

Поверхность. Рентгеновские, синхротронные и нейтронные исследования, 2022, № 10, стр. 93-101
Применение методов машинного обучения для аппроксимации энергии взаимодействия молекул со с поверхностью наночастиц Pd
А. А. Терещенко a, *, Д. М. Пашков a, b, А. А. Гуда a, **, С. А. Гуда a, b, Ю. В. Русалев a, А. В. Солдатов a
a Международный исследовательский институт интеллектуальных материалов,
Южный Федеральный университет
344090 Ростов-на-Дону, Россия
b Институт математики, механики и компьютерных наук им. И.И. Воровича,
Южный Федеральный университет
344058 Ростов-на-Дону, Россия
* E-mail: tereshch1@gmail.com
** E-mail: guda@sfedu.ru
Поступила в редакцию 29.01.2022
После доработки 07.03.2022
Принята к публикации 10.03.2022
- EDN: SKUTYC
- DOI: 10.31857/S1028096022100144
Аннотация
Исследована применимость методов машинного обучения для аппроксимации энергии связи адсорбатов монооксида углерода на наночастице палладия. Тренировку алгоритмов машинного обучения осуществляли с использованием выборки из структур, представляющих собой модели взаимодействия молекулы СО с разными участками нанокластера Pd55 с варьируемым расстоянием от молекулы до поверхности, для которых методами теории функционала плотности была рассчитана энергия. Для структур из этой выборки были рассчитаны функции радиального распределения атомов палладия относительно атома углерода молекулы СО. Используя эти функции и их участки в качестве дескрипторов, была проверена эффективность различных алгоритмов машинного обучения таких как “градиентный бустинг”, “гребневая регрессия”, “экстремально случайные деревья” и метод опорных векторов для расчета энергии связи. На основании трех различных метрик установлено, что погрешность определения энергии связи была наименьшей при использовании метода опорных векторов: среднее абсолютное отклонение составило 0.093 эВ. Приведено сравнение эффективности использования различных отдельных участков функции распределения в качестве дескрипторов. Установлено, что для корректной аппроксимации энергии наиболее критичен учет участка функции радиального распределения от 1.5 до 2.5 Å.
ВВЕДЕНИЕ
Наночастицы благородных металлов, в частности палладия – известные катализаторы множества химических реакций окисления [1] и восстановления [2]. Их каталитическая активность, помимо размера частиц [3] и материала подложи [4], также во многом определяется и формой наночастиц [5–8].
Одной из методик, позволяющих оценить доступность каталитически активных центров и изучить морфологию поверхности наночастиц, является инфракрасная спектроскопия адсорбированных молекул [9]. С помощью данной методики зондирующие молекулы, например, монооксида углерода, осаждают на поверхности катализатора и по инфракрасным спектрам, измеренным в ходе этого процесса, проводят оценку морфологии поверхности наночастиц. Это возможно благодаря тому, что энергия связи молекул СО с атомами катализатора в значительной мере определяет частоту колебаний адсорбированных молекул [10]. Например, для наночастиц палладия на инфракрасных спектрах можно различить линии, соответствующие колебаниям молекул СО, адсорбированных на дефектах наночастиц. Такими дефектами могут быть вершины и края кластеров, протяженные грани Pd(100) и Pd(111). Кроме того, по спектрам можно отличать друг от друга молекулы, связанные с одним, двумя или тремя атомами палладия [11]. Далее, по отношению площадей под соответствующими полосами поглощения возможно проведение как качественной [12, 13], так и количественной оценки размера наночастиц Pd [14].
Однако влияние температуры, давления газа или концентрации зондирующих молекул в общем потоке газа, проходящем через образец (в случае проведении эксперимента в проточной системе), геометрии эксперимента, инструментальных и иных экспериментальных факторов в сочетании с человеческим фактором осложняет анализ. Для дальнейшего развития описанной выше методики спектрального анализа адсорбции зондирующих молекул требуется разработка однозначной, быстрой и максимально автоматизированной процедуры извлечения информации о поверхности наночастиц из инфракрасных спектров адсорбированных молекул.
