

Российский физиологический журнал им. И.М. Сеченова, 2019, T. 105, № 1, стр. 3-23
Биофизические механизмы натриевых каналопатий в миокарде: cиндром удлиненного интервала QT и синдром Бругада
А. К. Зайцева 1, 2, *, А. В. Карпушев 1, Б. С. Жоров 1, 2, А. А. Костарева 1
1 Национальный медицинский исследовательский центр им. В.А. Алмазова
Минздрава России
Санкт-Петербург, Россия
2 Институт эволюционной физиологии и биохимии им. И.М. Сеченова РАН
Санкт-Петербург, Россия
* E-mail: zaytseva.anastasia.zak@gmail.com
Поступила в редакцию 02.11.2018
После доработки 30.11.2018
Принята к публикации 01.12.2018
Аннотация
Мутации в гене SCN5A, кодирующем потенциал-зависимый натриевый канал сердца Nav1.5, часто ассоциированы с синдромом удлиненного интервала QT подтипа 3 (LQT3) и синдромом Бругада подтипа 1 (BrS1). В последние годы достигнуты значительные успехи в понимании биофизических механизмов данных аритмий. Синдром LQT3 ассоциирован с увеличением активности натриевых каналов, которое может выражаться в ускорении активации, замедлении инактивации, появлении постоянного тока и/или ускорении восстановления из инактивированного состояния. Синдром Бругада, напротив, сопряжен со снижением натриевого тока, которое может быть вызвано дефектом транспорта каналов на мембрану, замедлением активации, ускорением развития быстрой и медленной инактивации, и/или замедлением восстановления из инактивации. Мутации, ассоциированные с развитием наследственных аритмий, также могут вызывать нарушение взаимодействия Nav1.5 с другими белками и/или его лигандами и приводить к развитию аномальной реакции в ответ на действие ряда фармакологических агентов. Кроме того, может изменяться характер посттрансляционных модификаций и чувствительность к изменению pH и температуры. В данном обзоре предпринята попытка систематизировать накопленные данные о биофизических механизмах развития синдрома Бругада и синдрома удлиненного интервала QT. Особое внимание уделено ограничениям исследований, выполненных в гетерологических системах экспрессии и причинам, по которым не удается однозначно установить корреляцию генотип-фенотип при мутациях в гене SCN5A1.
Потенциал-зависимый натриевый канал кардиомиоцитов Nav1.5 – это белковый комплекс, локализованный в цитоплазматической мембране сократительных клеток сердца. Основной функцией Nav1.5 является обеспечение входящего тока ионов натрия во время нулевой фазы потенциала действия (ПД) сердца [1]. Значение натриевого тока (INa) для нормальной электрической активности сердца иллюстрирует широкий спектр наследственных аритмогенных заболеваний, ассоциированных с мутациями в гене SCN5A, кодирующем порообразующую α-субъединицу Nav1.5, и в генах, кодирующих белки, регулирующие активность натриевых каналов [2]. Исследования, выполненные с помощью метода локальной фиксации потенциала (patch-clamp), дают представление о механизмах дисфункции Nav1.5. Регистрация биофизических характеристик генетических вариантов чаще всего осуществляется с помощью гетерологической системы экспрессии в клетках линий HEK293, CHO и др., трансфицированных вектором с мутантным SCN5A. Известно, что мутации в гене SCN5A, вызывающие увеличение активности Nav1.5, ассоциированы с синдромом удлиненного интервала QT подтипа 3 (LQT3), а мутации, связанные с уменьшением активности Nav1.5, ассоциированы с синдромом Бругада 1 подтипа (BrS1), болезнями проводящей системы сердца и другими аритмогенными заболеваниями.
α-субъединица Nav1.5 состоит из четырех доменов (I-IV), каждый из которых включает в себя 6 сегментов (S1–S6). Сегменты S1–S4 образуют потенциал-чувствительные модули, а сегменты S5–S6 вместе с соединяющими их внеклеточными Р-петлями формируют порообразующий модуль канала. Воротный механизм (гэйтинг) включает в себя переходы между разными конформационными состояниями белка канала и связан с движением потенциал-чувствительных сегментов S4, в которых находятся положительно заряженные аминокислотные остатки (рис. 1, А) [1]. При потенциале покоя каналы находятся в закрытом состоянии. Деполяризация мембраны вызывает переход канала в открытое состояние, что в свою очередь запускает механизм быстрой инактивации канала и его последующий переход в закрытое состояние. Длительная деполяризация вызывает переход канала в состояние медленной инактивации [1].
Рис. 1.
Биофизические механизмы синдрома Бругада и синдрома LQT3 А: упрощенная схема переходов между различными конформационными состояниями канала. Буквы в кружках обозначают состояния канала: О – открытое, З – закрытое, БИ – быстро инактивированное, МИ – медленно инактивированное. Буквы над стрелками обозначают, какие измеряемые характеристики (панели Б–Ж) влияют на процесс того или иного перехода. Б: Кривая стационарной активации. Кинетика, характерная для каналов дикого типа, обозначена черным. Красная линия (LQT) демонстрирует сдвиг в сторону гиперполяризации (стабилизация открытого или дестабилизация закрытого состояния). Синяя линия (BrS) сдвинута в сторону деполяризации (дестабилизация открытого состояния или стабилизация закрытого). В: Кривая стационарной инактивации. Для синдрома LQT3 характерна инактивация при более положительных потенциалах (дестабилизация инактивированных состояний), в то время как при мутациях в гене SCN5A, ассоциированных с синдромом Бругада, стационарная инактивация ускорена (стабилизация инактивированных состояний). Г: Развитие медленной инактивации. При синдроме Бругада развитие медленной инактивации в ответ на продолжительный деполяризующий импульс может быть ускорено, что отражает стабилизацию медленного инактивированного состояния. Д: Восстановление из инактивации. При мутациях, приводящих к развитию аритмий, может изменяться скорость выхода из инактивированного состояния. Так, при синдроме Бругада инактивированное состояние может быть более стабильно, что приводит к замедлению восстановления. При синдроме LQT наблюдается противоположная картина. Е. Изменение тока при аритмиях. Для синдрома Бругада характерно уменьшение плотности тока, в то время как при синдроме LQT наблюдается возникновение постоянного тока. Ж: Изменение ПД при аритмиях. Заболевания аритмогенной природы оказывают различное влияние на морфологию ПД. Так, дисфункция натриевых каналов при BrS1 приводит к замедлению нарастания деполяризации, в то время как гиперфункция Nav1.5 приводит к увеличению фазы плато и продолжительности ПД за счет повторной активации натриевых и кальциевых каналов.
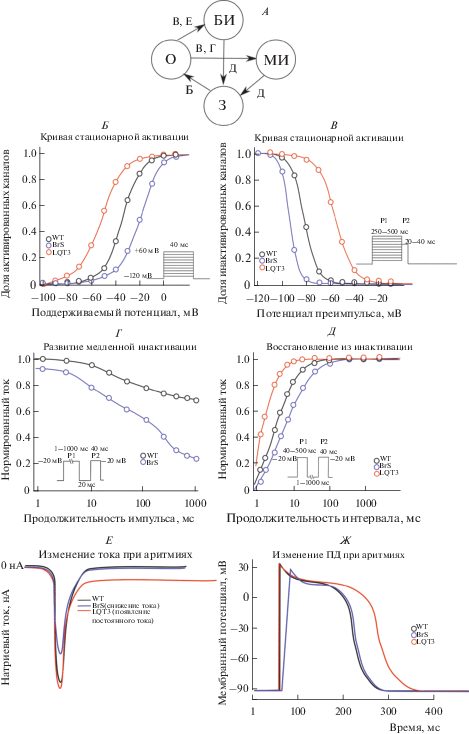
Патогенные генетические варианты могут быть ассоциированы со стабилизацией или дестабилизацией различных состояний канала. Влияние определенной аминокислотной замены на переход из закрытого в открытое состояние можно оценить с помощью кривой стационарной активации (рис. 1Б). Сдвиг кривой стационарной активации в сторону гиперполяризации может отражать стабилизацию открытого или дестабилизацию закрытого состояния канала, в то время как сдвиг данной кривой в сторону деполяризации означает дестабилизацию открытого или стабилизацию закрытого состояния. Для построения кривой стационарной инактивации (рис. 1В), как правило, используется преимпульс продолжительностью 250–500 мс. Потому такая кривая может отражать переходы в состояния как быстрой, так и в медленной инактивации. Стабилизация медленного инактивированного состояния, выявленная при ряде мутаций, ассоциированных с BrS1, может быть идентифицирована с помощью кривой возникновения медленной инактивации (рис. 1Г). График восстановления из инактивации (рис. 1Д) в зависимости от используемой продолжительности предимпульса может отражать изменение восстановления каналов из состояния быстрой и медленной инактивации. Кинетика восстановления из инактивированного состояния описывается с помощью биэкспоненциальной кривой, из уравнения которой можно вычислить быструю и медленную постоянную времени восстановления из инактивации. Нарушение перехода в состояние быстрой инактивации при синдроме LQT3 может приводить к возникновению постоянного тока ионов натрия через пору канала (рис. 1Е) [2]. Множество мутаций, ассоциированных с BrS1, приводят к уменьшению плотности INa (рис. 1Е), которое может быть как следствием изменения числа каналов в мембране, так и следствием изменения проводимости натриевого канала [2]. При синдроме LQT3 может наблюдаться стабилизация открытого состояния и дестабилизация закрытого и инактивированных состояний. Это приводит к удлинению интервала QT за счет повторной активации натриевых и кальциевых каналов и увеличения продолжительности фазы плато ПД (рис. 1Ж) [2]. Уменьшение вероятности открытого состояния и/или увеличение вероятности закрытого и инактивированных состояний, которые могут лежать в основе BrS1, приводят к замедлению нарастания деполяризации во время нулевой фазы ПД (рис. 1Ж) [2].
