходе численного решения уравнений Кона–Шэма для изолированных сферических атомов, дополненных набором слейтеровских орбиталей.
Спектры XANES были рассчитаны методом конечных разностей, реализованным в программе fdmnes [26–28]. Этот подход позволил избежать приближения muffin-tin и использовать “полный” потенциал системы для расчета спектров XANES. Данный подход хорошо зарекомендовал себя в случае систем с выраженными ковалентными связями или содержащих большие границы раздела – при исследовании пористых материалов на основе металл-органических каркасных структур [29] и метагидроксида кобальта [25].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
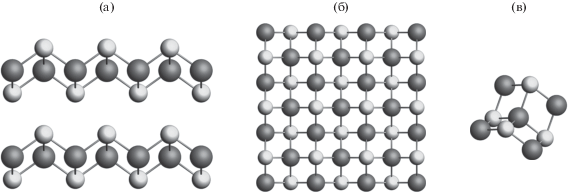
Макинавит (FeS) представляет собой тонкодисперсное, слоистое, нерастворимое в воде соединение. Он образует черные кристаллы тетрагональной сингонии, пространственная группа P4/nmm, параметры ячейки a = 0.36753 нм, c = = 0.50328 нм (база данных COD, № 9011800 [30]). Слоистость структуры наглядна при ее рассмотрении перпендикулярно оси с (рис. 1а). Расстояние между атомами серы двух соседних слоев составляет ~2.41 Å. Первая координационная сфера атома железа состоит из четырех атомов серы (рис. 1б), образующих почти идеальный тетраэдр, межатомные расстояния RFe–S = 2.25(6) Å. Вторая координационная сфера удалена на RFe–S = 2.95(8) Å и состоит из четырех атомов железа (рис. 1в). В случае рассмотрения двумерных нанолистов сульфида железа в воде практически весь материал представляет собой границу раздела между жидкостью и твердым телом. В данном случае существуют различные модели, описывающие активные центры наноструктурированных 2D катализаторов. Стоит отметить, что в чистом виде макинавит не проявляет выраженные каталитические свойства для реакции выделения водорода. Каталитические свойства обусловлены окислением макинавита [13], поэтому ожидается, что реакция выделения водорода будет проходить на атомах кислорода, замещающих атомы серы.
Рис. 1.
Изображение периодической структуры макинавита вдоль (a) и перпендикулярно (б) оси с. Изображение локальной атомной структуры окружения железа (в). Атомы железа – крупные, темно-серые шары, атомы серы – светло-серые шары.
Для расчетов геометрической и зонной структуры были построены три модели: структуры кристаллического макинавита, наноразмерного макинавита и окисленного наноразмерного макинавита. Структура кристаллического макинавита была взята из открытой кристаллографической базы данных [30]. Для создания модельной структуры наноразмерного макинавита была построена суперъячейка на основе кристаллической структуры. Для этого было проведено сечение по плоскости (001) таким образом, чтобы на поверхности находились атомы серы. Суперъячейка состояла из монослоев S–Fe–S, разделенных расстоянием 15 Å. Оптимизация геометрической структуры кристаллического и наноразмерного макинавита не показала существенных изменений. Расчеты запрещенной зоны в рамках теории функционала электронной плотности позволяют говорить об отсутствии запрещенной зоны в кристаллическом и наноразмерном макинавите. Каталитически активные центры предполагают окисление сульфида железа [13], вплоть до достижения соотношения сера : кислород 1 : 1. Поэтому модельная структура окисленного наноразмерного макинавита была получена путем замещения половины атомов серы атомами кислорода. Расчеты в приближении теории функционала плотности показали увеличение ширины запрещенной зоны на 0.37 эВ. Этот результат может объяснить появление каталитической активности у окисленного наноразмерного макинавита. На основании проведенного моделирования были построены структурные модели макинавита.