В качестве первого шага для развития такой процедуры необходимо c высокой точностью рассчитывать энергию связи адсорбатов и только потом определять частоты и интенсивности колебаний на энергетически выгодных центрах адсорбции. Обычно для такой задачи применяют времязатратные и требующие значительных ресурсов вычисления с помощью теории функционалов плотности [15]. Для наночастиц большого размера или наночастиц на подложках необходим поиск новых способов надежного и быстрого определения энергии адсорбции.
Машинное обучение уже продемонстрировало высокий потенциал в предсказании спектров рентгеновского поглощения [16] и их анализе, в частности, в предсказании координационных чисел атомов [17], функций радиального распределения [18] монометаллических наночастиц и реконструкции структуры биметаллических наночастиц с атомным разрешением [19]. Однако рентгеновская спектроскопия – это “объемный” метод, зачастую не чувствительный к процессам, происходящим на поверхности.
Ранее было показано, что для извлечения информации о микроструктуре наночастиц из инфракрасных спектров можно использовать полиномиальную регрессию с помощью нейросетевых ансамблей [20]. Авторы собрали выборку из теоретически рассчитанных (из первых принципов) спектров молекул СО и NО, адсорбированных на платиновых нанокатализаторах, и определили для них функции распределения вероятностей обобщенных координационных чисел. Чтобы установить соответствие спектров адсорбируемых молекул с микроструктурой наночастиц, на которых изучают адсорбцию, использовали дескрипторы – идентификаторы конкретных центров адсорбции, которые выделяют их из множества остальных. Дескрипторы должны обеспечивать инвариантность нахождения соответствия между спектром и центром адсорбции. В работе были выбраны частоты и интенсивности полос поглощения CO и Pt–C на ИК-спектрах в качестве дескрипторов микроструктуры, зависящих от типов связывания адсорбатов (на одном, двух, трех или четырех атомах платины) и обобщенных координационных чисел. [21] Таким образом, натренировав алгоритмы машинного обучения на выборке инфракрасных спектров для одиночных молекул CO и NO, адсорбированных на разных участках частицы платины, авторы смогли предсказывать для экспериментально измеренных спектров центры, где происходит адсорбция, и функции распределения обобщенных координационных чисел, тем самым устанавливая координацию поверхности.
Применение алгоритма “регрессии градиентного бустинга” [22] позволяет осуществить предсказания величины энергии адсорбции на различных участках наночастиц платины различного размера (0.2–1.5 нм). [23] Для данной цели авторами было использовано сочетание как структурных (обобщенные координационные числа центров адсорбции, размер кластера, длины связей), так и энергетических дескрипторов. В качестве последнего, в частности, выступало положение центра d-полосы. Это часто используемый дескриптор при изучении адсорбции молекул на переходных металлах в силу того, что взаимодействие происходит между валентными электронами адсорбата и s- и d-электронами поверхности переходного металла. Также авторы предложили использование в качестве дескриптора энергию “полностью замороженной структуры”, т.е. энергию, рассчитанную для случая, когда все атомы системы – кластера Pt и CO – неподвижны. Такой дескриптор является “недорогой” в вычислительном отношении величиной. Выбранный в работе подход позволил определить энергии связи адсорбатов с точностью, сопоставимой с результатами расчетов методами теории функционалов плотности.
В настоящей работе использовали функцию радиального распределения атомов палладия относительно атома углерода молекулы СО и ее участки как основной дескриптор для определения энергии адсорбции. Тренировку алгоритмов машинного обучения проводили на основе выборки энергий, рассчитанных с помощью теории функционалов плотности для молекул СО, находящихся на разном расстоянии от поверхности наночастицы Pd. Проверена эффективность различных алгоритмов машинного обучения для задачи аппроксимации энергии связи, таких как “градиентный бустинг”, “гребневая регрессия”, “экстремально случайные деревья” и метод опорных векторов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Составление тренировочной выборки
Оптимизация геометрии и расчет энергий связи адсорбированных молекул были проведены с помощью программного комплекса VASP 5.3.5 (Vienna Ab initio simulation package) [24, 25] методом псевдопотенциала для периодических структур. Расчеты проводили в прямом пространстве, используя разложение волновой функции по плоским волнам в рамках теории функционала плотности. В расчетах применяли обобщенное градиентное приближение [26] и обменно-корреляционный функционал Пердью–Берка–Эрнзерхофа [27].