СИНДРОМ УДЛИНЕННОГО ИНТЕРВАЛА QT
Синдром удлиненного интервала QT (LQTS) – это заболевание, характеризующееся удлинением интервала QT на электрокардиограмме (ЭКГ) и повышенным риском внезапной смерти в связи с желудочковой тахиаритмией (в т.ч. пируэтная тахикардия). Длительные интервалы QT отражают увеличение продолжительности потенциала действия в миоцитах желудочков и соответствуют задержке их реполяризации. Есть разные подтипы генетически обусловленных LQTS. Третий подтип связан с мутациями в гене SCN5A, которые прямо или косвенно влияют на формирование ПД [3]. Мутации в SCN5A, ассоциированные с синдромом LQT3, обнаруживаются у 10–13% всех генотипированных лиц с LQTS [4, 5].
МУТАЦИИ, АССОЦИИРОВАННЫЕ С СИНДРОМОМ LQT3
В настоящее время выявлено более 150 мутаций SCN5A у пациентов с LQT3, и около 50% этих мутаций охарактеризованы электрофизиологически (рис. 2). Большинство мутаций являются миссенс-мутациями, которые вызывают усиление активности Nav1.5 (gain-of-function phenotype) за счет нарушения быстрой инактивации и возникновения аномального постоянного натриевого тока (Isus) [2, 6]. Мутации, вызывающие Isus, в основном локализованы в сегментах Nav1.5, которые задействованы в процессе быстрой инактивации (т.е. IVS4, IIIS4-S5 и линкер III–IV) [4, 7–11]. Другие, менее распространенные механизмы действия мутаций SCN5A, вызывающие LQT3, включают замедление инактивации [12, 13], ускорение восстановления из инактивации [14, 15], увеличение пиковой плотности INa [16] и ускорение активации, а также различные сочетания данных механизмов. Важно отметить, что одна мутация SCN5A может вызывать несколько изменений в гэйтинге и/или экспрессии. Независимо от механизма, увеличение активности Nav1.5 нарушает баланс между деполяризующими и реполяризующими токами в пользу деполяризации. Полученная задержка в процессе реполяризации вызывает раннюю повторную деполяризацию (т.е. повторную активацию кальциевого канала L-типа во время фаз 2 или 3 ПД), особенно в миоцитах волокон Пуркинье, где длительность ПД изначально выше [17, 18], что лежит в основе развития тахикардии по типу пируэт (torsades de pointes) [19].
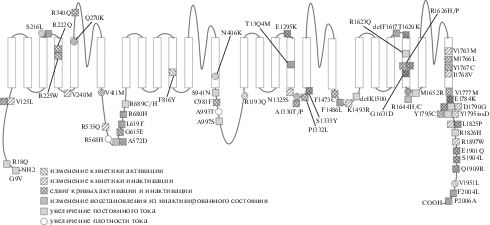
УВЕЛИЧЕНИЕ ПОСТОЯННОГО ТОКА
Появление постоянного (не инактивируемого) натриевого тока рассматривается как основной механизм развития синдрома LQT3. Для нескольких мутаций в цитоплазматических участках Nav1.5 описано увеличение Isus без существенных изменений других биофизических характеристик: G9V, R18Q (N-конец) [20], R689C, R689H (I–II) [21], S941N [13], A997S (II–III) [22], M1652R (IVS4–S5) [13], D1790G [23], Y1795C [23] и R1826H (C-конец) [22]. Такая же картина наблюдалась при мутации R1623Q, локализованной в IVS4 [24, 25].
Возникновение аритмии при LQT3 обычно наблюдается в состоянии покоя или во время сна, при низкой частоте сердечных сокращений. В соответствии с этим, Isus больше выражен при низкой частоте стимуляции, что позволяет предположить, что степень Isus может рассматриваться как ключевая детерминанта возникновения аритмии [26]. Это подтверждается тем, что у пациентов с мутацией в гомозиготном состоянии наблюдается большая степень удлинения интервала QT и большая вероятность развития ритма, чем в гетерозиготном. Это согласуется с данными биофизических экспериментов, показавших, что в гомозиготном состоянии экспрессия мутации приводит к увеличению Isus в большей степени, чем в гетерозиготном [27].
ЗАМЕДЛЕНИЕ СТАЦИОНАРНОЙ ИНАКТИВАЦИИ
Большинство мутаций в SCN5A, которые приводят к сдвигу стационарной инактивации в сторону деполяризации в отсутствие изменений других биофизических характеристик, локализованы в цитоплазматической части канала, за исключением V1763M [28] и I1768V (IVS6) [29]. Умеренные изменения инактивации были показаны для аминокислотных замен V125L (N-конец) [20], V240M (IS4–S5) [30], R535Q (I–II) [30], N1325S (IIIS4–S5) [31], K1493R (III–IV) [32], I1768V (IVS6) [29] и R1897W (C-конец) [33], в то время как мутации Y1795insD и L1825P (C-конец) приводили к более выраженному замедлению инактивации [34]. Для аминокислотной замены V1763M (IVS6) на модели кардиомиоцитов, дифференцированных из индуцированных плюрипотентных стволовых клеток (иПСК) пациента с синдромом LQT3, показан сдвиг кривой стационарной инактивации в сторону деполяризации на 16.8 мВ [28]. При мутации D1790G (C-конец) наблюдается нарушение регуляции инактивации со стороны вспомогательной β-субъединицы Nav1.5: в норме ко-экспрессия α-субъединицы дикого типа с β-субъединицей приводит к замедлению перехода в инактивированное состояние, в то время как при данной мутации ко-экспрессия с β-субъединицей не влияет на процесс инактивации [35].
УСКОРЕНИЕ ВОССТАНОВЛЕНИЯ ИЗ ИНАКТИВИРОВАННОГО СОСТОЯНИЯ
Ускорение восстановления из инактивированного состояния нарушает баланс деполяризации и реполяризации за счет увеличения доли каналов, активирующихся во время повторных деполяризаций. Для мутаций A572D и G615E (I–II) на модели ооцитов Xenopus l. было показано уменьшение быстрой постоянной времени восстановления из инактивации и небольшой сдвиг кривой стационарной активации в сторону гиперполяризации [14, 36]. Мутация V1951L (C-конец) приводила к уменьшению медленной постоянной времени восстановления из инактивации [37]. Аналогично на модели кардиомиоцитов, дифференцированных из иПСК пациента с синдромом LQT3 с мутацией R1644H (IVS4–S5), было показано ускорение восстановления из инактивированного состояния в три раза [38].
ПОВЫШЕНИЕ ПИКОВОЙ ПЛОТНОСТИ НАТРИЕВОГО ТОКА
В основе повышения плотности INa может лежать усиление активации или увеличение числа каналов на мембране вследствие дисбаланса экспрессии SCN5A, транспорта Nav1.5 на мембрану и его деградации. Увеличение плотности INa наблюдалось для мутаций S216L (IS3–S4) [33], N406K (IS6) [39], R568H (DI–DII) [36], F816Y (IIS4) [36], A993T (DII–DIII) [36], R1193Q (IIIS4) [40, 41] и V1951M (C-конец) [33]. В ряде случаев, например, при мутациях R568H и A993T, увеличение INa было единственным наблюдаемым изменением функциональных характеристик натриевого канала [36], в то время как для мутаций S216L, N406K, F816Y, V1951M было также обнаружено достоверное ускорение активации [33, 36, 39]. При мутации R1193Q INa возрастал в 4 раза и замедлялась инактивация [40]. Наличие только двух мутаций, которые приводят к повышению плотности тока без изменения других биофизических свойств канала, позволяет предположить, что изменение количества каналов в мембране – довольно редкий механизм развития синдрома LQT3 в отличие от BrS1.
КОМПЛЕКСНОЕ ИЗМЕНЕНИЕ БИОФИЗИЧЕСКИХ ХАРАКТЕРИСТИК
Как правило, мутации, ассоциированные с развитием синдрома LQT3, вызывают изменение сразу нескольких биофизических параметров. Ряд мутаций приводит к появлению Isus в сочетании с изменениями других электрофизиологических характеристик, однако встречаются и другие комбинации различных биофизических механизмов (табл. 1).
Таблица 1.
Комплексное изменение биофизических характеристик при мутациях в SCN5A, ассоциированных с LQT3
| Механизм | Мутация | Локализация | Ссылка |
|---|---|---|---|
| Ускорение восстановления из инактивированного состояния и увеличение постоянного тока | S216L | IS3–S4 | [36] |
| L619F | I–II | [41] | |
| R680H | I–II | [38] | |
| T1304M | IIIS4 | [32] | |
| F1486L | III–IV | [37] | |
| R1644C | IVS4–S5 | [37] | |
| F2004L | C-конец | [32] | |
| P2006A | C-конец | [42] | |
| Ускорение инактивации и увеличение постоянного тока | R1626P | IVS4 | [13] |
| Y1767C | IVS6 | [43] | |
| L1825P | C-конец | [44, 45] | |
| E1901Q | C-конец | [46] | |
| S1904L | C-конец | [47] | |
| Q1909R | C-конец | [46] | |
| Замедление активации и инактивации | R222Q | IS4 | [48] |
| R340Q | IS5–S6 | [32] | |
| P1332L | IIIS4–S5 | [13] | |
| V1777M | C-конец | [27] | |
| Замедление инактивации и ускорение восстановления из инактивации | V125L | N-конец | [20] |
| E1295K | IIIS3–S4 | [49] | |
| A1330P | IIIS4–S5 | [12] | |
| Увеличение плотности тока и ускорение активации | S216L | IS3–S4 | [32] |
| N406K | IS6 | [36] | |
| F816Y | IIS4 | [39] | |
| Ускорение активации, инактивации и восстановления из инактивации | S1333Y | IIIS4–S5 | [50] |
| T1620K | IVS3–S4 | [51] | |
| Замедление инактивации и ускорение активации | F1473C | III–IV | [52] |
| Увеличение постоянного тока, ускорение инактивации и восстановления из инактивации | G1631D | IVS4 | [53] |
| Замедление инактивации и увеличение постоянного тока | M1766L | IVS6 | [54] |
Отдельного упоминания заслуживает мутация E1784K (C-конец), которая приводит к развитию сложной клинической картины, включающей в себя проявления, характерные как для синдрома Бругада, так и для синдрома LQT. Для этой мутации было показано замедление активации и инактивации, ускорение восстановления из инактивации и появление постоянного тока [55].
