

Поверхность. Рентгеновские, синхротронные и нейтронные исследования, 2021, № 9, стр. 49-54
Поиск аналитических зависимостей между дескрипторами спектров рентгеновского поглощения и локальной атомной структурой вещества на основе машинного обучения
С. А. Гуда a, b, А. С. Алгасов b, А. А. Гуда a, *, А. Мартини a, А. Н. Кравцова a, А. Л. Бугаев a, Л. В. Гуда a, А. В. Солдатов a
a Международный исследовательский институт интеллектуальных материалов,
Южный федеральный университет
344090 Ростов-на-Дону, Россия
b Институт математики, механики и компьютерных наук им. И.И. Воровича,
Южный федеральный университет
344090 Ростов-на-Дону, Россия
* E-mail: guda@sfedu.ru
Поступила в редакцию 19.01.2021
После доработки 18.02.2021
Принята к публикации 25.02.2021
Аннотация
Разработана новая методика количественного анализа ближней области спектров рентгеновского поглощения, основанная на выделении дескрипторов спектра и машинном обучении. Использование дескрипторов (положение края, интенсивности и кривизна минимумов и максимумов, тангенс угла наклона края поглощения) позволяет решить проблему систематических отличий между теоретическими расчетами и экспериментальными данными, уменьшить размерность задачи и тем самым улучшить точность работы алгоритмов машинного обучения. Были получены аналитические зависимости между дескрипторами спектра и параметрами локальной атомной структуры вещества, которые расширяют область применимости эмпирического правила Натоли и анализа химического сдвига спектров на произвольные классы химических соединений.
ВВЕДЕНИЕ
Спектроскопия рентгеновского поглощения является эффективным методом исследования локальной атомной и электронной структуры вокруг поглощающего атома [1, 2]. Область энергии падающих фотонов в диапазоне 200 эВ за краем поглощения содержит информацию о дескрипторах структуры – расстояниях в первой координационной сфере, углах связи, типе ближайших соседей, степени окисления. Дескрипторы структуры влияют на дескрипторы спектра – положение края поглощения, высоту белой линии, положение минимумов и максимумов, расщепление пиков. Специалист в области спектроскопии может легко отличить спектр поглощения оксидов 3d- и 4d-металлов по их форме, а также увидеть характерные особенности плотноупакованных решеток металлов. На этапе становления теоретических моделей расчета спектров поглощения было открыто полуэмпирическое правило Натоли [3], связывающее положения максимумов в спектре поглощения с межатомными расстояниями. Правило химического сдвига, наоборот, связывает степень окисления атома металла со сдвигом края поглощения [4].
В последние несколько лет активное развитие получили алгоритмы машинного обучения для количественного спектрального анализа. Так, авторы [5] использовали модель случайного леса, натренированную на всех точках спектра, для классификации симметрии локального окружения атомов 3d-металла. Сверточная нейронная сеть была применена для оценки координационных чисел в первой координационной сфере атомов меди кластеров оксидов меди [6] и для предсказания первых трех координационных чисел платины платиновых наночастиц с целью определения их формы и размера [7]. Глубокая нейронная сеть может предсказывать функции радиального распределения атомов по спектру рентгеновского поглощения и, наоборот, спектр по заданным дескрипторам структуры [8]. Для оптимизации работы алгоритмов машинного обучения уменьшается размерность подаваемых на вход данных. Например, по 3N атомным координатам для N атомов может быть построена кулоновская матрица, а также рассчитаны обобщенные функции радиального или углового распределения, учитывающие массы атомов [9, 10]. С помощью анализа основных компонент и величины нормы структурные параметры могут быть отсортированы по их влиянию на форму спектров поглощения XANES [11]. Сотни значений коэффициента поглощения, измеренные с малым шагом по энергии, в одном спектре можно сократить до нескольких дескрипторов. Такой подход часто применяется для анализа предкраевой области спектров. Рассчитывают центр масс предкрая и его площадь после вычитания фона, которые исследовали в [12] для анализа степени окисления и координационных чисел. Авторы [13] продемонстрировали, что проекции на главные компоненты обучающей выборки теоретических спектров могут быть использованы для классификации четырех-, пяти- и шестикоординированного окружения атома 3d-металла, а также типа функциональных групп для легких атомов [14]. Недавно в [15] было продемонстрировано построение дескрипторов на основе аппроксимации участков спектра с помощью полиномов разного порядка.
Целью настоящей работы было расширение методики вычисления дескрипторов спектров для тренировки алгоритмов машинного обучения и последующего получения аналитических зависимостей между дескрипторами спектра и структурными параметрами.