Была осуществлена оптимизация позиций атомов структуры нанокластера Pd55, помещенного в центр ячейки размером 30 × 30 × 30 Å. Далее был проведен расчет энергии при моделировании взаимодействия молекулы СО с различными участками поверхности нанокластера Pd55 (рис. 1) в зависимости от расстояний Pd–C в диапазоне 0.5–5 Å с шагом 0.1 Å, от наибольших расстояний с последующим приближения к поверхности. Положение всех атомов при расчетах на каждом отдельном шаге было зафиксировано, причем межатомное расстояние С–О составляло всегда 1.128 Å. Также был проведен расчет энергии при перемещении молекулы СО вдоль поверхностей Pd(100) и Pd(111). Таким образом, был составлен набор структур, каждая из которых представляет собой один и тот же нанокластер Pd55 и одиночную молекулу СО, с их различным взаимным расположением. Из полученного набора энергий был проведен расчет энергии связи Pd–CO, с использованием формулы:
(1)
${{E}_{{{\text{связи}}}}} = {{E}_{{{\text{Pd}}\,\,{\kern 1pt} + \,\,{\kern 1pt} {\text{CO}}}}} - \left( {{{E}_{{{\text{Pd}}}}} + {{E}_{{{\text{CO}}}}}} \right).$Рис. 1.
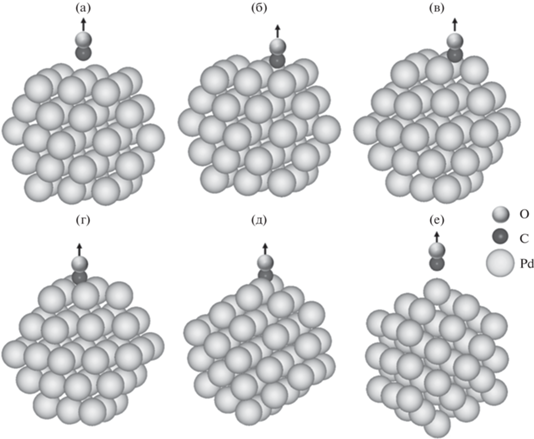
Конфигурации, используемые для расчета потенциальной энергии при удалении молекулы СО от выбранных участков кластера Pd55: СО адсорбирован (а) на одном атоме грани Pd(100); (б) на двух атомах грани Pd(100); (в) на трех атомах грани Pd(111); (г) на двух атомах грани Pd(111); (д) на двух атомах на краю на стыке поверхностей Pd(111) и Pd(100); (е) на одном атоме на вершине.
Далее для каждой структуры из вышеуказанного набора был проведен расчет функций радиального распределения. Эти функции, известные также как парные корреляционные функции, нужны для описания изменения плотности частиц в системе в зависимости от расстояния от эталонной частицы (в нашем случае атомов палладия относительно атома углерода) и рассчитываются по формуле:
(2)
$g\left( r \right) = \frac{V}{{4\pi {{r}^{2}}{{N}^{2}}}}\sum\limits_{i\,{\kern 1pt} = \,{\kern 1pt} 1}^N {\frac{{\Delta {{n}_{i}}\left( r \right)}}{{\Delta r}}} ,$Расчет производили с использованием пакета Pymatgen для Python, в диапазоне значений r от 0.00 до 7.00 Å с шагом 0.01 Å. После была проведена свертка рассчитанных функций с использованием Гауссовой функции и параметром размытия, влияющим на уширение пиков функций радиального распределения, равным 0.1 (значение по умолчанию в Pymatgen). Выборка итоговых функций радиального распределения показана на рис. 2.
Рис. 2.
Пример функций радиального распределения, рассчитанных для структур из выборки. Оттенками серого показана энергия связи СО (в эВ), рассчитанная для этих структур. Индикация величины энергии меняется от черного к белому при ее увеличении.
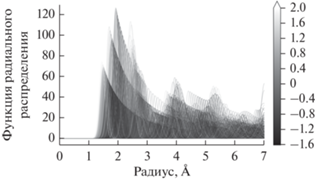
Таким образом, была собрана выборка из более чем 1000 конфигураций нанокластеров Pd55 с разным положением СО относительно кластера, для каждой из которых известна энергия связи и построена функция радиального распределения. После того как из полученной выборки были исключены значения энергии, соответствующие сильному отталкиванию (энергия связи положительна и превышает 2 эВ), тренировочная выборка была представлена набором из 878 структур.