Для моделирования структуры каталитически активных центров для реакции выделения водорода на базе структуры наноразмерного макинавита были построены три модели, включающие в себя дефекты замещения серы кислородом. Первая модель – замещение атомов серы атомами кислорода в транс-конфигурации, вторая – замещение в цис-конфигурации, третья модель – замещение в цис-конфигурации, в которой на атомах кислорода находятся атомы водорода. Для построенных моделей были вычислены спектры XANES. Сравнение теоретических спектров кристаллического и наноразмерного макинавита приведено на рис. 2. В случае наноразмерного макинавита наблюдается увеличение интенсивности основного максимума (7122 эВ) и сдвиг плеча 7145 эВ в сторону меньших значений энергии. При рассмотрении моделей с дефектами замещения атомов серы атомами кислорода (рис. 3) следует отметить уменьшение интенсивности основного максимума (7122 эВ) и его смещение на 1.5 эВ в область высоких энергий, что согласуется с окислением. Также уменьшается интенсивность максимума при 7145 эВ, что можно объяснить снижением симметрии. Следует обратить внимание на появление предкраевого пика (7117 эВ). Отдельного внимания требует спектр структуры с дефектом замещения двух атомов серы двумя атомами кислорода в цис-конфигурации со связанными атомами водорода. Можно заметить еще большее снижение интенсивности основного максимума (7124 эВ) и интенсивности предкраевого пика (7114 эВ).
Рис. 2.
Результаты теоретического расчета спектра XANES для кристаллического (1) и наноразмерного (2) макинавита.
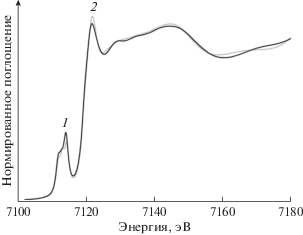
Рис. 3.
Результаты теоретического расчета спектра XANES для модельных структур наноразмерного макинавита (1) и дефектов замещения двух атомов серы атомами кислорода в транс- (2) и цис-конфигурации (3), а также модели, учитывающей атомы водорода, связанные с атомами кислорода в цис-конфигурации (4).

Стоит отметить, что аналогичные работы по теоретическому анализу спектров XANES на границе раздела двумерных наноструктурированных материалов практически отсутствуют. Основным преимуществом теоретически предсказанных спектров рентгеновского поглощения XANES является возможность провести моделирование принципиально новых типов структур, которые могут реализовываться как статически, так и в ходе динамических процессов, в том числе в ходе реакций в реальных технологических условиях (в режиме operando). Данный метод позволяет определять параметры локальной атомной структуры материалов с высокой точностью при сравнении теоретических данных с экспериментом. Данный метод является наиболее быстрым среди имеющихся в мировой практике аналогов, что позволяет использовать его для изучения процессов преобразования локальной атомной структуры материалов в ходе быстропротекающих процессов.
ВЫВОДЫ
В ходе работы проведены расчеты атомной и электронной структур нанолистов сульфида железа, которые являются новыми перспективными двумерными нанокатализаторами для реакции выделения водорода. Установлено расширение запрещенной зоны для модельной структуры активного катализатора на основе макинавита в результате замещения половины атомов серы атомами кислорода по сравнению с наноразмерным макинавитом. Рассчитанные спектры XANES показывают чувствительность к изменению локального окружения атома железа, особенно его первой координационной сферы. Рассчитанные модельные структуры позволят определить особенности локальной атомной и электронной структуры на поверхности макинавита в ходе реакции выделения водорода.
Список литературы
Walther G.R., Post E., Convey P. et al. // Nature. 2002. V. 416. № 6879. P. 389. https://doi.org/10.1038/416389a
Parmesan C. // Annu. Rev. Ecology, Evolution, and Systematics. 2006. V. 37. P. 637.