СИНДРОМ БРУГАДА
Синдром Бругада – это генетически обусловленное нарушение ритма, которое является причиной 4–12% случаев внезапной смерти вследствие желудочковой тахикардии [56–58]. На электрокардиограмме синдром Бругада проявляется как феномен Бругада, который представляет собой подъем сегмента ST в правых грудных отведениях. Данный ЭКГ-паттерн высоко динамичен и часто не проявляется постоянно; он может проявляться под действием таких факторов, как повышение температуры, воздействие блокаторов INa и препаратов, оказывающих ваготоническое действие [59–62]. Клиническая картина синдрома Бругада сильно варьирует от бессимптомного течения до внезапной смерти. Клинические события, как правило, случаются в состоянии покоя, вследствие нарушений баланса активности симпатической и парасимпатической нервной системы, а также могут провоцироваться гормональными и метаболическими факторами [63–65].
МУТАЦИИ, АССОЦИИРОВАННЫЕ С СИНДРОМОМ БРУГАДА
Известно более 200 мутаций, ассоциированных с синдромом Бругада. Из них электрофизиологически охарактеризовано менее 50% (рис. 3). Данные биофизических исследований позволяют предложить несколько основных механизмов синдрома Бругада: уменьшение INa, замедление активации, ускорение инактивации, ускорение медленной инактивации и замедление восстановления из быстрого или медленного инактивированных состояний. Наряду с изменениями биофизических характеристик, мутации в SCN5A способны менять характер взаимодействия канала с другими белками, влиять на его регуляцию, нарушать транспорт канала в мембрану и способствовать его деградации. Большинство мутаций в SCN5A, ассоциированных с синдромом Бругада, являются миссенс-мутациями [8]. В случае нонсенс-мутаций (Q55X, E473X, R535X, W822X, W1191X, R1623X) и мутаций со сдвигом рамки считывания (K1578fs, A1680fs) чаще всего наблюдается формирование функционально не активного канала [62, 66–71]. Мутации встречаются во всех участках канала, однако их наиболее высокая концентрация наблюдается в области P-петель [8].
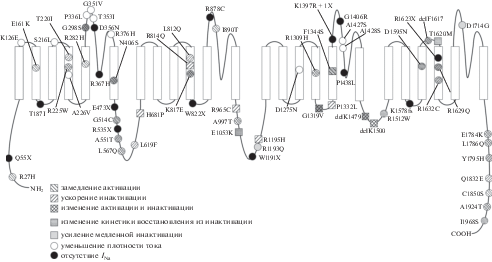
УМЕНЬШЕНИЕ ПЛОТНОСТИ ТОКА
Уменьшение плотности тока может свидетельствовать о снижении экспрессии SCN5A, нарушении транспорта канала в мембрану или регуляции со стороны сигнальных процессов, а также быть вторичным изменением, обусловленным замедлением активации. Снижение INa может возникать вследствие как одиночной мутации, так и двух гетерозиготных мутаций, имеющих аддитивный эффект на плотность INa, как в случае двойной мутации P336L + I1660V (IS5–S6, IVS5) [72]. Миссенс-мутации, приводящие к отсутствию тока или к его сильному падению, могут быть разделены на две группы: (1) аминокислотные замены, приводящие к формированию функционально неактивного белка или значительному снижению проводимости для натрия; и (2) замены, вызывающие нарушение доставки канала на мембрану.
Отсутствие функциональной активности было показано для мутаций R367H (IS5–S6) [73], R878C (IIS5–S6) [74], G1406R (IIIS5–S6) (идентифицирована у пациентов с синдромом Бругада и с прогрессирующей проводящей болезнью сердца) [75, 76] и P1438L [77]. Все вышеперечисленные мутации локализованы в P-петлях канала и, наиболее вероятно, блокируют ток натрия через пору канала.
С синдромом Бругада ассоциирован ряд мутаций, приводящих к сильному падению плотности INa, но не влияющих на другие биофизические характеристики. Часть подобных мутаций локализована в Р-петлях доменов I (G351V, R376H) [78, 79] и III (A1427S, A1428S) [80, 81]. Это позволяет предположить, что уменьшение плотности тока вызвано затрудненным прохождением ионов через пору канала. Однако значительное снижение регистрируемого тока было также показано для замены A226V (IS4) [82], локализованной в сенсоре напряжения, а также для мутаций K126E (IS1–S2) [20] и S216L (IS3–S4) [83], локализованных во внеклеточных петлях.
Мутации, влияющие на транспорт канала, локализованы в различных частях белка и включают в себя как мутации, нарушающие фолдинг, так и мутации, затрагивающие сайты связывания с различными внутриклеточными белками, заякоривающими канал на мембране. В ряде случаев мутантные каналы преимущественно задерживаются внутри клетки, как продемонстрировано для мутаций T353I (IS5–S6) [84], P336L + I1660V (IS5–S6, IVS5) [72], D1275N (IIIS3) [85], R1432G (IIIS5–S6) [86] и G1743R (IVS5–S6) [87]. Мутация R1432G при экспрессии в клетках линии tsA201 вызывала ко-локализацию с кальнексином в ЭПР при отсутствии детектируемых токов [86]. Однако при экспрессии мутантной формы белка в ооцитах Xe-nopus l. регистрировался ток нормальной амплитуды. Предполагается, что доставке канала на мембрану в клетках tsA-201 препятствует нарушение фолдинга. При мутациях R104W, R121W и R104K (N-терминальный домен) также нарушается доставка каналов на мембрану [88].
Дефект транспорта канала на мембрану может быть результатом нарушения взаимодействий с белками, которые необходимы для правильной локализации канала. В случае мутации E1053K (II–III) анкирин-связывающий мотив нарушен, и доставка мутантных каналов на плазматическую мембрану кардиомиоцитов резко снижена [89]. Интересно, что в клетках линии HEK-293 мутантные каналы успешно доставляются на мембрану и не найдено значимых различий по плотности INa. Это свидетельствует о том, что мутация E1053K не затрагивает процесс фолдинга. Данный пример показывает, что регистрируемый в гетерологической системе ток необязательно свидетельствует о нормальном транспорте канала в кардиомиоцитах и что тканеспецифичные свойства могут влиять на регуляцию транспорта Nav1.5.
Мутация R526H (I–II) располагается в консервативном сайте узнавания протеинкиназы А (PKA) и приводит к нарушениям сигнальных путей и доставки канала на мембрану, оказывая эффект аналогичный аминокислотной замене S528A, элиминирующей сайт фосфорилирования PKA [90]. При этом кинетические характеристики мутантной формы белка не отличаются от таковых для канала дикого типа [90]. В ряде случаев изменяется ответ мутантной формы белка на различные внешние стимулы. Мутация R1512W (III–IV) приводит к снижению пиковой плотности натриевого тока на 30% при физиологических значениях pH и на 50% при ацидозе (pH 7.0) [91]. Исходя из вышеперечисленных примеров, можно сделать вывод, что образование функционально неактивных каналов и снижение плотности INa является широко распространенным механизмом синдрома Бругада при миссенс-мутациях.
ЗАМЕДЛЕНИЕ АКТИВАЦИИ
Сдвиг кривой стационарной активации в сторону деполяризации отражает увеличение порога активации натриевых каналов и приводит к замедлению инициации и распространения ПД сердца. Такое изменение возбудимости сердца может быть ответственно за небольшое удлинение интервала H-V и блокаду правой ножки пучка Гиса у пациентов с BrS1 [92]. Наиболее выраженное замедление активации было показано для мутаций, локализованных в сенсорах напряжения E161K (IS4) [93] и K817E (IIS4) [94]. Менее выраженные изменения описаны для мутаций, расположенных в петлях S3–S4 и S4–S5. Например, мутация G1319V (IIIS4–S5) вызывала небольшое замедление активации [95], а мутации T1620M (IVS3–S4) и F1344S (IIS5) приводили к выраженному замедлению развития активации при повышении температуры [92, 96]. В эту группу мутаций также можно отнести замену G514C (I–II), которая может препятствовать переходу канала в открытое состояние [97]. Сдвиг кривой активации приводит к укорочению времени пикового тока и к уменьшению INa в момент, соответствующий началу 1 фазы ПД в правом желудочке [2]. Считается, что снижение INa приводит к невозможности активации быстрого выходящего калиевого тока [2].
УСКОРЕНИЕ ИНАКТИВАЦИИ
Сдвиг кривой инактивации в сторону гиперполяризации свидетельствует об уменьшении количества каналов, способных к активации при определенных потенциалах. Это приводит к замедлению нарастания деполяризации во время фазы 0 ПД сердца. Ускорение инактивации в отсутствие изменений других биофизических характеристик было показано для мутаций T220I (IS5) [33], R965C [98] и R1195H (II–III) [99]. Позиция R965 интересна тем, что вместе с соседними аминокислотами формирует амфифильную спираль с регулярно расположенными положительно заряженными аминокислотами и оказывает влияние на кинетику инактивации канала [98]. Стабилизация инактивированного состояния натриевых каналов обуславливает уменьшение INa во время фаз деполяризации и реполяризации ПД, снижает скорость нарастания ПД и приводит к замедлению проведения в предсердиях и желудочках, что нарушает баланс входящих и выходящих токов во время фазы 1 ПД и сокращает фазу плато [100].