Тренировка алгоритмов машинного обучения и оценка качества предсказаний
Задача аппроксимации энергии по функциям радиального распределения является в терминах машинного обучения типичной задачей регрессии. В качестве признаков каждого объекта используют значения функции радиального распределения атомов для каждого значения радиуса в заданном интервале от 0.10 до 7.00 Å с шагом 0.01 Å. Таким образом, каждый объект обучающей выборки был представлен 690 значениями. Целевыми значениями являлись энергии, распределенные в диапазоне от –2.64 до 1.97 эВ. Предсказание энергии связи по функциям радиального распределения проводили двумя способами: по всем значениям функции или по ее отдельным отрезкам.
Для решения задачи были применены 4 разных метода машинного обучения, среди них ансамблевые методы: “экстремально случайные деревья” и “градиентный бустинг”, метод опорных векторов, а также линейная модель – регрессия с регуляризацией (“гребневая регрессия”).
Обучение и тестирование моделей машинного обучения проводили с помощью метода перекрестной проверки (известного как “cross-validation”), в ходе которого вся обучающая выборка делится случайным образом на k частей, (k – 1) из которых используются для обучения модели, а оставшаяся для тестирования. Данный процесс повторяется k раз, благодаря чему каждая из частей обучающей выборки в свою очередь будет использована для тестирования. Такая процедура тестирования позволяет наиболее объективно оценить качество предсказаний. Процедуру перекрестной проверки проводили с разделением выборки на 10 частей с предварительным перемешиванием, так что для обучения модели и тестирования использовали структуры из выборки со случайным взаимным расположением СО и нанокластера Pd55 (в случайном порядке).
Оценку качества предсказаний проводили с помощью трех метрик: коэффициента детерминации R 2, средней абсолютной ошибки (MAE) и среднеквадратичной ошибки (MSE):
(3)
${{R}^{2}} = 1 - \frac{{\sum\limits_{i\,{\kern 1pt} = \,\,{\kern 1pt} 1}^N {{{{\left\| {{{E}_{i}} - \widehat {{{E}_{i}}}} \right\|}}^{2}}} }}{{\sum\limits_{i\,{\kern 1pt} = \,\,{\kern 1pt} 1}^N {{{{\left\| {{{E}_{i}} - \left\langle E \right\rangle } \right\|}}^{2}}} }},$(4)
$MAE\left( {E,\hat {E}} \right) = \frac{1}{N}\sum\limits_{i\,{\kern 1pt} = \,{\kern 1pt} 1}^N {\left| {{{E}_{i}} - \widehat {{{E}_{i}}}} \right|} ,$(5)
$MSE\left( {E,\hat {E}} \right) = \frac{1}{N}\sum\limits_{i{\kern 1pt} \, = \,{\kern 1pt} 1}^N {{{{\left( {{{E}_{i}} - \widehat {{{E}_{i}}}} \right)}}^{2}}} ,$РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Среди расчетов, проведенных для составления тренировочной выборки, можно выделить шесть серий, соответствующих процессу адсорбции молекулы СО в зависимости от того, к какому участку поверхности кластера происходит ее приближение. Так можно выделить модели линейных карбонилов, образуемых при связи СО с одним атомом на поверхности Pd(100) или одним атомом на вершине наночастицы, а также модели различных мостиковых карбонилов: при связи с двумя атомами на поверхности Pd(100); двумя атомами на поверхности Pd(111); двумя атомами на стыке поверхностей Pd(111) и Pd(100); тремя атомами на поверхности Pd(111).
Согласно этим сериям расчетов (рис. 3), сильнее всего молекула СО связана с участками, состоящими из трех атомов на поверхности Pd(111) (энергия связи равна –2.64 эВ), затем идут позиции, где молекула СО связана с двумя атомами Pd (от –2.40 эВ для краевых позиций до –2.28 эВ на Pd(100)). Наименее прочная связь молекулы СО обнаружена при взаимодействии с одиночными атомами палладия. В частности, при адсорбции на атоме, расположенном на вершине кластера, энергия связи составляет –1.85 эВ, а на атоме на поверхности Pd(100) энергия связи равна –1.74 эВ. Полученные данные согласуются с результатами, описанными в литературе [10].