МЕТОДЫ
Спектры рентгеновского поглощения рассчитывали в рамках метода конечных разностей и полного потенциала [16], реализованного в программном обеспечении FDMNES [17]. Волновую функцию фотоэлектрона вычисляли на трехмерной сетке точек в сфере радиусом 6 Å вокруг поглощающего атома. Расстояние между точками сетки составляло 0.25 Å. Для учета конечного времени жизни фотоэлектрона теоретические спектры были обработаны с помощью операции свертки для уширения линий. Зависимость ширины ядра лоренциана для свертки аппроксимировали с помощью функции арктангенса.
На рис. 1 показан пример четырехкоординированного кластера для расчета спектра рентгеновского поглощения за K-краем железа (центральный атом). Моделирование проводили для структур силикатов Fe(SiO4)N с координационными числами (КЧ) N = 2–6. Для каждого КЧ варьировали межатомные расстояния d1, d2 в пределах 1.8–2.2 Å и углы связи O–Fe–O α, β в интервале 70°–110°. В пространстве структурных параметров точки для расчета спектров выбирали методом улучшенного латинского гиперкуба (IHS) в количестве 700 штук. Таким образом, для всех КЧ были рассчитаны 3500 спектров рентгеновского поглощения, которые использовали для тренировки алгоритма машинного обучения на основе радиальных базисных функций с линейным ядром. Таким образом, в терминах области машинного обучения, набор рассчитанных спектров составлял обучающую выборку.
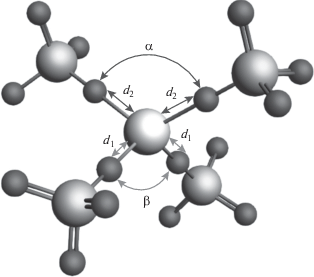
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
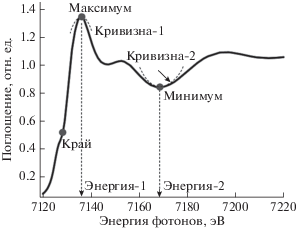
Рассчитанные спектры для КЧ = 4 показаны на рис. 2. Изменение межатомных расстояний приводит как к сдвигу края поглощения, так и к изменению положений минимумов и максимумов в спектре. Как правило, теоретический спектр XANES содержит около 100 энергетических точек. Распространенным подходом к повышению эффективности алгоритмов машинного обучения является уменьшение размерности такого объекта путем извлечения только информативных особенностей, т.е. соответствующих спектральных дескрипторов [15]. На рис. 3 показан набор дескрипторов, рассчитанный для каждого отдельного спектра: положение края поглощения, положение и интенсивность основного максимума, положение и интенсивность главного минимума, кривизна минимумов и максимумов. Для оценки положения края поглощения использована аппроксимация всего спектра функцией arctg, параметры арктангенсоиды тоже использованы в качестве дескрипторов – положение центра арктангенсоиды и тангенса угла наклона в центре. Для устойчивого вычисления дескрипторов была проведена дополнительная “размазка” спектров поглощения на величину 5 эВ перед расчетом кривизны и подгонки арктангенсоидой.
Рис. 2.
Теоретические спектры XANES, рассчитанные в рамках метода конечных разностей за K-краем Fe для кластера Fe(SiO4)4.
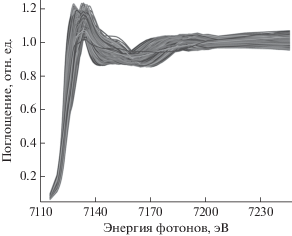
Рис. 3.
Набор используемых дескрипторов для анализа XANES FeK-края кластера Fe(SiO4)4. Точками отмечены участки спектра, в которых производится вычисление дескрипторов.