УСИЛЕНИЕ МЕДЛЕННОЙ И ПРОМЕЖУТОЧНОЙ ИНАКТИВАЦИИ
Усиление промежуточной и медленной инактивации также может рассматриваться как механизм уменьшения функциональной активности Nav1.5 при синдроме Бругада; этот механизм был впервые описан для мутации 1795insD [101]. Усиление медленной инактивации обуславливает увеличение числа инактивированных каналов и, как следствие, уменьшение входящего INa, в особенности на высоких частотах стимуляции т.е. при высокой частоте ударов сердца. Это было подтверждено значительным увеличением подъема сегмента ST во время физической нагрузки у носителей 1795insD [101]. По причине того, что 1795insD каналы генерируют фракцию Isus, значительное увеличение интервала QT было зарегистрировано только при удлиненных RR интервалах. Это объясняется тем, что успешно восстановившиеся из инактивированного состояния каналы приводят к появлению не инактивирующейся фракции каналов. Усиление медленной инактивации было показано только для небольшого числа мутантных каналов и ни в одном случае не являлось единственным изменением биофизических свойств. Так, ускорение развития медленной инактивации продемонстрировано для мутаций G298S (IS5–S6) [102], N406S (IS6) [103], E1053K (II–III) [89], R1193Q (II–III) [40], G1319V (IIIS4–S5) [95], K1527R + A1569P (III–IV, IVS2) [104], D1595N (IVS3) [102], T1620M (IVS3–S4) [105], R1629Q (IVS4) [106], S1710L (IVS5–S6) [107], D1714G (IVS5–S6) [108] и Y1795H (C-конец) [16]. Однако усиление входа в состояние промежуточной или медленной инактивации не может рассматриваться в качестве распространенного механизма развития синдрома Бругада вследствие того, что внезапная смерть наступает во время сна при низкой (ЧСС), когда промежуточная и медленная инактивация наименее вероятны [2]. Тем не менее, даже небольшое по величине уменьшение активности каналов при низкой ЧСС может вносить вклад в проявления клинической картины заболевания.
ЗАМЕДЛЕНИЕ ВОССТАНОВЛЕНИЯ ИЗ ИНАКТИВИРОВАННОГО СОСТОЯНИЯ
Замедление восстановления из инактивированного состояния. может вносить вклад в развитие аритмии за счет уменьшения INa во время повторных деполяризующих импульсов. Как правило, замедление восстановления из инактивации наблюдается в сочетании с другими изменениями биофизических характеристик. Исключение составляет мутация G35S, локализованная в N-терминальном домене [20].
КОМПЛЕКСНОЕ ИЗМЕНЕНИЕ БИОФИЗИЧЕСКИХ ХАРАКТЕРИСТИК
Большинство мутаций, ассоциированных с BrS1, вызывают комплексное изменение биофизических характеристик. Часто встречается ускорение стационарной инактивации в сочетании с изменениями других электрофизиологических свойств натриевых каналов (табл. 2). Поскольку охарактеризовано лишь около 50% мутаций, ассоциированных с синдромом Бругада, и для подавляющего их числа не регистрировались все электрофизиологические параметры, некоторые сочетания изменений биофизических характеристик обнаружены только у единичных мутаций.
Таблица 2.
Комплексные изменения биофизических характеристик при мутациях, ассоциированных с BrS1
| Механизм | Мутация | Локализация | Ссылка |
|---|---|---|---|
| Уменьшение плотности тока, замедление активации и ускорение инактивации | R27H | N-конец | [109] |
| R225W | IS4 | [109] | |
| H681P | линкер I–II | [110] | |
| A997T | II–III | [111] | |
| R1309H | IIIS4 | [112] | |
| L1786Q | C-конец | [113] | |
| A1924T | C-конец | [11] | |
| Уменьшение плотности тока и ускорение инактивации | T220I | IS4 | [32, 85] |
| L812Q | IIS4 | [100] | |
| R1023H | II–III | [114] | |
| Q1832E | C-конец | [115] | |
| Замедление активации и усиление медленной инактивации | N406S | IS6 | [116] |
| G752R | IIS2 | [117] | |
| S1710L | IVS5–S6 | [118] | |
| Уменьшение плотности тока и замедление активации | E161K | IS2 | [93] |
| R282H | IS5–S6 | [119] | |
| L325R | IS5–S6 | [62] | |
| Уменьшение плотности тока и замедление восстановления из инактивации | A551T | [120] | |
| delF1617 | IVS4 | [121] | |
| R1632C | IVS3 | [122] | |
| Замедление активации и замедление восстановления из инактивации | A735V | IIS1 | [123] |
| delK1479 | III–IV | [124] | |
| Замедление активации и ускорение инактивации | A1649V | IVS4–S5 | [125] |
| delK1500 | III–IV | [126] | |
| Ускорение инактивации, усиление промежуточной инактивации и замедление восстановления из инактивации | R1629Q | IVS4 | [106] |
| Уменьшение плотности тока и усиление медленной инактивации | G298S | IS5–S6 | [102] |
| Уменьшение плотности тока, усиление медленной инактивации и замедление восстановления из инактивации | D1595N | IVS3 | [102] |
| Замедление активации, ускорение инактивации и замедление восстановления из инактивации | L567Q | I–II | [127] |
ФАКТОРЫ, ОГРАНИЧИВАЮЩИЕ ПРИМЕНЕНИЕ БИОФИЗИЧЕСКИХ МЕТОДОВ
Следует принять во внимание ограничения, которые в ряде случаев не позволяют установить взаимосвязь между генотипом и фенотипом пациента.
Во-первых, небольшое число носителей конкретной мутации не позволяет однозначно определить генетический вариант SCN5A как причину развития заболевания и исключить влияние других факторов, таких как возраст, пол и гормональный статус пациента [8].
Во-вторых, гетерологическая система экспрессии не воспроизводит тканеспецифичные особенности кардиомиоцитов, которые могут влиять на экспрессию SCN5A, транспорт и регуляцию Nav1.5. В частности, мутации могут затрагивать взаимодействие с такими белками сердца как анкирин-G, анкирин-B, кальмодулин, FHF1B, убиквитин-лигазы, синтрофин и др [89, 128]. Величина постоянного тока для нескольких мутантных каналов может зависеть от активности протеинкиназ A и C [23, 129].
В-третьих, данная модель не позволяет оценить долговременный эффект экспрессии мутации в SCN5A в миокарде. Обнаружено изменение экспрессии 33 генов у трансгенной мыши с мутацией N1325S, в их числе транскрипционный фактор STAT1, вовлеченный в процессы апоптоза и интерферонового ответа [130]. Можно предположить, что нарушение функции SCN5A способно вызвать комплексный клеточный ответ, влияющий на фенотип. Эта гипотеза согласуется с данными о структурных изменениях проводящей системы, ассоциированных с гетерозиготными мутациями W156X и R225W [131].
В-четвертых, как правило, не учитывается влияние полиморфизмов в гене SCN5A на кинетику и транспорт мутантного канала. Биофизические свойства каналов с сочетанием генетических вариантов исследованы in vitro только для небольшого числа случаев [132–134]. Кроме того, альтернативный сплайсинг также может приводить к изменению электрофизиологических свойств канала [37, 76, 135, 136].
В-пятых, биофизические исследования часто проводятся при температурах ниже физиологической, что может затруднить выявление дефектов гэйтинга и транспорта канала. Так дефекты воротного механизма при повышенной температуре были выявлены при мутациях delKPQ [137]; T1620M [92]; Y1795H [16]; G1935S [138]; Y1795C, A1330P [139]. Наконец, на активность Nav1.5 могут влиять такие факторы как ацидоз [37, 91], действие ксенобиотиков [70] и др.
ЗАКЛЮЧЕНИЕ
Электрофизиологические исследования на моделях гетерологической экспрессии являются актуальным и перспективным подходом для выявления корреляции генотип-фенотип. Важность данных исследований для клинической практики подчеркивается тем, что метод локальной фиксации потенциала (patch-clamp) позволяет выявлять чувствительность пациента к различным антиаритмикам и факторам внешней среды. Таким образом, биофизические исследования позволяют подобрать комплекс индивидуальных терапевтических мер для пациентов с конкретной мутацией в гене SCN5A, что подчеркивает перспективы данного направления в области персонализированной медицины. Кроме того, при мутациях, вызывающих смешанную клиническую картину, которая включает в себя проявления LQT3 и BrS1 (как, например, при мутации E1784K, описанной выше), выявлены как изменения биофизических свойств, характерные для увеличения, так и для уменьшения активности Nav1.5. Таким образом, данные исследования позволяют объяснить феномен смешанных фенотипов.
Изучение влияния патогенных мутаций на биофизические характеристики INa позволяет установить роль конкретных аминокислотных остатков в процессах перехода Nav1.5 между различными конформационными состояниями, и, таким образом, вносит существенный вклад в понимание механизмов функционирования натриевого канала в норме и патологии.
Список литературы
Balser J.R. The cardiac sodium channel: gating function and molecular pharmacology. J. Mol. Cell. Cardiol. 33(4): 599–613.2001.
Amin A.S., Asghari-Roodsari A., Tan H.L. Cardiac sodium channelopathies. Pflugers Arch. Eur. J. Physiol. 460(2): 223–237.2009.
Morita H., Wu J., Zipes D.P. The QT syndromes: long and short. Lancet. 372(9640): 750–763. 2008.
Kapplinger J.D., Tester D.J., Salisbury B.A., Carr J.L., Harris-Kerr C., Pollevick G.D., Wilde A.A.M., Ackerman M.J. Spectrum and prevalence of mutations from the first 2500 consecutive unrelated patients referred for the FAMILION® long QT syndrome genetic test. Hear. Rhythm. 6(9): 1297–1303. 2009.
Wang Q., Shen J., Splawski I., Atkinson D., Li Z., Robinson J.L. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 80(5): 805–811.1995.
Bennett P.B., Yazawa K., Makita N., George A.L. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 376(6542): 683–685.1995.
Kapplinger J.D., Giudicessi J.R., Ye D., Tester D.J., Callis T.E., Valdivia C.R., Makielski J.C., Wilde A. A., Ackerman M.J. Enhanced classification of brugada syndrome–associated and long-QT syndrome–associated genetic variants in the SCN5A-encoded Na v 1.5 cardiac sodium channel: clinical perspective. Circ. Cardiovasc. Genet. 8(4): 582–595. 2015.
Zimmer T., Surber R. SCN5A channelopathies - An update on mutations and mechanisms. Prog. Biophys. Mol. Biol. 98(2–3): 120–136.2008.
Tester D.J., Will M.L., Haglund C.M., Ackerman M.J. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Hear. Rhythm. 2(5): 507–517.2005.