Lesk C., Rowhani P., Ramankutty N. // Nature. 2016. V. 529. № 7584. P. 84. https://doi.org/10.1038/nature16467
Kang D., Kim T.W., Kubota S.R. et al. // Chem. Rev. 2015. V. 115. № 23. P. 12839. https://doi.org/10.1021/acs.chemrev.5b00498
Huang J.H., Shang Q.C., Huang Y.Y. et al. // Ang. Chem. 2016. V. 55. № 6. P. 2137. https://doi.org/10.1002/anie.201510642
Dhawale D.S., Kim S., Park D.H. et al. // ChemElectroChem. 2015. V. 2. № 4. P. 497. https://doi.org/10.1002/celc.201402365
Miao R., Dutta B., Sahoo S. et al. // J. Am. Chem. Soc. 2017. V. 139. № 39. P. 13604. https://doi.org/10.1021/jacs.7b07044
Faber M.S., Lukowski M.A., Ding Q. et al. // J. Phys. Chem. C. 2014. V. 118. № 37. P. 21347. https://doi.org/10.1021/jp506288w
Giovanni C.D., Reyes-Carmona Á., Coursier A. et al. // ACS Catal. 2016. V. 6. № 4. P. 2626. https://doi.org/10.1021/acscatal.5b02443
Di Giovanni C., Wang W.-A., Nowak S. et al. // ACS Catal. 2014. V. 4. № 2. P. 681. https://doi.org/10.1021/cs4011698
Wu Z., Li X., Liu W. et al. // ACS Catal. 2017. V. 7. № 6. P. 4026. https://doi.org/10.1021/acscatal.7b00466
Heift D. // Inorganics. 2019. V. 7. № 6. P. 75. https://doi.org/10.3390/inorganics7060075
Zou X., Wu Y., Liu Y. et al. // Chem. 2018. V. 4. № 5. P. 1139. https://doi.org/10.1016/j.chempr.2018.02.023
Wang D.Y., Gong M., Chou H.L. et al. // J. Am. Chem. Soc. 2015. V. 137. № 4. P. 1587. https://doi.org/10.1021/ja511572q
Martini A., Guda S.A., Guda A.A. et al. // Comput. Phys. Commun. 2020. V. 250. P. 107064. https://doi.org/10.1016/j.cpc.2019.107064
Franchini M., Philipsen P.H.T., van Lenthe E. et al. // J. Chem. Theory Comput. 2014. V. 10. № 5. P. 1994. https://doi.org/10.1021/ct500172n
Franchini M., Philipsen P.H.T., Visscher L. // J. Comput. Chem. 2013. V. 34. № 21. P. 1819. https://doi.org/10.1002/jcc.23323
Velde G.T., Baerends E.J. // Phys. Rev. B. 1991. V. 44. № 15. P. 7888. https://doi.org/10.1103/PhysRevB.44.7888
Wiesenekker G., Baerends E.J. // J. Phys.: Condes. Matt. 1991. V. 3. № 35. P. 6721. https://doi.org/10.1088/0953-8984/3/35/005
Wiesenekker G., Tevelde G., Baerends E.J. // J. Phys. C. 1988. V. 21. № 23. P. 4263. https://doi.org/10.1088/0022-3719/21/23/012
Kadantsev E.S., Klooster R., De Boeij P.L. et al. // Mol. Phys. 2007. V. 105. № 19–22. P. 2583. https://doi.org/10.1080/00268970701598063
Becke A.D. // Phys. Rev. A. 1988. V. 38. № 6. P. 3098. https://doi.org/10.1103/PhysRevA.38.3098
Lee C.T., Yang W.T., Parr R.G. // Phys. Rev. B. 1988. V. 37. № 2. P. 785. https://doi.org/10.1103/PhysRevB.37.785
Tao J.M., Perdew J.P., Staroverov V.N. et al. // Phys. Rev. Lett. 2003. V. 91. № 14. P. 4. https://doi.org/10.1103/PhysRevLett.91.146401
Soldatov M.A., Medvedev P.V., Wei S. et al. // J. Surf. Invest.: X-Ray, Sychrotron Neutron Tech. 2019. V. 13. № 6. P. 1028. https://doi.org/10.1134/s1027451019060193
Joly Y. // Phys. Rev. B. 2001. V. 63. № 12. P. 125120. https://doi.org/10.1103/PhysRevB.63.125120
Joly Y. // J. Synchr. Rad. 2003. V. 10. P. 58. https://doi.org/10.1107/s0909049502017211
Joly Y., Grenier S. // X-Ray Absorption and X-Ray Emission Spectroscopy: Theory and Applications / Ed. van Bokhoven J.A., Lamberti C. Chichester: John Wiley & Sons, 2016. P. 73.
Guda S.A., Guda A.A., Soldatov M.A. et al. // J. Chem. Theory Comput. 2015. V. 11. № 9. P. 4512. https://doi.org/10.1021/acs.jctc.5b00327
Lennie A.R., Redfern S.A.T., Schofield P.F. et al. // Mineral. Magazine. 1995. V. 59. № 397. P. 677. https://doi.org/10.1180/minmag.1995.059.397.10
Дополнительные материалы отсутствуют.
Инструменты
Поверхность. Рентгеновские, синхротронные и нейтронные исследования