В начале 80-х годов прошлого столетия Натоли сформулировал эмпирическое правило [3], которое устанавливает зависимость между положениями пиков в спектре XANES и межатомными расстояниями для структур с аналогичной симметрией. Данное правило применимо, например, к металлам, кристаллические решетки которых относятся к одной и той же пространственной группе (например, ОЦК Nb и Mo) или к структурам, которые претерпевают объемное расширение, например, палладий при поглощении водорода [1, 18, 19]. В [20] показан другой пример эмпирического анализа спектров рентгеновского поглощения. Авторы вывели аналитическую зависимость между положениями максимумов в спектрах поглощения за L3-краем уранильных комплексов и расстояниями между ураном и атомами кислорода в первой координационной сфере. Рассмотренные примеры основаны на ограниченном количестве дескрипторов спектра и не могут быть распространены на большее количество структурных параметров. Остается открытым вопрос и об области применимости методики для других комплексов металлов. В настоящей работе описана методология получения аналитических связей между любым набором спектральных дескрипторов и структурных параметров с использованием алгоритма машинного обучения. В общем случае аналитическая зависимость между известными параметрами x1…xn и целевой переменной y определяется с помощью линейной регрессии:
Более сложные зависимости основаны на многочленах более высокого порядка с перекрестным произведением параметров x1…xn. Нас интересуют простые аналитические решения с хорошим качеством аппроксимации. Целевым критерием в данном случае выступает отсутствие больших коэффициентов ${{a}_{i}}$ и минимально возможное количество ненулевых коэффициентов. Для задачи целочисленных отношений такая оптимизация достигается применением специальных алгоритмов ортогонализации (например, [21] или [22]). В случае с рациональными коэффициентами используем свойства алгоритма эластичной сети [23] в сочетании с полуэмпирическими рассуждениями. Для построения аналитических зависимостей ограничимся полиномом второй степени с параметрами x1…xn и их попарными произведениями:
(2)
$y = \mathop \sum \limits_{i\, = \,1}^n {{a}_{i}}{{x}_{i}} + \mathop \sum \limits_{i\, = \,1}^n \mathop \sum \limits_{j\, = \,1}^n {{a}_{{ij}}}{{x}_{i}}{{x}_{j}}.$На первом шаге была проведена нормировка значений всех дескрипторов, с тем чтобы среднее значение по выборке было равно нулю, а стандартные отклонения значений для всех наборов дескрипторов были одинаковыми. Только при таком условии можно сравнивать между собой величины с разной размерностью, например, положение краев поглощения и кривизну минимумов и максимумов. Для аппроксимации был использован метод эластичной сети, который включает алгоритм LASSO [24] и гребневую регрессию. В случае группы сильно коррелированных переменных алгоритм LASSO имеет тенденцию выбирать одну переменную из группы и игнорировать другие, т.е. осуществляется рациональный выбор особенностей. Если линейная формула, возвращаемая эластичной сетью, слишком сложна, можно попытаться упростить ее за счет точности модели. Для этого сортируем коэффициенты (wi, wij), возвращаемые алгоритмом эластичной сети, по их абсолютным значениям и пытаемся построить линейную модель на основе подмножеств функций с наибольшими абсолютными коэффициентами. Анализ проводился для подмножеств каждого размера: 1, 2, 3, …, и для всех них оценивалась величина качества аппроксимации. В табл. 1 представлены выбранные аналитические соотношения между дескрипторами спектров и структурными параметрами некристаллических соединений силикатов железа.
Таблица 1.
Аналитические отношения между дескрипторами спектров и структурными параметрами соединений некристаллических силикатов железа (за оценку качества аппроксимации выбран параметр R2 score)
| № | Дескриптор | Аналитическая формула | Качество аппроксимации |
|---|---|---|---|
| Дескрипторы структуры | |||
| 1 | КЧ (N) | 0.59EdgeE – 0.56Maxcurv | 0.85 |
| 2 | 0.58EdgeE – 0.43Maxcurv + 0.22Maxint | 0.88 | |
| 3 | Среднее расстояние Fe–O, (Dist) | –0.77(MinE – MaxE) − 0.40Maxcurv | 0.88 |
| 4 | –0.29MaxE + 0.54EdgeE – 0.94MinE | 0.93 | |
| 5 | Стандартное отклонение Dist от среднего (Dev) | +0.99EdgeE – 0.19Edgeslope – 1.12Maxcurv – 0.42MaxE + + 0.38Maxint – 0.94Max – Minslope + 0.37Mincurv + 1.49Minint | 0.66 |
| Дескрипторы спектра | |||
| 1 | Edgeslope | −0.26N + 0.36N2 + 0.80Dist – 0.36 | 0.84 |
| 2 | Maxcurv | −0.51Dist – 0.69N | 0.78 |
| 3 | −0.82N – 0.55Dist + 0.35Dev | 0.88 | |
| 4 | MinE | 0.34N – 0.90Dist | 0.89 |
| 5 | Minint | −0.96N + 0.63Dev | 0.84 |
Примечание. Обозначение “Dist” используется для среднего расстояния Fe–O в первой координационной оболочке. “Dev” используется для стандартного отклонения расстояний Fe–O от среднего, параметр, который измеряет беспорядок в первой координационной сфере. EdgeE – положение края поглощения, Edgeslope – тангенс угла наклона края поглощения, Maxcurv – кривизна спектра в точке основного максимума, MinE – энергия основного минимума, Minint – интенсивность спектра в точке минимума. Перед построением аналитических зависимостей дескрипторы обучающей выборки были нормированы по нулевому среднему и единичному стандартному отклонению.