Napolitano C., Priori S.G., Schwartz P.J., Bloise R., Ronchetti E., Nastoli J., Bottelli G., Cerrone M., Leonardi S. Genetic Testing in the Long QT Syndrome. JAMA. 294(23): 2975.2005.
Potet F., Chagot B., Anghelescu M., Viswanathan P.C., Stepanovic S.Z., Kupershmidt S., Chazin, Walter J.Balser, Jeffrey R. Functional interactions between distinct sodium channel cytoplasmic domains through the action of calmodulin. J. Biol. Chem. 284(13): 8846–8854. 2009.
Wedekind H., Smits J.P.P., Schulze-Bahr E., Arnold R., Veldkamp M.W., Bajanowski T., Borggrefe M., Brinkmann B., Warnecke I., Funke H., Bhuiyan Z.A., Wilde A.A.M., Breithardt G., Haverkamp W. De novo mutation in the SCN5A gene associated with early onset of sudden infant death. Circulation. 104(10): 1158–1164. 2001.
Ruan Y., Liu N., Bloise R., Napolitano C., Priori S.G. Gating properties of SCN5A mutations and the response to mexiletine in long-QT syndrome type 3 patients. Circulation. 116(10): 1137–1144. 2007.
Albert C.M., Nam E.G., Rimm E.B., Jin H.W., Hajjar R.J., Hunter D.J., MacRae C.A., Ellinor P.T. Cardiac sodium channel gene variants and sudden cardiac death in women. Circulation. 117(1): 16–23. 2008.
Clancy C.E., Tateyama M., Liu H., Wehrens X.H.T., Kass R.S. Non-equilibrium gating in cardiac Na+ channels: an original mechanism of arrhythmia. Circulation. 107(17): 2233–2237. 2003.
Rivolta I., Abriel H., Tateyama M., Liu H., Memmi M., Vardas P., Napolitano C., Priori S.G., Kass R.S. Inherited brugada and long QT-3 syndrome mutations of a single residue of the cardiac sodium channel confer distinct channel and clinical phenotypes. J. Biol. Chem. 276(33): 30 623–30 630. 2001.
Dun W., Boyden P.A. The Purkinje cell; 2008 style. J. Mol. Cell. Cardiol. 45(5): 617–624. 2008.
Haissaguerre M., Extramiana F., Hocini M., Cauchemez B., Jaïs P., Cabrera J.A., Farré, J., Farre G., Leenhardt A., Sanders P., Scavée, C., Hsu L.F., Weerasooriya R., Shah D.C., Frank R., Maury P., Delay M., Garrigue S., Clémenty J. Mapping and ablation of ventricular fibrillation associated with long-QT and brugada syndromes. Circulation. 108(8): 925–928. 2003.
Yan G.X., Wu Y., Liu T., Wang J., Marinchak R.A., Kowey P.R. Phase 2 early afterdepolarization as a trigger of polymorphic ventricular tachycardia in acquired long-QT syndrome : direct evidence from intracellular recordings in the intact left ventricular wall. Circulation. 103(23): 2851–2856. 2001.
Gütter C., Benndorf K., Zimmer T. Characterization of n-terminally mutated cardiac Na+ channels associated with long QT syndrome 3 and brugada syndrome. Front. Physiol. 4(6): 1–12. 2013.
Sottas V., Rougier J.S., Jousset F., Kucera J.P., Shestak A., Makarov L.M., Zaklyazminskaya E.V., Abriel H. Characterization of 2 genetic variants of Nav1.5-arginine 689 found in patients with cardiac arrhythmias. J. Cardiovasc. Electrophysiol. 24(9): 1037–1046.2013.
Ackerman M.J. Postmortem molecular analysis of SCN5A defects in sudden infant death syndrome. JAMA. 286(18): 2264.2001.
Tateyama M., Rivolta I., Clancy C.E., Kass R.S. Modulation of cardiac sodium channel gating by protein kinase a can be altered by disease-linked mutation. J. Biol. Chem. 278(47): 46 718–46 726. 2003.
Makita N., Shirai N., Nagashima M., Matsuoka R., Yamada Y., Tohse N., Kitabatake A. A de novo missense mutation of human cardiac Na+ channel exhibiting novel molecular mechanisms of long QT syndrome. FEBS Lett. 423(1): 5–9.1998.
Liu H., Clancy C., Cormier J., Kass R. Mutations in cardiac sodium channels: clinical implications. Am. J. Pharmacogenomics. 3(3): 173–179.2003.
Schwartz P.J., Priori S.G., Spazzolini C., Moss A.J., Vincent G.M., Napolitano C., Denjoy I., Guicheney P., Breithardt G., Keating M.T., Towbin J.A., Beggs A.H., Brink P., Wilde A.A., Toivonen L., Zareba W., Robinson J.L.,Timothy K.W., Corfield V., Wattanasirichaigoon D., Corbett C., Haverkamp W., Schulze-Bahr E., Lehmann M.H., Schwartz K., Coumel P., Bloise R. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 103(1): 89–95. 2001.
Lupoglazoff J.M., Cheav T., Baroudi G., Berthet M., Denjoy I., Cauchemez B., Extramiana F., Chahine M., Guicheney P. Homozygous SCN5A mutation in long-QT syndrome with functional two-to-one atrioventricular block. Circ. Res. 89(2): E16–21. 2001.
Chang C., Acharfi S., Wu M., Chiang F., Wang J., Sung T., Chanine M. A novel SCN5A mutation manifests as a malignant form of long QT syndrome with perinatal onset of tachycardia/bradycardia. Cardiovasc. Res. 64(2): 268–278. 2004.
Rivolta I., Clancy C.E., Tateyama M., Liu H., Priori S.G., Kass R.S. A novel SCN5A mutation associated with long QT-3: altered inactivation kinetics and channel dysfunction. Physiol. Genomics. 10(3): 191–197.2002.
Fatima A., Kaifeng S., Dittmann S., Xu G., Gupta M.K., Linke M., Zechner U., Nguemo F., Milting H., Farr M., Hescheler J., Šarić T. The disease-specific phenotype in cardiomyocytes derived from induced pluripotent stem cells of two long qt syndrome type 3 patients. PLoS One. 8(12): 1–11. 2013.
Yong S.L., Ni Y., Zhang T., Tester D.J., Ackerman M.J., Wang Q.K. Characterization of the cardiac sodium channel SCN5A mutation, N1325S, in single murine ventricular myocytes. Biochem. Biophys. Res. Commun. 352(2): 378–383. 2007.
Li Q., Huang H., Liu G., Lam K., Rutberg J., Green M.S., Birnie D.H., Lemery R., Chahine M., Gollob M.H. Gain-of-function mutation of Nav1.5 in atrial fibrillation enhances cellular excitability and lowers the threshold for action potential firing. Biochem. Biophys. Res. Commun. 380(1): 132–137. 2009.
Olesen M.S., Yuan L., Liang B., Hols A.G., Nielsen N., Nielsen J.B., Hedley P. L., Christiansen M., Olesen S.P., Haunsø S., Schmitt N., Jespersen T., Svendsen J.H. High prevalence of long QT syndrome-associated SCN5A variants in patients with early-onset lone atrial fibrillation. Circ. Cardiovasc. Genet. 5(4): 450–459. 2012.
Gardill B.R., Rivera-Acevedo R.E., Tung C.C., Okon M, McIntosh L.P., Van Petegem F. The voltage-gated sodium channel EF-hands form an interaction with the III–IV linker that is disturbed by disease-causing mutations. Sci. Rep. 8(1): 1–13. 2018.
An R.H., Wang X.L., Kerem B., Benhorin J., Medina A., Goldmit M., Kass, R.S. Novel LQT3 Mutation Affects Na+ Channel Activity Through Interactions Between a- and b1-Subunits. Circ. Res. 83: 141–146. 1998.
Ortiz-Bonnin B., Rinné S., Moss R., Streit A.K., Scharf M., Richter K., Stöber A., Pfeufer A., Seemann G., Kääb S., Beckmann B.M., Decher N. Electrophysiological characterization of a large set of novel variants in the SCN5A-gene: identification of novel LQTS3 and BrS mutations. Pflugers. Arch. Eur. J. Physiol. 468(8): 1375–1387. 2016.
Wang D.W., Desai R.R., Crotti L, Arnestad M., Insolia R., Pedrazzini M., Ferrandi C., Vege A., Rognum T., Schwartz P.J., George A.L. Cardiac sodium channel dysfunction in sudden infant death syndrome. Circulation. 115(3): 368–376.2007.
Malan D., Zhang M., Stallmeyer B., Müller J., Fleischmann B.K., Schulze-Bahr E., Sasse P., Greber B. Human iPS cell model of type 3 long QT syndrome recapitulates drug-based phenotype correction. Basic. Res. Cardiol. 111(2): 1–11. 2016.
Loussouarn G., Sternberg D., Nicole S., Marionneau C., Le Bouffant F., Toumaniantz G., Barc J., Malak O.A., Fressart V., Péréon Y., Baró I., Charpentier F. Physiological and pathophysiological insights of Nav1.4 and Nav1.5 comparison. Front. Pharmacol. 6(1): 214. 2016.
Wang Q., Chen S., Chen Q., Wan X., Shen J., Hoeltge G.A., Timur A.A., Keating M.T., Kirsch G.E. The common SCN5A mutation R1193Q causes LQTS-type electrophysiological alterations of the cardiac sodium channel. J. Med. Genet. 41(5): 1–7. 2004.
Wehrens X.H.T., Rossenbacker T., Jongbloed R.J., Gewillig M., Heidbüchel H., Doevendans P., Vos M. A., Wellens H.J.J.,Kass R.S. A novel mutation L619F in the cardiac Na+ channel SCN5A associated with long-QT syndrome (LQT3): a role for the I-II linker in inactivation gating. Hum. Mutat. 21(5): 552. (2003).
Frustaci A., Priori S.G., Pieroni M., Chimenti C., Napolitano C., Rivolta I., Sanna T., Bellocci F., Russo M.A. Cardiac Histological Substrate in Patients With Clinical Phenotype of Brugada Syndrome.Circulation. 112(24): 3680–3687. 2005.