Рис. 3.
Кривые потенциальной энергии, рассчитанные при удалении молекул СО от участков на поверхности кластера Pd55: 1 – на одном атоме грани Pd(100); 2 – на двух атомах грани Pd(100); 3 – на трех атомах грани Pd(111); 4 – на двух атомах грани Pd(111); 5 – на двух атомах на краю наночастицы; 6 – на одном атоме на вершине. Расстояние Pd–C рассчитывали от атома углерода до ближайшего к нему атома палладия.
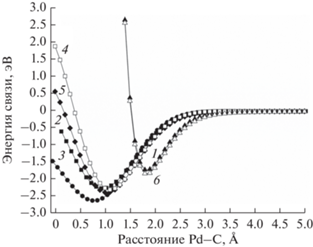
Известно, что относительно частоты колебаний свободных молекул СО наименьший сдвиг испытывают линейно-адсорбированные молекулы, сильнее сдвинуты частоты колебаний молекул СО, адсорбированных на двух атомах палладия, и наибольший сдвиг характерен адсорбатам на трех атомах Pd [14, 28]. Таким образом, чем сильнее связь адсорбат–адсорбент, тем сильнее величина сдвига частоты колебания. Для иллюстрации данного вывода, проведено измерение ИК-спектра поглощения молекул СО, адсорбированных на коммерческом катализаторе Pd/Al2O3 (Chimet SpA) со средним размером наночастиц 2.5 ± 0.5 нм. Пробоподготовка, измерение и анализ данных осуществляли по методике, описаной ранее [9] с единственным отличием: напуск молекул СО осуществляли при комнатной температуре. Полученный спектр приведен на рис. 4.
Рис. 4.
Инфракрасный спектр поглощения Pd/Al2O3, измеренный после адсорбции CO при комнатной температуре (полужирная сплошная линия). Тонкими линиями показаны функции Гаусса, с помощью которых осуществлялась аппроксимация спектра, результирующий спектр показан штриховой линией. Вертикальные серые линии показывают положение центров тяжести пиков. Центру полосы поглощения СО в газовой фазе соответствует значение волнового числа 2125 см–1.

Центр полосы поглощения СО в газовой фазе был расположен на 2143 см–1. Наиболее близкий пик (2125 см–1) соответствовал колебаниям молекул СО, адсорбированных на ионах Pd+. Далее были обнаружены пики, связанные с линейной адсорбцией на Pd(100) (2090 см–1) и атомах-вершинах кластера (2070 см–1). Сильнее сдвинута относительно полосы поглощения газообразного СО группа полос, связанная с адсорбцией на 2, 3 и 4 атомах палладия. Среди них были выявлены пики, отнесенные к адсорбции СО на 2 атомах (мостиковые карбонилы) на поверхности Pd(100) и на стыке Pd(111) и Pd(100) (1982 см–1), а также на поверхности Pd(111) (1950 см–1). Далее был выявлен широкий пик, связанный с адсорбцией на 3 атомах Pd(111) (1905 см–1). Пик, выявленный на 1705 см–1, был отнесен к адсорбирции СО на границах раздела металл/подложка.
Таким образом, рассчитанные энергии связи согласуются с результатами эксперимента, и величина энергии связи кореллирует с величиной сдвига полос поглощения адсорбированных молекул CO относительно полосы поглощения молекул CO в газовой фазе.
Результаты предсказания всех моделей машинного обучения, тренировка которых была проведена по всей функции радиального распределения, представлены на рис. 5. Значения метрик качества для полученных предсказаний приведены в табл. 1. По значениям данных параметров можно сделать вывод, что наилучший результат в определении энергии по полному интервалу функций распределения (0.1–7.0 Å) показал метод опорных векторов, со средней абсолютной ошибкой в 0.093 эВ, что сопоставимо с погрешностью расчетов с помощью теории функционала плотности. Более того, этот результат превосходит точность аппроксимации энергии связи CO с кластерами Pd, указанной в работе [23] (средняя абсолютная ошибка была равна 0.12 эВ). Несколько менее эффективны для предсказания энергии ансамблевые методы – “экстремально случайные деревья” и “градиентный бустинг” – они позволили предсказать энергию с точностью 0.114 и 0.132 эВ соответственно. Их более низкая эффективность по сравнению с методом опорных векторов может быть связана с недостаточно большим размером обучающей выборки (менее 1000 объектов). Наименьшую эффективность показал метод “гребневой регрессии” (абсолютное значение ошибки составило 0.373 эВ), что может быть обусловлено сложной зависимостью энергии связи от функций радиального распределения, из-за чего аппроксимации линейной моделью недостаточно.