Аналитические соотношения между дескрипторами могут быть получены для любого количества спектральных характеристик и структурных параметров. Хотя в целом алгоритмы машинного обучения работают как черный ящик, в таблице дана наглядная интерпретация лучших комбинаций дескрипторов. Так, качество предсказания до 93% может быть достигнуто для межатомных расстояний, если учитывать энергетические положения края, первого максимума и минимума. Важно отметить, что, в отличие от эмпирического правила Натоли, оценка точности, приводимая в настоящей работе, распространяется на все структуры с КЧ = 2–6 и большими вариациями структурным параметров.
Интенсивность основного максимума изменяется вместе с сокращением длины связей Fe–O, поэтому данный дескриптор может помочь различить сдвиги, связанные со степенью окисления или изменениями объема. Формулы для КЧ зависят от кривизны главного максимума, что согласуется с общим поведением EXAFS-колебаний, амплитуда которых пропорциональна КЧ. Как показывают зависимости в таблице, для надежной оценки КЧ следует использовать и положение края поглощения.
Также было смоделировано изменение степени окисления атома железа путем применения энергетического сдвига к каждому спектру выборки. Для классификации спектров по зарядовому состоянию первоочередное значение играет положение края поглощения. Однако использование только одного этого дескриптора обеспечивает точность предсказания хуже 60%. Химический сдвиг всего спектра может быть неверно истолкован из-за сдвига края при изменении расстояний. Этот эффект частично компенсируется при учете дескриптора интенсивности главного максимума (Maxint), который в совокупности с положением края поглощения приводит к правильной классификации в 75% случаев. Дальнейшее повышение точности предсказания возможно при применении ограничений на диапазон возможных расстояний в структурах Fe2+ и Fe3+.
Вторая часть таблицы (дескрипторы спектра) отражает обратную зависимость особенностей спектра XANES, если рассматривать геометрические параметры. Так, тангенс угла наклона края поглощения зависит от средних расстояний и координационного числа. Кривизна белой линии коррелирует с беспорядком в первой координационной сфере железа (Dev). Больший разброс расстояний приводит к уширению главного максимума. Положение первого минимума (MinE) – довольно важная характеристика в спектре, хотя его реже анализируют по сравнению с положениями максимумов. Эта особенность почти на 90% обусловлена КЧ и расстоянием Fe–О. Его интенсивность определяется КЧ и разбросом длин связей в первой координационной сфере.
ЗАКЛЮЧЕНИЕ
В настоящей работе рассмотрен новый подход количественного анализа спектров рентгеновского поглощения с помощью алгоритмов машинного обучения. Для функции μ(E) выделены ключевые особенности, а именно энергетическое положение края, минимумов, максимумов, интенсивность главного максимума и минимума, кривизна функции в экстремумах, угол наклона края поглощения. С помощью алгоритма радиальных базисных функций были выбраны такие комбинации дескрипторов, которые обеспечивают наилучшую точность предсказания структурных параметров вокруг поглощающего атома. Обычно алгоритм машинного обучения работает для исследователей как “черный ящик”. Показана работа универсального метода для построения аналитических соотношений между дескрипторами спектра и структурными параметрами, такими как координационные числа, межатомные расстояния, степень окисления и стандартные отклонения межатомных расстояний.
Список литературы
Bugaev A.L., Guda A.A., Lomachenko K.A., Srabionyan V.V., Bugaev L.A., Soldatov A.V., C. Lamberti, Dmitriev V.P., van Bokhoven J.A. // J. Phys. Chem. C. 2014. V. 118. P. 10416.https://doi.org/10.1021/jp500734p
Van Bokhoven J.A., Lamberti C. X-ray Absorption and X-Ray Emission Spectroscopy: Theory and Applications. John Wiley & Sons, 2016.
Natoli C.R. Distance Dependence of Continuum and Bound State of Excitonic Resonances in X-Ray Absorption Near Edge Structure (XANES), in EXAFS and Near Edge Structure III / Ed. Hodgson K.O., Hedman B., Penner-Hahn J.E. Berlin: Springer, 1984. Springer Proc. Phys. V. 2. P. 38–42.
García J., Subías G., Blasco J. XAS Studies on Mixed Valence Oxides, in X-Ray Absorption and X-Ray Emission Spectroscopy: Theory and Applications / Ed. van Bokhoven J.A., Lamberti C. Chichester: John Wiley & Sons, 2016. P. 459–484.
Zheng C., Chen C., Chen Y., Ong S.P. // Patterns. 2020. V. 1. 100013.https://doi.org/10.1016/j.patter.2020.100013
Liu Y., Marcella N., Timoshenko J., Halder A., Yang B., Kolipaka L., Pellin M.J., Seifert S., Vajda S., Liu P., Frenkel A.I. // J. Chem. Phys. 2019. V. 151. 164201.https://doi.org/10.1063/1.5126597
Timoshenko J., Lu D.Y., Lin Y.W., Frenkel A.I. // J. Phys. Chem. Lett. 2017. V. 8. P. 5091.https://doi.org/10.1021/acs.jpclett.7b02364
Rankine C.D., Madkhali M.M.M., Penfold T.J. // The J. Physical Chemistry A. 2020. V. 124. P. 4263.https://doi.org/10.1021/acs.jpca.0c03723
Martini A., Guda S.A., Guda A.A., Smolentsev G., Algasov A., Usoltsev O., Soldatov M.A., Bugaev A., Rusalev Y., Lamberti C., Soldatov A.V. // Comput. Phys. Commun. 2019. V. 250. P. 107064.https://doi.org/10.1016/j.cpc.2019.107064
Schmidt J., Marques M.R.G., Botti S., Marques M.A.L. // npj Comput. Mater. 2019. V. 5. P. 83. https://doi.org/10.1038/s41524-019-0221-0
Trejo O., Dadlani A.L., De La Paz F., Acharya S., Kravec R., Nordlund D., Sarangi R., Prinz F.B., Torgersen J., Dasgupta N.P. // Chem. Mater. 2019. V. 31. P. 8937.https://doi.org/10.1021/acs.chemmater.9b03025
Wilke M., Farges F., Petit P.E., Brown G.E., Martin F. // Am. Mineral. 2001. V. 86. P. 714.https://doi.org/10.2138/am-2001-5-612
Carbone M.R., Yoo S., Topsakal M., Lu D.Y. // Phys. Rev. Mater. 2019. V. 3. P. 033604.https://doi.org/10.1103/PhysRevMaterials.3.033604
Carbone M.R., Topsakal M., Lu D.Y., Yoo S. // Phys. Rev. Lett. 2020. V. 124. P. 156401.https://doi.org/10.1103/PhysRevLett.124.156401
Torrisi S.B., Carbone M.R., Rohr B.A., Montoya J.H., Ha Y., Yano J., Suram S.K., Hung L. // npj Comput. Mater. 2020. V. 6. P. 109. https://doi.org/10.1038/s41524-020-00376-6
Joly Y. // Phys. Rev. B. 2001. V. 63. P. 125120.https://doi.org/10.1103/PhysRevB.63.125120
Guda S.A., Guda A.A., Soldatov M.A., Lomachenko K.A., Bugaev A.L., Lamberti C., Gawelda W., Bressler C., Smolentsev G., Soldatov A.V., Joly Y. // J. Chem. Theory Comput. 2015. V. 11. P. 4512.https://doi.org/10.1021/acs.jctc.5b00327
Bugaev A.L., Srabionyan V.V., Soldatov A.V., Bugaev L.A., van Bokhoven J.A. // J. Phys.: Conf. Ser. 2013. V. 430. P. 012028.https://doi.org/10.1088/1742-6596/430/1/012028
Bugaev A.L., Guda A.A., Lomachenko K.A., Lazzarini A., Srabionyan V.V., Vitillo J.G., Piovano A., Groppo E., Bugaev L.A., Soldatov A.V., Dmitriev V.P., Pellegrini R., van Bokhoven J.A., Lamberti C. // J. Phys.: Conf. Ser. 2016. V. 712. P. 012032.https://doi.org/10.1088/1742-6596/712/1/012032
Zhang L.J., Zhou J., Zhang J.Y., Su J., Zhang S., Chen N., Jia Y.P., Li J., Wang Y., Wang J.Q. // J. Synchr. Rad. 2016. V. 23. P. 758.https://doi.org/10.1107/S1600577516001910
Bailey D.H. // Comput. Sci. Eng. 2000. V. 2. P. 24.https://doi.org/10.1109/5992.814653
Bailey D.H., Borwein J., Calkin N., Luke R., Girgensohn R., Moll V. Experimental Mathematics in Action. CRC Press, 2007.
Zou H., Hastie T. // J. Royal Stat. Soc. B. 2005. V. 67. P. 301.https://doi.org/10.1111/J.1467-9868.2005.00503.X
Tibshirani R. // J. Royal Stat. Soc. B. 1996. V. 58. P. 267.https://doi.org/10.1111/j.2517-6161.1996.tb02080.x
Дополнительные материалы отсутствуют.
Инструменты
Поверхность. Рентгеновские, синхротронные и нейтронные исследования