Huang H., Priori S.G., Napolitano C., O’Leary M.E., Chahine M. Y1767C, a novel SCN5A mutation, induces a persistent Na + current and potentiates ranolazine inhibition of Na v 1.5 channels. Am. J. Physiol. Circ. Physiol. 300(1): H288–H299. 2011.
Liu K., Yang T., Viswanathan P.C., Roden D.M. New mechanism contributing to drug-induced arrhythmia: rescue of a misprocessed LQT3 mutant. Circulation. 112(21): 3239–3246. 2005.
Makita N., Horie M., Nakamura T., Ai T., Sasaki K., Yokoi H., Sakurai M., Sakuma I., Otani H., Sawa H., Kitabatake A. Drug-induced long-QT syndrome associated with a subclinical SCN5A mutation. Circulation. 106(10): 1269–1274. 2002.
Yan H., Wang C., Marx S.O, Pitt G.S. Calmodulin limits pathogenic Na + channel persistent current. J. Gen. Physiol. 149(2): 277–293. 2017.
Bankston J.R., Sampson K.J., Kateriya S., Glaaser I.W., Malito D.L., Chung W.K., Kass R.S. A novel LQT3 mutation disrupts an inactivation gate complex with distinct rate-dependent phenotypic consequences. Channels. 1(4): 273–280. 2007.
Mann S.A., Castro M.L., Ohanian M., Guo G., Zodgekar P., Sheu A., Stockhammer K., Thompson T., Playford D., Subbiah R., Kuchar D., Aggarwal A., Vandenberg J.I., Fatkin D. R222Q SCN5A mutation is associated with reversible ventricular ectopy and dilated cardiomyopathy. J. Am. Coll. Cardiol. 60(16): 1566–1573. 2012.
Abriel H., Cabo C., Wehrens X.H.T., Rivolta I., Motoike H.K., Memmi M., Napolitano C., Priori S.G., Kass, R.S. Novel arrhythmogenic mechanism revealed by a long-QT syndrome mutation in the cardiac Na+channel. Circ. Res. 88(7): 740–745. 2001.
Huang H., Millat G., Rodriguez-Lafrasse C., Rousson R., Kugener B., Chevalier P., Chahine M. Biophysical characterization of a new SCN5A mutation S1333Y in a SIDS infant linked to long QT syndrome. FEBS Lett. 583(5): 890–896. 2009.
Surber R., Hensellek S., Prochnau D., Werner G.S., Benndorf K., Figulla H.R., Zimmer T. Combination of cardiac conduction disease and long QT syndrome caused by mutation T1620K in the cardiac sodium channel. Cardiovasc. Res. 77(4): 740–748. 2008.
Bankston J.R., Yue M., Chung W., Spyres M., Pass R.H., Silver E., Sampson K.J., Kass R.S. A novel and lethal De Novo LQT3 mutation in a newborn with distinct molecular pharmacology and therapeutic response. PLoS One. 2(12): e1258. 2007.
Wang D.W., Crotti L., Shimizu W., Pedrazzini M., Cantu F., De Filippo P., Kishiki K., Miyazaki A., Ikeda T., Schwartz P.J., George A.L. Malignant perinatal variant of long-QT syndrome caused by a profoundly dysfunctional cardiac sodium channel. Circ. Arrhythm. Electrophysiol. 1(5): 370–378. 2008.
Valdivia C.R., Ackerman M.J., Tester D.J., Wada T., McCormack J., Ye B., Makielski, J.C. A novel SCN5A arrhythmia mutation, M1766L, with expression defect rescued by mexiletine. Cardiovasc. Res. 55(2): 279–89. 2002.
Makita N., Behr E., Shimizu W., Horie M., Sunami A., Crotti L., Schulze-bahr E., Fukuhara S., Mochizuki N., Makiyama T., Itoh H., Christiansen M., Mckeown P., Miyamoto K., Kamakura S., Tsutsui H., Schwartz P.J., Roden D.M. The E1784K mutation in SCN5A is associated with mixed clinical phenotype of type 3 long QT syndrome. J. Clin. Invest. 118(6): 2219–2229. 2008.
Antzelevitch C., Brugada P., Brugada J., Brugada R. Brugada syndrome: from cell to bedside. Curr. Probl. Cardiol. 6(9): 2166–2171. 2005.
Priori S.G., Wilde A.A., Horie M., Cho Y., Behr E.R., Berul C., Blom N., Brugada J., Chiang C., Huikuri H., Kannankeril P., Krahn A., Leenhardt A., Moss A., Schwartz P.J., Shimizu W., Tomaselli G., Tracy C. Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Hear. Rhythm. 10(12): e85–e108. 2013.
Antzelevitch C., Brugada P., Borggrefe M., Brugada J., Brugada R., Corrado D. Gussak I., LeMarec H., Nademanee K., Perez Riera A.R., Shimizu W., Schulze-Bahr E., Tan H., Wilde A. Brugada syndrome: report of the second consensus conference: endorsed by the heart rhythm society and the european heart rhythm association. Circulation. 111(5): 659–670. 2005.
Manohar S., Dahal B.R., Gitler B. Fever-induced brugada syndrome. J. Investig. Med. high impact case reports. 3(1): 2 324 709 615 577 414. 2015.
Miyazaki T., Mitamura H., Miyoshi S., Soejima K., Aizawa Y., Ogawa S. Autonomic and antiarrhythmic drug modulation of ST segment elevation in patients with Brugada syndrome. J. Am. Coll. Cardiol. 27(5): 1061–1070. 1996.
Peters C.H., Abdelsayed M., Ruben P.C. Triggers for arrhythmogenesis in the Brugada and long QT 3 syndromes. Prog. Biophys. Mol. Biol. 120(1–3): 77–88. 2016.
Keller D.I., Rougier J.S., Kucera J.P., Benammar N., Fressart V., Guicheney P., Madle A., Fromer M., Schläpfer J., Abriel H. Brugada syndrome and fever: Genetic and molecular characterization of patients carrying SCN5A mutations. Cardiovasc. Res. 67(3): 510–519. 2005.
Matsuo K., Kurita T., Inagaki M., Kakishita M., Aihara N., Shimizu W., Taguchi A., Suyama K., Kamakura S., Shimomura K. The circadian pattern of the development of ventricular fibrillation in patients with Brugada syndrome. Eur. Heart J. 20(6): 465–470. 1999.
Mizumaki K., Fujiki A., Tsuneda T., Sakabe M., Nishida K., Sugao M., Inoue H. Vagal activity modulates spontaneous augmentation of ST elevation in the daily life of patients with brugada syndrome. J. Cardiovasc. Electrophysiol. 15(6): 667–673. 2004.
Зайцева А.К., Карпушев А.В., Михайлов Е.Н., Жоров Б.С., Костарева А.А. Молекулярные механизмы синдрома бругада подтипа 1. Трансл. Мед. 4(4): 23–35. 2017. [Zaytseva A.K., Karpushev A.V., Mikhaylov E.N., Zhorov B.S., Kostareva A.A. Molecular mechanisms of brugada syndome subtype 1. Transl. Med. 4(4): 23–35. 2017. (In Russ.)].
Makita N., Sumitomo N., Watanabe I., Tsutsui H. Novel SCN5A mutation (Q55X) associated with age-dependent expression of Brugada syndrome presenting as neurally mediated syncope. Hear. Rhythm. 4(4): 516–519. 2007.
Makiyama T., Akao M., Tsuji K., Doi T., Ohno S., Takenaka K., Kobori A., Ninomiya T., Yoshida H., Takano M., Makita N., Yanagisawa F., Higashi Y., Takeyama Y., Kita T., Horie M. High risk for bradyarrhythmic complications in patients with Brugada syndrome caused by SCN5A gene mutations. J. Am. Coll. Cardiol. 46(11): 2100–2106. 2005.
Keller D.I., Barrane F.Z., Gouas L., Martin J., Pilote S., Suarez V., Osswald S., Brink M., Guicheney P., Schwick N., Chahine M. A novel nonsense mutation in the SCN5A gene leads to Brugada syndrome and a silent gene mutation carrier state. Can. J. Cardiol. 21(11): 925–931. 2005.
Baroudi G., Napolitano C., Priori S.G., Del Bufalo A., Chahine M. Loss of function associated with novel mutations of the SCN5A gene in patients with Brugada syndrome. Can. J. Cardiol. 20(4): 425–430. 2004.
Kranjec D., Bergoveg M., Rouger J.S., Raguz M., Pavlovic S., Jesperson T., Castella V., Keller D.I., Abriel H. Brugada syndrome unmasked by accidental inhalation of gasoline vapors. Pacing Clin. Electrophysiol. 30(10): 1294–1298. 2007.
Shin D.J., Kim E., Park S.B., Jang W.C., Bae Y., Han J., Jang Y., Joung B., Lee M.H., Kim S.S., Huang H., Chahine M., Yoon S.K. A novel mutation in the SCN5A gene is associated with Brugada syndrome. Life Sci. 80(8): 716–724. 2007.
Cordeiro J.M., Barajas-Martinez H., Hong K., Burashnikov E., Pfeiffer R., Orsino A.M., Wu Y.S., Hu D., Brugada J., Brugada P., Antzelevitch C., Dumaine R., Brugada R. Compound heterozygous mutations P336L and I1660V in the human cardiac sodium channel associated with the Brugada syndrome. Circulation. 114(19): 2026–2033. 2006.
Takehara N., Makita N., Kawabe J., Sato N., Kawamura Y., Kitabatake A., Kikuchi, K. A cardiac sodium channel mutation identified in Brugada syndrome associated with atrial standstill. J. Intern. Med. 255(1): 137–142. 2004.
Zhang Y., Wang T., Ma A., Zhou X., Gui J., Wan H., Shi R., Huang C., Grace A.A., Huang, C.L.H., Trump D., Zhang H., Zimmer T., Lei M. Correlations between clinical and physiological consequences of the novel mutation R878C in a highly conserved pore residue in the cardiac Na+ channel. Acta Physiol. 194(4): 311–323. 2008.