Рис. 5.
Результаты определения энергии связи с помощью различных алгоритмов машинного обучения: (а) “экстремально случайные деревья”; (б) “градиентный бустинг”; (в) метод опорных векторов; (г) “гребневая регрессия”. Штриховой линией показано идеальное совпадение теоретической и предсказанной энергии.
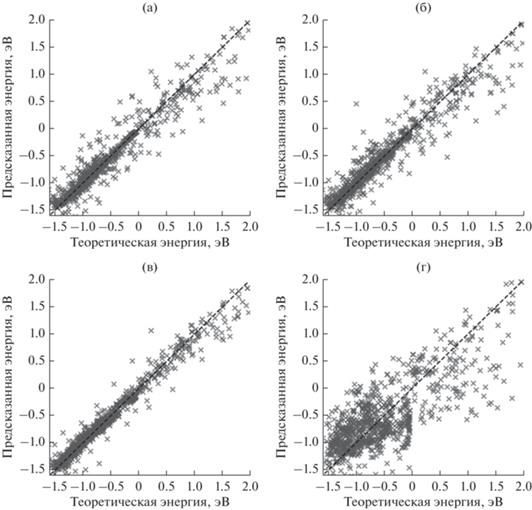
Таблица 1.
Сравнение используемых алгоритмов машинного обучения по их эффективности для предсказания энергии связи по всему интервалу рассчитанных функций радиального распределения
| Алгоритм машинного обучения | Метрика | ||
|---|---|---|---|
| среднее абсолютное отклонение, эВ | среднеквадратичное отклонение, эВ | коэффициент детерминации R2 | |
| “Экстремально случайные деревья” | 0.114 | 0.042 | 0.924 |
| “Градиентный бустинг” | 0.132 | 0.047 | 0.916 |
| Метод опорных векторов | 0.093 | 0.019 | 0.966 |
| “Гребневая регрессия” | 0.373 | 0.229 | 0.595 |
Для модели, показавшей наилучший результат (метод опорных векторов), было проведено обучение на отрезках функции радиального распределения различной длины (табл. 2). Сокращение интервала с 0.1–7.0 до 0.1–3.0 Å слабо повлияло на точность предсказания, которая, однако, резко уменьшилась при уменьшении интервала до 0.1–2.0 Å. Практически на всем отрезке от 0 до 1.5 Å функции радиального распределения имеют нулевые или близкие к нулю значения, поэтому обучение невозможно. При дроблении функции радиального распределения на отрезки длиной в 1, 1.5, 2, 2.5 и 3 Å наибольшую точность аппроксимации энергии обеспечили отрезки 1.5–2.5, 1.5–3.0, 1.5–3.5, 1.5–4.0 и 1.5–4.5 Å, соответственно, тем самым подтверждая влияния локального окружения на энергию связи адсорбата и применимость вышеописанного подхода для ультрамалых частиц. Тем не менее, даже исключение части функции радиального распределения, лежащей в диапазоне 1.5–3.0 Å приводило к увеличению погрешности определения энергии приблизительно в 2 раза, свидетельствуя о возможности восстановления информации об участке, на котором происходит адсорбция, и по удаленным атомам наночастицы.
Таблица 2.