Kyndt F., Probst V., Potet F., Demolombe S., Chevallier J.C., Baro I., Moisan J.P., Boisseau P., Schott J.J., Escande, D. Le Marec H. Novel SCN5A mutation leading either to isolated cardiac conduction defect or Brugada syndrome in a large french family. Circulation. 104(25): 3081–3086. 2001.
Tan B.H., Valdivia C.R., Song C., Makielski J.C. Partial expression defect for the SCN5A missense mutation G1406R depends on splice variant background Q1077 and rescue by mexiletine. Am. J. Physiol. Heart Circ. Physiol. 291(4): H1822–H1828. 2006.
Six I., Hermida J.S., Huang H., Gouas L., Fressart V., Benammar N., Hainque B., Denjoy I., Chahine M., Guicheney P. The occurrence of Brugada syndrome and isolated cardiac conductive disease in the same family could be due to a single SCN5A mutation or to the accidental association of both diseases. Europace. 10(1): 79–85. 2008.
Vatta M., Dumaine R., Antzelevitch C., Brugada R., Li H., Bowles N.E., Nademanee K., Brugada J., Brugada P., Towbin J.A. Novel mutations in domain I of SCN5A cause Brugada syndrome. Mol. Genet. Metab. 75(4): 317–324. 2002.
Rossenbacker T., Carroll S.J., Liu H., Kuipéri C., de Ravel T.J.L., Devriendt K., Carmeliet P., Kass R.S., Heidbüchel H. Novel pore mutation in SCN5A manifests as a spectrum of phenotypes ranging from atrial flutter, conduction disease, and Brugada syndrome to sudden cardiac death. Hear. Rhythm. 1(5): 610–615. 2004.
Xiong Q., Cao L., Hu J., Maria A.J., Hong K. A Rare loss-of-function SCN5A variant is associated with lidocaine-induced ventricular fibrillatio. Pharmacogenomics. 26(12): 16–25. 2015.
Zhu J.F., Du L.L., Tian Y., Du Y.M., Zhang L., Zhou T., Tian L. Novel heterozygous mutation c.4282G>T in the SCN5A gene in a family with Brugada syndrome. Exp. Ther. Med. 9(5): 1639–1645. 2015.
Tan B.Y., Yong R.Y.Y., Barajas-Martinez H., Dumaine R., Chew Y.X., Wasan P.S., Ching C.K., Ho K.L., Gan L.S.H., Morin N., Chong A.P.L., Yap S.H., Neo J.L., Yap E.P.H., Moochhala S., Chong D.T.T., Chow W., Seow S.C., Hu D., Uttamchandani M., Teo W.S.A Brugada syndrome proband with compound heterozygote SCN5A mutations identified from a Chinese family in Singapore. Europace. 18(6): 897–904. 2016.
Marangoni S., Di Resta C., Rocchetti M., Barile L., Rizzetto R., Summa A., Severi S., Sommariva E., Pappone C., Ferrari M., Benedetti S., Zaza A. A Brugada syndrome mutation (p.S216L) and its modulation by p.H558R polymorphism: Standard and dynamic characterization. Cardiovasc. Res. 91(4): 606–616. 2011.
Pfahnl A.E., Viswanathan P.C., Weiss R., Shang L.L., Shamrendra S., Shusterman V., Kornblit C., London B., Dudley S.C. A sodium channel pore mutation causing Brugada syndrome. Hear. Rhythm. 27(4): 590–609. 2007.
Olson T.M., Michels V.V., Ballew J.D., Reyna S.P., Karst M.L., Herron K.J., Horton Steven C., Rodeheffer R.J, Anderson J.L. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA. 293(4): 447. 2005.
Baroudi G., Pouliot V., Denjoy I., Guicheney P., Shrier A., Chahine M. Novel mechanism for Brugada syndrome surface defective mutant scna R1432G. Circ. Res. 88: e78–e83. 2001.
Valdivia C.R., Tester D.J., Rok B.A., Porter C.B.J., Munger T.M., Jahangir A., Makielski J.C., Ackerman M.J. A trafficking defective, Brugada syndrome-causing SCN5A mutation rescued by drugs. Cardiovasc. Res. 62(1): 53–62. 2004.
Clatot J., Ziyadeh-Isleem A., Maugenre S., Denjoy I., Liu H., Dilanian G., Hatem S.N., Deschênes I., Coulombe A., Guicheney P., Neyroud N. Dominant-negative effect of SCN5A N-terminal mutations through the interaction of Nav1.5 α-subunits. Cardiovasc. Res. 96(1): 53–63. 2012.
Mohler P.J., Rivolta I., Napolitano C., LeMaillet G., Lambert S., Priori S.G., Bennett V. Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes. Proc. Natl. Acad. Sci.USA. 101(50): 17 533–17 538. 2004.
Aiba T., Farinelli F., Kostecki G., Hesketh G.G., Edwards D., Biswas S., Tung L., Tomaselli G.F. A mutation causing brugada syndrome identifies a mechanism for altered autonomic and oxidant regulation of cardiac sodium currents. Circ. Cardiovasc. Genet. 7(3): 249–256. 2014.
Zheng J., Zhou F., Su T., Huang L., Wu Y., Yin K., Qiuping W., Tang S., Makielski J.C., Cheng J. The biophysical characterization of the first SCN5A mutation R1512W identified in Chinese sudden unexplained nocturnal death syndrome. Medicine. 95(23): e3836. 2016.
Baroudi G., Carbonneau E., Pouliot V., Chahine M. SCN5A mutation (T1620M) causing Brugada syndrome exhibits different phenotypes when expressed in Xenopus oocytes and mammalian cells. FEBS Lett. 467(1): 12–16. 2000.
Smits J.P.P., Koopmann T.T., Wilders R., Veldkamp M.W., Opthof T., Bhuiyan Z.A., Mannens M.M., Balser J.R., Tan H.L., Bezzina C.R., Wilde A.A.M. A mutation in the human cardiac sodium channel (E161K) contributes to sick sinus syndrome, conduction disease and Brugada syndrome in two families. J. Mol. Cell Cardiol. 38(6): 969–981. 2005.
Kinoshita K., Takahashi H., Hata Y., Nishide K., Kato M., Fujita H., Yoshida S., Murai K., Mizumaki K., Nishida K., Yamaguchi Y., Kano M., Tabata T., Nishida N. SCN5A(K817E), a novel Brugada syndrome-associated mutation that alters the activation gating of NaV1.5 channel. Hear. Rhythm. 13(5): 1113–1120. 2016.
Casini S., Tan H.L., Bhuiyan Z.A., Bezzina C.R., Barnett P., Cerbai E., Mugelli A., Wilde. A.A.M., Veldkamp M.W. Characterization of a novel SCN5A mutation associated with Brugada syndrome reveals involvement of DIIIS4-S5 linker in slow inactivation. Cardiovasc. Res. 76(3): 418–429 (2007).
Keller D.I., Huang H., Zhao J., Frank R., Suarez V., Delacrétaz E., Brink M., Osswald S., Schwick N., Chahine M. A novel SCN5A mutation, F1344S, identified in a patient with Brugada syndrome and fever-induced ventricular fibrillation. Cardiovasc. Res. 70(3): 521–529. 2006.
Tan H.L., Bink-Boelkens M.T.E., Bezzina C.R., Viswanathan P.C., Beaufort-Krol G.C.M., van Tintelen P.J., van den Berg M.P., Wilde A.A.M., Balser J.R. A sodium-channel mutation causes isolated cardiac conduction disease. Nature. 409(6823): 1043–1047. 2001.
Hsueh C.H., Chen W.P., Lin J.L., Tsai C.T., Liu Y.B., Juang J.M., Tsao H.M., Su M.J., Lai L.P. Distinct functional defect of three novel Brugada syndrome related cardiac sodium channel mutations. J. Biomed. Sci. 16(1): 23. 2009.
Medeiros-Domingo A., Tan B.H., Torres P.I., Tester D.J., Luna T., Makielski J.C., Ackerman M.J. Unique mixed phenotype and unexpected functional effect revealed by novel compound heterozygosity. Hear. Rhythm. 6(8): 1170–1175. 2009.
Wang L., Meng X., Yuchi Z., Zhao Z., Xu D., Fedida D., Wang Z., Huang C. De novo mutation in the SCN5A gene associated with brugada syndrome. Cell. Physiol. Biochem. 36(6): 2250–2262. 2015.
Veldkamp M.W., Viswanathan P.C., Bezzina C., Baartscheer A., Wilde A.A., Balser J.R. Two distinct congenital arrhythmias evoked by a multidysfunctional Na(+) channel. Circ. Res. 86(9): E91–E97. 2000.
Wang D.W., Viswanathan P.C., Balser J.R., George A.L., Benson D.W. Clinical, genetic, and biophysical characterization of SCN5A mutations associated with atrioventricular conduction Block. Circulation. 341–346. 2002.
Itoh H, Tsuji K, Sakaguchi T, Nagaoka I, Oka Y, Nakazawa Y, Yao T., Jo H., Ashihara T., Ito M., Horie M., Imoto K. A paradoxical effect of lidocaine for the N406S mutation of SCN5A associated with Brugada syndrome. Int. J. Cardiol. 121(3): 239–248. 2007.
Yokoi H., Makita N., Sasaki K., Takagi Y., Okumura Y.., Nishino T, Makiyama T., Kitabatake A., Horie M., Watanabe I., Tsutsui H. Double SCN5A mutation underlying asymptomatic Brugada syndrome. Hear. Rhythm. 2(3): 285–292. 2005.
Wang D.W., Makita N., Kitabatake A., Balser J.R., George A.L. Enhanced Na(+) channel intermediate inactivation in Brugada syndrome. Circ. Res. 87(8): E37–E43. 2000.
Zeng Z., Zhou J., Hou Y., Liang X., Zhang Z., Xu X., Xie Q., Li W., Huang Z. Electrophysiological characteristics of a SCN5A voltage sensors mutation R1629Q associated with brugada syndrome. PLoS One. 8(10): 1–8. 2013.