Влияние диапазона функции радиального распределения, используемого для определений энергий связи методом опорных векторов
| Диапазон, Å | Метрика | ||
|---|---|---|---|
| среднее абсолютное отклонение, эВ | среднеквадратичное отклонение, эВ | коэффициент детерминации R2 | |
| Варьирование длины отрезка | |||
| 0.1–7.0 | 0.094 | 0.019 | 0.966 |
| 0.1–6.0 | 0.09 | 0.018 | 0.968 |
| 0.1–5.5 | 0.091 | 0.018 | 0.968 |
| 0.1–5.0 | 0.095 | 0.018 | 0.968 |
| 0.1–4.5 | 0.097 | 0.018 | 0.968 |
| 0.1–4.0 | 0.1 | 0.02 | 0.965 |
| 0.1–3.5 | 0.109 | 0.023 | 0.959 |
| 0.1–3.0 | 0.119 | 0.028 | 0.95 |
| 0.1–2.5 | 0.184 | 0.08 | 0.859 |
| 0.1–2.0 | 0.329 | 0.291 | 0.488 |
| С использованием различных интервалов длиной 1 Å | |||
| 0.5–1.5 | 0.45 | 0.369 | 0.35 |
| 1.0–2.0 | 0.329 | 0.29 | 0.488 |
| 1.5–2.5 | 0.184 | 0.079 | 0.862 |
| 2.0–3.0 | 0.336 | 0.318 | 0.439 |
| 2.5–3.5 | 0.456 | 0.438 | 0.227 |
| 3.0–4.0 | 0.484 | 0.516 | 0.091 |
| 3.5–4.5 | 0.435 | 0.442 | 0.221 |
| 4.0–5.0 | 0.432 | 0.436 | 0.231 |
| 4.5–5.5 | 0.43 | 0.397 | 0.301 |
| 5.0–6.0 | 0.445 | 0.437 | 0.23 |
| 5.5–6.5 | 0.509 | 0.518 | 0.086 |
| 6.0–7.0 | 0.496 | 0.526 | 0.072 |
Таким образом, можно сделать вывод о высокой эффективности функций радиального распределения и их частей в качестве дескрипторов для задачи аппроксимации энергии связи нанокластеров палладия, где локальное окружение атомов адсорбатов имеет решающее значение.
ЗАКЛЮЧЕНИЕ
Использование функций радиального распределения и их частей в качестве дескрипторов и применение методов машинного обучения, таких как “градиентный бустинг”, “гребневая регрессия”, “экстремальные случайные деревья” и метод опорных векторов, позволили с высокой точностью провести аппроксимацию энергии связи молекул СО с нанокластером Pd. Данные подходы не требуют больших вычислительных мощностей после того, как проведена генерация тренировочной выборки, и в дальнейшем могут быть расширены для задач расчета энергий адсорбции на наночастицах разной формы и размеров.
Развитие этого подхода в дальнейшем может привести к быстрому и точному предсказанию не только энергий связи, но и частот и интенсивностей колебаний атомов, что, в свою очередь, позволит создать однозначную автоматизированную методику извлечения информации о поверхности наночастиц из инфракрасных спектров адсорбированных молекул.
Список литературы
Pakhare D., Spivey J. // Chem. Soc. Rev. 2014. V. 43. № 22. P. 7813. https://www.doi.org/10.1039/C3CS60395D
Pareek V., Bhargava A., Gupta R., Jain N., Panwar J. // Adv. Sci. Eng. Med. 2017. V. 9. № 7. P. 527. https://www.doi.org/10.1166/asem.2017.2027
Kinoshita K. // J. Electrochem. Soc. 2017. V. 137. № 3. https://www.doi.org/10.1149/1.2086566
Rojluechai S., Chavadej S., Schwank J.W., Meeyoo V. // Catal. Commun. 2007. V. 8. № 1. P. 57. https://www.doi.org/10.1016/j.catcom.2006.05.029
DeSantis C.J., Peverly A.A., Peters D.G., Skrabalak S.E. // Nano Lett. 2011. V. 11. № 5. P. 2164. https://www.doi.org/10.1021/nl200824p
Sun C., Cao Z., Wang J., Lin L., Xie X. // New J. Chem. 2019. V. 43. № 6. P. 2567. https://www.doi.org/10.1039/C8NJ05152F
Vatti S.K., Ramaswamy K.K., Balasubramanaian V. // J. Adv. Nanomat. 2017. V. 2. № 2. P. 127. https://www.doi.org/10.22606/jan.2017.22006.
Cuenya B.R. // Thin Solid Films. 2010. V. 518. № 12. P. 3127. https://www.doi.org/10.1016/j.tsf.2010.01.018
Терещенко А., Поляков В., Гуда А., Булгаков А., Тарасов А., Кустов Л., Бутова В., Тригуб А., Солдатов А. // Поверхность. Рентген. синхротр. и нейтрон. исслед. 2020. № 5. С. 17. https://www.doi.org/10.31857/s1028096020050180
Yudanov I.V., Sahnoun R., Neyman K.M., Rösch N., Hoffmann J., Schauermann S., Johanek V., Unterhalt H., Rupprechter G., Libuda J. // J. Chem. Phys. B. 2003. V. 107. № 1. P. 255. https://www.doi.org/10.1021/jp022052b
Wang X., Wu G., Guan N., Li L. // Appl. Catal. B-Environ. 2012. V. 115. № P. 7. https://www.doi.org/10.1016/j.apcatb.2011.12.011
Fan Q., He S., Hao L., Liu X., Zhu Y., Xu S., Zhang F. // Sci. Rep. 2017. V. 7. № 1. P. 1. https://www.doi.org/10.1038/srep42172
Sheu L.L., Karpinski Z., Sachtler W.M. // J. Chem. Phys. 1989. V. 93. № 12. P. 4890. https://www.doi.org/10.1021/j100349a042
Tereshchenko A., Guda A., Polyakov V., Rusalev Y., Butova V., Soldatov A. // Analyst. 2020. №. https://www.doi.org/10.1039/D0AN01303J
Cuenya B.R., Behafarid F. // Surf. Sci. Rep. 2015. V. 70. № 2. P. 135. https://www.doi.org/10.1016/j.surfrep.2015.01.001
Rankine C.D., Madkhali M.M., Penfold T.J. // J. Chem. Phys. A. 2020. V. 124. № 21. P. 4263. https://www.doi.org/10.1021/acs.jpca.0c03723
Timoshenko J., Lu D., Lin Y., Frenkel A.I. // J. Phys. Chem. Lett. 2017. V. 8. № 20. P. 5091. https://www.doi.org/10.1021/acs.jpclett.7b02364
Timoshenko J., Anspoks A., Cintins A., Kuzmin A., Purans J., Frenkel A.I. // Phys. Rev. Lett. 2018. V. Thin Solid Films. № 22. P. 225502. https://www.doi.org/10.1103/PhysRevLett.120.225502
Tupy S.A., Karim A.M., Bagia C., Deng W., Huang Y., Vlachos D.G., Chen J.G. // ACS Catal. 2012. V. 2. № 11. P. 2290. https://www.doi.org/10.1021/cs3004227
Lansford J.L., Vlachos D.G. // Nat. Commun. 2020. V. 11. № 1. P. 1. https://www.doi.org/10.1038/s41467-020-15340-7
Calle-Vallejo F., Martínez J.I., García-Lastra J.M., Sautet P., Loffreda D. // Angew. Chem. Int. Edit. 2014. V. 53. № 32. P. 8316. https://www.doi.org/10.1002/anie.201402958
Takigawa I., Shimizu K.-i., Tsuda K., Takakusagi S. // RSC Adv. 2016. V. 6. № 58. P. 52 587. https://www.doi.org/10.1039/C6RA04345C
Gasper R., Shi H., Ramasubramaniam A. // J. Chem. Phys. C. 2017. V. 121. № 10. P. 5612. https://www.doi.org/10.1021/acs.jpcc.6b12800
Kresse G., Furthmüller J. // Phys. Rev. B. 1996. V. 54. № 16. P. 11169
Kresse G., Furthmüller J. // Comp. Mater. Sci. 1996. V. 6. № 1. P. 15
Grimme S. // J. Comput. Chem. 2006. V. 27. № 15. P. 1787
Perdew J., Burke K., Ernzerhof M. // Phys. Rev. Lett. 1998. V. 80. № 4. P. 891.
Lamberti C., Zecchina A., Groppo E., Bordiga S. // Chem. Soc. Rev. 2010. V. 39. № 12. P. 4951. https://www.doi.org/10.1039/C0CS00117A
Дополнительные материалы отсутствуют.
Инструменты
Поверхность. Рентгеновские, синхротронные и нейтронные исследования