Shirai N., Makita N., Sasaki K., Yokoi H., Sakuma I. A mutant cardiac sodium channel with multiple biophysical defects associated with overlapping clinical features of Brugada syndrome and cardiac conduction disease. Cardiovasc. Res 53: 348–354. 2002.
Amin A.S., Verkerk A.O., Bhuiyan Z.A., Wilde A.A.M., Tan H.L. Novel Brugada syndrome-causing mutation in ion-conducting pore of cardiac Na+ channel does not affect ion selectivity properties. Acta Physiol. Scand. 185(4): 291–301. 2005.
Moreau A., Gosselin-Badaroudine P., Delemotte L., Klein M.L., Chahine M. Gating pore currents are defects in common with two Na v 1.5 mutations in patients with mixed arrhythmias and dilated cardiomyopathy. J. Gen. Physiol. 145(2): 93–106. 2015.
Mok N.S., Priori S.G., Napolitano C., Chan N.Y., Chahine M., Baroudi G. A newly characterized SCN5A mutation underlying Brugada syndrome unmasked by hyperthermia. J. Cardiovasc. Electrophysiol. 14(4): 407–411. 2003.
Beyder A., Mazzone A., Strege P.R., Tester D.J., Saito Y.A., Bernard C.E., Enders F.T., Ek W.E., Schmidt PT., Lindberg G., Karling P., Ohlsson B., Gazouli M., Cuomo R., Usai-satta P., Galeazzi F., Neri M., Portincasa P., Bellini M., Barbara G., Camilleri M., Richard G., Talley N.J., Amato M.D., Ackerman M.J. Loss-of-function of the voltage-gated sodium channel NaV1.5 (Channelopathies) in Patients with Irritable Bowel Syndrome. Gastroenterology. 146(7): 1659–1668. 2015.
Wang H.G., Zhu W., Kanter R.J., Silva J.R., Honeywell C., Gow R.M., Pitt G.S. A novel NaV1.5 voltage sensor mutation associated with severe atrial and ventricular arrhythmias. J. Mol. Cell. Cardiol. 92: 52–62. 2016.
Kanters J.K., Yuan L., Hedley P.L., Stoevring B., Jons C, Bloch Thomsen P.E., Grunnet M., Christiansen M., Jespersen T. Flecainide provocation reveals concealed brugada syndrome in a long QT syndrome family with a novel L1786Q mutation in SCN5A. Circ. J. 78(5): 1136–1143. 2014.
Hoshi M., Du X.X., Shinlapawittayatorn K., Liu H., Chai S., Wan X., Ficker E., Deschênes I. Brugada syndrome disease phenotype explained in apparently benign sodium channel mutations. Circ. Cardiovasc. Genet. 23(1): 1–7. 2014.
Gando I., Morganstein J., Jana K., McDonald T.V., Tang Y., Coetzee W.A. Infant sudden death: mutations responsible for impaired Nav1.5 channel trafficking and function. Pacing Clin. Electrophysiol. 2017.
Itoh H., Shimizu M., Takata S., Mabuchi H., Imoto K. A novel missense mutation in the SCN5A gene associated with Brugada syndrome bidirectionally affecting blocking actions of antiarrhythmic drugs. J. Cardiovasc. Electrophysiol. 16(5): 486–493. 2005.
Hoogendijk M.G., Potse M., Linnenbank A.C., Verkerk A.O., den Ruijter H.M., van Amersfoorth S.C.M., Klaver E.C., Beekman L., Bezzina C.R., Postema P.G., Tan H.L., Reimer A.G., van der Wal A.C., ten Harkel A.D.J., Dalinghaus M., Vinet A., Wilde A.A.M., de Bakker J.M.T., Coronel R. Mechanism of right precordial ST-segment elevation in structural heart disease: Excitation failure by current-to-load mismatch. Hear. Rhythm. 7(2): 238–248. 2010.
Akai J., Makita N., Sakurada H., Shirai N., Ueda K., Kitabatake A., Nakazawa K., Kimura A., Hiraoka M. A novel SCN5A mutation associated with idiopathic ventricular fibrillation without typical ECG findings of Brugada syndrome. FEBS Lett. 479: 29–34. 2000.
Itoh H., Shimizu M., Mabuchi H., Imoto K. Clinical and electrophysiological characteristics of Brugada syndrome caused by a missense mutation in the S5-pore site of SCN5A. J. Cardiovasc. Electrophysiol. 16(4): 378–383. 2005.
Chiang K.C., Lai L.P., Shieh R.C. Characterization of a novel Nav1.5 channel mutation, A551T, associated with Brugada syndrome. J. Biomed. Sci. 16: 76. 2009.
Nakajima T., Kaneko Y., Saito A., Ota M., Iijima T., Kurabayashi M. Enhanced fast-inactivated state stability of cardiac sodium channels by a novel voltage sensor SCN5A mutation, R1632C, as a cause of atypical Brugada syndrome. Hear. Rhythm. 12(11): 2296–2304. 2015.
Chen T., Inoue M., Sheets M.F. Reduced voltage dependence of inactivation in the SCN5A sodium channel mutation delF1617. Am. J. Physiol. Circ. Physiol. 288(6): H2666–H2676. 2005.
Vatta M., Dumaine R., Varghese G., Richard T.A., Shimizu W., Aihara N., Nademanee K., Brugada R., Brugada J., Veerakul G., Li H., Bowles N.E., Brugada P., Antzelevitch C., Towbin J.A. Genetic and biophysical basis of sudden unexplained nocturnal death syndrome (SUNDS), a disease allelic to Brugada syndrome. Hum. Mol. Genet. 11(3): 337–345. 2002.
Zhang Z.S., Tranquillo J., Neplioueva V., Bursac N., Grant A.O. Sodium channel kinetic changes that produce Brugada syndrome or progressive cardiac conduction system disease. Am. J. Physiol. Circ. Physiol. 292(1): H399–H407. 2007.
Liang P., Liu W., Hu D., Li C., Tao W., Li L. Novel SCN5A gene mutations associated with Brugada syndrome: V95I, A1649V and delF1617. Zhonghua Xin Xue Guan Bing Za Zhi. 34(7): 616–9. 2006.
Grant A.O., Carboni M.P., Neplioueva V., Starmer C.F., Memmi M., Napolitano C., Priori S. Long QT syndrome, Brugada syndrome, and conduction system disease are linked to a single sodium channel mutation. J. Clin. Invest. 110(8): 1201–1209. 2002.
Wan X., Chen S., Sadeghpour A., Wang Q., Kirsch G.E. Accelerated inactivation in a mutant Na(+) channel associated with idiopathic ventricular fibrillation. Am. J. Physiol. Heart Circ. Physiol. 280(1): H354–H360. 2001.
Balse E., Eichel C. The cardiac sodium channel and its protein partners. Springer Int. Publ. 2017.
Tateyama M., Kurokawa J., Terrenoire C., Rivolta I., Kass R.S. Stimulation of protein kinase C inhibits bursting in disease-linked mutant human cardiac sodium channels. Circulation. 107(25): 3216–3222. 2003.
Wu L., Archacki S.R., Zhang T., Wang Q.K. Induction of high STAT1 expression in transgenic mice with LQTS and heart failure. Biochem. Biophys. Res. Commun. 358(2): 449–454. 2007.
Bezzina C.R., Rook M.B., Groenewegen W.A., Herfst L.J., van der Wal A.C., Lam J., Jongsma H.J., Wilde A.A.M., Mannens M.M. Compound heterozygosity for mutations (W156X and R225W) in SCN5A associated with severe cardiac conduction disturbances and degenerative changes in the conduction system. Circ. Res. 92(2): 159–168. 2003.
Ackerman M.J., Splawski I., Makielski J.C., Tester D.J., Will M.L., Timothy K.W., Keating M.T., Jones G., Chadha M., Burrow C.R., Stephens J.C., Xu C., Judson R., Curran M.E. Spectrum and prevalence of cardiac sodium channel variants among black, white, Asian, and Hispanic individuals: Implications for arrhythmogenic susceptibility and Brugada/long QT syndrome genetic testing. Hear. Rhythm. 1(5): 600–607. 2004.
Viswanathan P.C., Benson D.W., Balser J.R. A common SCN5A polymorphism modulates the biophysical effects of an SCN5A mutation. J. Clin. Invest. 111(3): 341–346. 2003.
Ye B., Valdivia C.R., Ackerman M.J., Makielski J.C. A common human SCN5A polymorphism modifies expression of an arrhythmia causing mutation. Physiol.Genomics. 12(3): 187–193. 2003.
Tan B.H., Valdivia C.R., Rok B.A., Ye B., Ruwaldt K.M., Tester D.J., Ackerman M.J., Makielski J.C. Common human SCN5A polymorphisms have altered electrophysiology when expressed in Q1077 splice variants. Hear. Rhythm. 2(7): 741–747. 2005.
Makielski J.C., Ye B., Valdivia C.R., Pagel M.D., Pu J., Tester D.J., Ackerman M.J. A Ubiquitous splice variant and a common polymorphism affect heterologous expression of recombinant human SCN5A heart sodium channels. Circ. Res. 93(9): 821–828. 2003.
Nagatomo T., Fan Z., Ye B., Tonkovich G.S., January C.T., Kyle J.W., Makielski J.C. Temperature dependence of early and late currents in human cardiac wild-type and long Q-T DeltaKPQ Na+ channels. Am. J. Physiol. 275(6 Pt 2): H2016–H2024. 1998.
Coronel R., Casini S., Koopmann T.T., Wilms-Schopman F.J.G., Verkerk A.O., de Groot J.R., Bhuiyan Z., Bezzina C.R., Veldkamp M.W., Linnenbank A.C., van der Wal A.C., Tan H.L., Brugada P., Wilde A.A.M., de Bakker J.M.T. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: a combined electrophysiological, genetic, histopathologic, and computational study. Circulation. 112(18): 2769–2777. 2005.
Berecki G., Zegers J.G., Bhuiyan Z.A., Verkerk A.O., Wilders R., Van Ginneken A.C.G. Long-QT syndrome-related sodium channel mutations probed by the dynamic action potential clamp technique. J. Physiol. 570(2): 237–250. 2006.
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова