

Российский физиологический журнал им. И.М. Сеченова, 2019, T. 105, № 9, стр. 1067-1095
Незаслуженно забытые: место глиальных клеток в гипотезах возникновения болезни Альцгеймера
Л. А. Кушнирёва 1, *, Э. А. Коркотян 2, А. В. Семьянов 3
1 Пермский государственный национальный исследовательский университет
Пермь, Россия
2 Weizmann Institute of Science
Rehovot, Israel
3 Институт биоорганической химии им. академиков М.М. Шемякина
и Ю.А. Овчинникова РАН
Москва, Россия
* E-mail: kushnireva@neuro.nnov.ru
Поступила в редакцию 09.07.2019
После доработки 09.07.2019
Принята к публикации 09.07.2019
Аннотация
Болезнь Альцгеймера – наиболее распространенная на сегодняшний день нейродегенеративная патология, характеризующаяся прогрессирующей потерей памяти и ухудшением когнитивных процессов. Современные терапевтические методы лечения способны лишь ненадолго замедлить ее развитие, но не излечить. До ближайшего десятилетия, в большинстве исследований, посвященных нейродегенеративным заболеваниям, изучались, в основном, нейрональные процессы. Такой подход, на наш взгляд, не может дать полной картины патологии. Нейроны, астроциты и микроглия реагируют на изменения в мозге согласованным и взаимозависимым образом. Помимо нейродегенеративных процессов, при болезни Альцгеймера наблюдается множество биохимических, структурных и пролиферативных изменений в астроцитах. Кроме того, клетки микроглии увеличивают свою плотность и переходят к активированным фенотипам. Исходные причины этих изменений до сих пор не выяснены. Некоторые исследования указывают на кальциевые сигналы в нейронах и астроцитах как на важный элемент этиологии заболевания. Воспалительный ответ астроцитов и микроглии, высвобождающий нейротоксические факторы, также нарушает баланс в ЦНС, усугубляя болезнь. В данном обзоре рассматриваются характерные для патологии процессы, связанные с утратой синапсов и нейронов, образованием нейрофибриллярных клубков и амилоидных отложений, но основное внимание сосредоточено на отклонениях в функционировании глиальных клеток при болезни Альцгеймера.
Болезнь Альцгеймера (БА), описанная в 1907 г. немецким психиатром Алоизиусом (Алоисом) Альцгеймером [1], является длительно текущим нейродегенеративным заболеванием мозга, в ходе которого наблюдается прогрессирующая потеря памяти и снижение когнитивных способностей, с последующей утратой основных функций организма и летальным исходом. На сегодняшний день БА является самым распространенным нейродегенеративным заболеванием в мире. По данным Alzheimer’s Disease International на 2015 г. около 46.8 млн человек страдало от этого заболевания, и, по прогнозам, к 2050 г. это число достигнет 131.5 млн [2]. Заболевание приносит огромные финансовые потери государствам, а также социальные и психологические проблемы для семей заболевших. Было проведено значительное количество исследований, посвященных патологическим процессам в нейронах, связанных с утратой синапсов, образованием нейрофибриллярных клубков и амилоидных отложений. Благодаря этим исследованиям, на сегодняшний день удается временно замедлить прогрессирование заболевания с помощью лекарственных препаратов, но до сих пор не разработано терапевтических методик, позволяющих полностью излечить больных или хотя бы остановить нейродегенерацию. Выглядит несколько удивительным тот факт, что БА, как и другие нейродегенеративные заболевания, продолжают рассматривать, в первую очередь, с нейроцентрической точки зрения, хотя патологический вклад глиальных клеток в нейродегенерацию был признан уже в начале XX века и отмечался самим Алоисом Альцгеймером [3]. Экспериментальные данные последних десятилетий указывают на значимость глиальных клеток, а именно, астроцитов и микроглии, в патогенезе БА [4–6]. Как правило, глиальным реакциям отводится скромная роль во второстепенных событиях, которые, несмотря на защитную направленность, могут привести к ухудшению состояния мозговой ткани. Переход микроглии в активированное состояние в ответ на нейротоксическое воздействие бета-амилоида (Aβ) влечет за собой обширную иммунную реакцию. Астроциты тоже активируются ближайшими скоплениями Aβ, а также и цитокинами и хемокинами, вырабатываемыми микроглией, в свою очередь, высвобождая провоспалительные факторы и усиливая активацию микроглии [7]. Окружая отложения Aβ, астроциты, совместно с микроглией, снижают опосредованную Aβ нейротоксичность посредством его деградации и фагоцитоза. Однако воспаление – обоюдоострый процесс, так как попытка удаления Aβ способствует нейротоксичности через чрезмерное высвобождение цитокинов и влечет за собой нейродегенерацию.
Но глиальные клетки способны выступать инициатором и более ранних стадий заболевания. В ряде исследований сообщается о нарушениях кальциевого баланса и кальциевых колебаний в астроцитах из мозга мышей с моделью БА [8–10]. Как известно, кальций является универсальным внутриклеточным посредником, выполняя множество фундаментальных регуляторных и сигнальных функций в клетке. Изменения концентрации ионов кальция, ограниченные во времени и пространстве, влияют на высвобождение трансмиттеров/гормонов, синаптическую передачу и пластичность, в итоге приводя к изменениям в работе нейронов и глиальных клеток. Дисрегуляция кальциевого гомеостаза, развивающаяся при БА, поддерживает гипотезу “хронической кальцитопатии” [11], но она не различает нейрональные и глиальные изменения кальциевой динамики. Механизмы Aβ-индуцированного увеличения уровня кальция в большинстве своем описаны в нейронах. Необходимо исследовать, действуют ли те же пути в глиальных клетках и особенно в астроцитах, которые по праву могут считаться участниками процесса обработки информации в мозге, благодаря своим внутренним кальциевым осцилляциям и генерации межклеточных сигналов кальция. Уточнение биофизических, биохимических и физиологических механизмов, лежащих в основе регуляции кальциевых сигналов в глиальных клетках, откроет исследователям дополнительные, а возможно, и основные рычаги воздействия на патологический механизм развития болезни Альцгеймера и других нейродегенераций.
ХАРАКТЕРИСТИКА БОЛЕЗНИ АЛЬЦГЕЙМЕРА
Описательная симптоматика БА, регулярно применявшаяся в течение нескольких десятилетий, имеет ограниченную ценность, так как не позволяет выделить ее из ряда других, особенно цереброваскулярных заболеваний. Отграничение БА от широкого спектра деменций только на основе когнитивных нарушений затруднено. К счастью, в последние годы стало возможным прижизненное выявление БА на основе позитронно-эмиссионной томографии (ПЭТ) и определения уровня биомаркеров к ряду характерных белков в цереброспинальной жидкости.
Так или иначе, основным симптомом БА на ранних стадиях является прогрессирующее ухудшение кратковременной памяти и познавательных способностей. На более поздних стадиях происходит потеря долговременной памяти и нарушение элементарных функций организма (локомоция, дыхание, пищеварение), контролируемых ЦНС, что, в конечном итоге, приводит к летальному исходу [12]. Патологическими признаками БА является амилоидоз – отложение внеклеточных агрегатов, “бляшек” Aβ во внеклеточном пространстве головного мозга, в основном в гиппокампе и энторинальной коре. Согласно амилоидной гипотезе, перепроизводство Aβ является следствием нарушения гомеостатических процессов, которые регулируют протеолитическое расщепление белка предшественника Aβ (amyloid precursor protein, APP). Установление корреляции между невропатологическими данными от пациентов с БА и лиц среднего возраста с синдромом Дауна (трисомия хромосомы 21) привело к идентификации гена, который кодирует APP в той же хромосоме [13]. Также внутри нейронов образуются нейрофибриллярные клубки тау-белка [14]. Поздняя нейродегенерация поглощает височную, лобную и теменную доли коры головного мозга, где серое вещество подвергается значительной атрофии, что клинически проявляется тяжелой формой деменции [15]. Активация микроглии и реактивный астроглиоз также являются признаками поздних стадий заболевания [16].
ФОРМЫ И ФАКТОРЫ РИСКА
В зависимости от возраста больных различают БА с ранним началом (пресенильная форма) и с поздним началом (сенильная или спорадическая форма). Доля пациентов с ранним началом БА составляет приблизительно 6% от всех случаев, возраст заболевших при этом колеблется от 30 до 60–65 лет [12]. Около 60% случаев раннего начала БА являются семейными (т.е., наследуемыми генетически, проявляющейся не менее, чем в двух поколениях и у нескольких членов семьи), 13% от этих случаев наследуется по аутосомно-доминантному типу [17]. Разделяют три формы семейной БА с ранним началом, основываясь на лежащих в основе генетических механизмах: форму БА3, вызванную мутацией гена PSEN1 в хромосоме 14, кодирующего пресенелин-1 (PS1) (30–70% случаев); форму БА1, которая наблюдается в 10–15% случаев и вызвана мутацией гена APP, кодирующего белок-предшественник амилоида; и форму БА4, связанную с мутацией гена PSEN2 в хромосоме 1, кодирующего пресенелин-2 (PS2), встречающуюся крайне редко [17]. Наблюдаются и случаи аутосомно-доминантной семейной БА без этих мутаций, что указывает на вовлечение некоторых других генов. Наиболее часто встречается БА с поздним началом (форма БА2, свыше 90% пациентов), возраст начала заболевания – от 60–65 лет [12]. На сегодняшний день основным генетическим фактором риска для позднего начала заболевания является ген APOE в хромосоме 19, в частности, аллель ε4, кодирующая изоформу белка аполипротеина E4 (ApoE4) [12]. ApoE участвует в обмене липидов и холестерина в мозге и синтезируется, в основном, астроцитами и микроглией, а также нейронами, но в условиях повреждения ЦНС [18]. Изоформа АpоE4 характеризуется сниженной способностью к стимуляции деградации Aβ микроглией и астроцитами, усиленным замедлением транспорта растворимых форм Aβ через гематоэнцефалический барьер в сравнении с изоформами E2 и E3, и другими негативными эффектами [18]. ApoE4 филогенетически является самой старой из трех изоформ, но она не была полностью исключена из генофонда, несмотря на существование более адаптивных аллелей APOE [19].
Гораздо реже встречаются генетические факторы риска, связанные с фосфатидилинозитол-связывающим белком сборки клатрина (phosphatidylinositol binding clathrin assembly protein, PICALM) [20]; антигеном миелоидной клеточной поверхности CD33 (или Siglec-3, sialic acid binding Ig-like lectin 3), который экспрессируется, в основном, в микроглиальных клетках [21, 22]; белком АТФ-связывающего кассетного транспортера (ATP-binding cassette transporter, подсемейство A член 7), ABCA7; кластерином (CLU, или аполипопротеином J, ApoJ) [23] и рядом других: предполагается, что они играют роль в молекулярных путях удаления Aβ. Особый интерес представляет ген TREM2 (triggering receptor expressed on myeloid cells 2), врожденного иммунного рецептора TREM2, который экспрессируется в миелоидных клетках (в мозге – в клетках микроглии). TREM2 способствует микроглиальному фагоцитозу Aβ, подавляет выработку воспалительных цитокинов и усиливает транскрипцию противовоспалительных цитокинов [24]. Гетерозиготные миссенс-мутации в TREM2 являются факторами риска развития БА [25]. Экспрессия TREM2 увеличивается в мозге пациентов с БА, что расценивается как попытка микроглиальных клеток поддержать баланс и/или усилить воспалительный ответ в условиях нейродегенерации, возможно с участием эпигенетических механизмов [24, 26]. В результате мутаций фагоцитарная функция TREM2 нарушается, что может ускорить течение заболевания, снижая возраст начала и сокращая продолжительность БА [27].
К факторам риска БА также следует отнести сосудистые дисфункции, вызванные ожирением [28], высоким уровнем холестерина [29], сахарным диабетом [30], а также курение [31], недостаточную физическую активность, низкий уровень образования и социальную изоляцию [32]. Комплекс профилактических мер, включающий в себя диету, физические нагрузки, когнитивный тренинг и снижение риска сердечно-сосудистых заболеваний показал некоторую эффективность, хоть и не предотвращал БА [33].
Данные мета-анализов говорят о том, что спорадическая форма БА встречается в любых географических популяциях мира примерно с равной частотой и имеет примерно одинаковую возрастную распространенность в разных регионах [2, 34, 35]. Некоторые свидетельства влияния расовых различий на риск развития БА и других деменций являются предметом споров и могут быть объяснены различной степенью предрасположенности к сердечно-сосудистым заболеваниям, а также социокультурными факторами [34, 36, 37]. Есть данные, указывающие на значительно более высокую распространенность заболевания среди женщин [36, 38]. Cообщалось, что у женщин аллель APOE-ε4 в большей степени повышает риск БА, чем у мужчин [39]. Исследования по выявлению половых различий в заболеваемости БА часто связывают эту закономерность с уровнем эстрогенов и влиянием перименопаузы [40, 41].
Возраст является наибольшим фактором риска развития БА, поскольку после 65 лет наблюдается почти экспоненциальный рост заболеваемости БА [34]. Однако некоторые посмертные оценки указывают на тенденцию, при которой распространенность заболевания достигает пика в возрасте ≈95 лет, а затем снижается [42]. Если эти данные подтвердятся в будущих исследованиях, это позволит отбросить предположение, что БА является неизбежным следствием старения и будет свидетельствовать в пользу генетической этиологии заболевания и/или о наличии у определенной группы людей фактора, препятствующего его развитию.
АМИЛОИД β
Генетические, возрастные, экологические и другие факторы могут способствовать нарушению процессов, которые регулируют протеолитическое расщепление APP [43]. APP представляет собой трансмембранный интегральный гликопротеин с массой 110–130 кДа и является одним из самых распространенных белков в ЦНС [44]. Он повсеместно экспрессируется в человеческих тканях и располагается в плазматической мембране клеток, а также в некоторых органеллах, таких как эндоплазматический ретикулум (ЭР), аппарат Гольджи и митохондрии [45]. Предполагается, что в нормальных физиологических условиях APP участвует в миграции клеток, удлинении аксонов, формировании синапсов, сигнальной трансдукции и синаптической пластичности [33, 46, 47], но его точные физиологические функции все еще изучаются. APP метаболизируется двумя различными взаимоисключающими путями: секреторным путем (или “неамилоидогенным”, α-секретазным) и “амилоидогенным” путем. Aβ пептиды образуются в “амилоидогенном” пути последовательного расщепления APP β-секретазой (β-site amyloid precursor protein cleaving enzyme 1, BACE-1) с образованием C-концевого фрагмента длиной 99 аминокислот (С99), который затем расщепляется γ-секретазой, являющейся частью многосубъединичного комплекса с пресенелинами [43, 48] (рис. 1А). Aβ является нормальным продуктом метаболизма APP и генерируется в нейронах и других типах клеток на протяжении всей жизни человека [33]. Впоследствии Aβ транспортируется и разлагается изоформами ApoE2/E3 и полностью удаляется через гематоэнцефалический барьер вместе с ферментом, разлагающим инсулин (insulin degrading enzyme, IDE) или путем неприлизин-зависимой протеолитической деградации, однако связывание Aβ с изоформой ApoE4 в значительной степени способствует его агрегации во внеклеточном пространстве [47]. Агрегация Aβ проходит через несколько конформационных состояний, включая димеры, сферические олигомеры, состоящие из 10–24 мономеров, и цепочки олигомеров (протофибриллы), прежде чем, наконец, принять нерастворимую фибриллярную конформацию [49]. В здоровом мозге во время производства Aβ реализуется механизм обратной связи, который высвобождает внутриклеточный С-концевой домен APP и повышает уровень эндопептидазы неприлизина, способствующего удалению Aβ [47]. Мутации и изменения в экспрессии APP, BACE1, IDE, APOE приводят к перепроизводству и прогрессирующему накоплению Aβ [50]. Некоторые данные указывают на весьма быструю кинетику производства и удаление Aβ с периодом полураспада около 9 ч у здоровых людей [51]. Причины снижения клиренса амилоида у пациентов с БА доподлинно неизвестны, предполагается, что микроглия и астроциты теряют способность эффективно устранять токсичные формы Aβ (см. ниже).
Рис. 1.
Схема патологичеких процессов при болезни Альцгеймера: А – метаболизм APP идет преимущественно по амилоидогенному пути, в результате образуются токсичные формы Aβ; Б – активация микроглии в ответ на отложения Aβ сопровождается активным фагоцитозом (фенотип M2) и воспалительной реакцией (фенотип M1); В – гипертрофированный активированный астроцит вблизи агрегатов амилоида сменяет профиль функций на иммунно-ориентированный, а регуляторные функции деградируют; Г – снижение обратного захвата глутамата перисинаптическими отростками приводит к эксайтотоксичности и утрате синапсов; Д – дистальная атрофия астроцитов, при которой нарушается их взаимодействие с синапсами и нейронами в подконтрольной области, появляется на ранней стадии заболевания и может являться следствием аберрантных кальциевых сигналов (возможно, вызванных растворимыми формами Aβ), которые могут передаваться соседним астроцитам; Е – скопления гиперфосфорилированного тау нарушают цитоскелет и транспорт по аксону, что приводит к деградации нейрона.

Существует несколько видов Aβ, которые варьируют в зависимости от количества и последовательности аминокислотных остатков (от 39 до 43), с несколько различающимися свойствами: Aβ40 и Aβ42 являются наиболее распространенными в головном мозге [52]. Изоформа Aβ42 более склонна к агрегации и является основным компонентом амилоидных бляшек [53], следовательно, играет ключевую роль в патогенезе БА [52]. Большинство мутаций в гене APP приводят к тому, что отношение Aβ42 к Aβ40 увеличивается [54] (рис. 1А). К этому же приводят мутации PS1 и PS2 [55]. Зарегистрирована также мутация в APP, снижающая продукцию Aβ, она обладает защитным эффектом от поздних форм БА [56].
Предполагалось, что нейротоксический потенциал Aβ является результатом биохимических свойств, которые способствуют его агрегации в нерастворимые олигомеры и протофибриллы, последние взаимодействуют с нейронами и глиальными клетками, что приводит к активации провоспалительных каскадов, митохондриальной дисфункции и повышенному окислительному стрессу [57, 58], нарушению внутриклеточных сигнальных путей и кальциевого метаболизма, повреждению синапсов, увеличению фосфорилирования белка тау и деградации нейронов [44]. Амилоидоз наблюдается в головном мозге некоторых пациентов уже на третьем или четвертом десятилетии жизни с увеличением накопления в позднем среднем возрасте и наибольшим количеством в пожилом возрасте [43]. Но несмотря на весомые свидетельства, подтверждающие существенную роль Aβ-пептидов, ни одно исследование, основанное на амилоидной гипотезе, не привело к прорыву в лечении БА. Увеличенный уровень Aβ встречается в мозге у людей без когнитивных нарушений и, наоборот, у некоторых пациентов с диагнозом БА амилоидные бляшки отсутствовали или их уровень был недостаточен для соответствия невропатологическим диагностическим критериям заболевания [59, 60]. Невропатологические исследования не обнаружили строгой корреляции между плотностью и количеством бляшек в мозге и тяжестью деменции, которая значительнее коррелирует с плотностью нейрофибриллярных клубков [42, 61]. Это свидетельствует о том, что Aβ может быть необязательным или недостаточным компонентом для инициации клеточной дисфункции. Амилоидная гипотеза обретает все новые формы, акцентируя внимание на токсичности растворимых Aβ-олигомеров, участии Aβ в генерации свободных радикалов, нейровоспалении и других патологических механизмах [62–64].
НЕЙРОФИБРИЛЛЯРНЫЕ КЛУБКИ
Помимо амилоидных отложений между клетками, внутри нейронов при БА наблюдаются нейрофибриллярные клубки (neurofibrillary tangles, NFTs) являющиеся скоплениями гиперфосфорилированного белка тау [14]. Тау (microtubule-associated protein tau, MAPT) – белок, связанный с субъединицами микротрубочек, обнаруженный в большинстве тканей, в ЦНС широко представлен в нейронах, особенно в аксонах и дендритах, а также в астроцитах [65]. Тау является важным компонентом цитоскелета: взаимодействуя с α- и β-тубулином, участвует в сборке и стабилизации микротрубочек. Взаимодействие между тау-белком и тубулином является динамическим процессом, в котором тау способствует своей собственной полимеризации и ингибирует быструю деполимеризацию тубулина. Этот процесс регулируется балансом между фосфорилированием и дефосфорилированием участков тау-белка на остатках серина и треонина [66]. Гиперфосфорилирование тау внутриклеточными киназами препятствует его способности связываться с тубулином, дестабилизируя микротрубочки, что приводит к нарушениям аксонного транспорта, деградации цитоскелета и, в конечном итоге, к потере жизнеспособности клеток [67]. Аномальное отсоединении тау от микротрубочек может быть вызвано как увеличением скорости фосфорилирования, так и снижением скорости дефосфорилирования, в результате чего возрастает концентрация свободного тау в цитозоле и повышается вероятность его полимеризации, принятия β-конформации и дальнейшей агрегации в NFT [68].
Возможные причины патологического разобщения тау с микротрубочками включают мутации гена MAPT и дисбаланс между тау-киназами и фосфатазами [69]. Другие патологические события, такие как амилоидная нейротоксичность, окислительный стресс и воспаление также могут способствовать Тау-опосредованной нейродегенерации, однако их точное расположение в каскаде событий, приводящем к потере нейронов, до сих пор выясняется [69–71]. Общий уровень NFT хорошо коррелирует со степенью когнитивного нарушения при БА [42]. NFT могут напрямую вызвать физическое нарушение нормального функционирования клеток из-за относительно большого размера фибриллярного материала, который накапливается внутри нейронов [72] (рис. 1Е). Однако дефекты аксонного транспорта, потеря синапсов и нейровоспаление могут быть ранними признаками нейродегенерации, являющимися результатом токсичности растворимых форм тау, тогда как NFT – проявления более поздней стадии, которые лишь способствуют прогрессированию заболевания. Некоторые исследователи считают гиперфосфорилированный тау ответственным за митохондриальную дисфункцию, которая приводит к увеличению продукции активных форм кислорода и снижению уровня АТФ [58, 73].
ХОЛИНЕРГИЧЕСКИЕ НЕЙРОНЫ
Наиболее сильным коррелятом когнитивного спада при прогрессировании БА является утрата синапсов [74] и холинергический дефицит [75, 76]. Хотя при конечной стадии БА снижается количество различных типов нейронов, наиболее значительно в течение прогрессирования заболевания дегенерируют холинергические нейроны базального переднего мозга (сholinergic basal forebrain, CBF) [77]. Известно, что холинергическая система в сочетании с другими системами регуляции (например, ГАМК-ергической) вовлекается в формирование ритмов головного мозга, которые связаны с основными физиологическими состояниями, такими как сон и бодрствование, а также с обучением и некоторыми формами памяти [78–80]. Холинергическая передача реализуется путем высвобождения нейромедиатора ацетилхолина (ACh), связывающегося с мускариновыми или никотиновыми ацетилхолиновыми рецепторами, для оказания множественных эффектов на процессы памяти и синаптическую пластичность, в частности, на долговременную потенциацию (LTP) в гиппокампе [81, 82]. Повреждение холинергической системы в немалой доле ответственны за когнитивные нарушения, наблюдаемые при нейродегенерации [81].
Холинергические нейроны медиальной перегородки (medial septum, MS), вертикальной ветви диагональной полоски Брока (vertical limb of the diagonal band of Broca, VDB) и базального ядра Мейнерта (nucleus basalis of Meynert, NBM) посылают холинергические проекции в гиппокамп и неокортекс. Септо-гиппокампальный путь, возникающий из медиального ядра перегородки и ядра диагональной полоски, является основной структурой центральной холинергической системы и источником холинергической иннервации для гиппокампальной формации [83]. При БА дегенерация нейронов CBF распространяется на энторинальную кору и гиппокамп, затем на височные, теменные и, в конце концов, лобные ассоциативные зоны коры. Подмечено, что холинергические нейроны NBM наиболее чувствительны к нейрофибриллярной дегенерации и демонстрируют NFT на ранней стадии заболевания [84]. Подобные цитоскелетные изменения в нейронах СBF наблюдаются и у здоровых субъектов пожилого возраста [84], а их количество и размер снижаются на протяжении всей жизни человека, это позволяет предположить, что холинергическая дегенерация при БА может происходить на фоне возрастной атрофии [86–88]. В остающихся холинергических нейронах транскрипция холин-ацетилтрансферазы (СhAT), фермента, катализирующего реакцию образования нейромедиатора ацетилхолина, значительно снижается [89].
Возможно, что уязвимость CBF-нейронов и их склонность к дегенерации связана с достаточно большой протяженностью их аксонов, поскольку обслуживание таких длинных корковых проекций требует высоких энергозатрат на рост, обслуживание и репарацию, особенно в связи с аксонным транспортом, чувствительным к биохимическим нарушениям, вызванным, например, токсичными формами Aβ и тау [90] (рис. 1Г, Е). Отсрочка холинергической нейродегенерации или минимизация ее последствий является механизмом действия большинства используемых в настоящее время препаратов для лечения когнитивной дисфункции при БА. Препараты-ингибиторы ацетилхолинэстеразы (AChE), фермента, катализирующего гидролиз ацетилхолина, могут замедлять распад ацетилхолина в синаптической щели и тем самым увеличивают холинергическую нейротрансмиссию, что частично улучшает когнитивные функции и качество жизни у пациентов как с легкой, так и с тяжелой формой БА [89]. И хотя эти препараты имеют положительный эффект только в течение короткого периода времени, от 1 до 3 лет, и не могут остановить дальнейшее прогрессирование заболевания, в поддержку холинергической гипотезы свидетельствует тот факт, что до настоящего времени лекарства, нацеленные на холинергическую систему, показали наибольшую эффективность. Разработка новых препаратов (в рамках этой гипотезы) все же могут дать многообещающие результаты.
РОЛЬ GSK3β
Были исследованы и другие биохимические пути, связанные с патофизиологией БА. Так, киназа-3β гликогенсинтетазы (GSK3β), серин/треонин-протеинкиназа, участвующая во множестве физиологических процессов, рассматривается как молекулярная связь между двумя гистопатологическими признаками заболевания: бляшками Aβ и NFT. GSK3β является одной из тау-протеинкиназ и обладает способностью фосфорилировать тау по нескольким сайтам [91]. Согласно GSK3β-гипотезе БА, усиление активности GSK3β является ранним патологическим событием в патофизиологии БА, вызывая каскад молекулярных взаимодействий, приводящих к увеличению продуцирования Aβ, гиперфосфорилированию тау-белка (рис. 1А, Е), индукции апоптоза, нарушению нейрогенеза и синаптической пластичности. Патологической активации GSK3β способствует сам Aβ, продукция которого впоследствие еще больше увеличивается за счет воздействия GSK3β на γ-секретазную активность [92] (рис. 1Г, Е). Сверхактивная GSK3β также наблюдается при PS1-мутациях [93]. Исследования in vitro и in vivo показали, что ингибиторы GSK3β блокируют производство и накопление Aβ, предотвращает образование NFT и дегенерацию нейронов [92, 93]. GSK3β также задействована в метаболизме холина, про- и антиапоптотических механизмах, формировании NMDAR (N-метил-D-аспартатный рецептор) -зависимой долговременной депрессии (long-term depression, LTD) с эндоцитозом рецепторов α-амино-3-гидрокси-5-метил-4-изоксазолпропионовой кислоты (AMPA) и, кроме того, способствует производству провоспалительных цитокинов [93, 94]. Недавнее исследование впервые показало, что индуцированная Aβ патологическая передача кальциевых сигналов через G-белковые Сa2+-чувствительные рецепторы (calcium-sensing receptor, CaSR) непосредственно усиливает активность GSK3β для увеличения продукции Aβ42 и фосфорилированного тау астроцитами коры головного мозга у взрослого человека [65]. Но несмотря на изящность этой гипотезы, которая пытается объединить сразу несколько патологических механизмов БА, а также положительные результаты экспериментов, подавляющих активность GSK3β, следует учесть, что киназа необходима для нормальной жизнедеятельности, и ее ингибирование может нарушить правильное функционирование клеток [91].
Sig-1 РЕЦЕПТОРЫ, МИТОХОНДРИИ И МАМ
На роль терапевтической мишени при БА претендуют сигма-1-рецепторы (S-ig‑1Rs, σ1), которые фигурируют и в других нейродегенеративных заболеваниях. Сигма-1-рецептор представляет собой небольшой (28 кДа), высококонсервативный трансмембранный белок, локализующийся как в нейронах, так и в глиальных клетках по всей ЦНС и располагается, в основном, на связанных с митохондриями мембранах эндоплазматического ретикулума (ЭР) (mitochondria-associated membranes, MAM) [95, 96] (рис. 2). МАМ участвуют в процессах, происходящих на границе между митохондриями и ЭР, обеспечивая связь между двумя органеллами. Наиболее важные функции MAM включают высокоэффективную передачу Ca2+ из ЭР в митохондрии для стимуляции окислительного метаболизма, а также транспорт липидов и контроль апоптоза [97]. Передача Ca2+-сигналов и их регуляция в MAM важны для нормального функционирования митохондрий, особенно в отношении роли последних в биоэнергетике и апоптозе [98]. Доставка Ca2+ в митохондрии имеет решающее значение для согласования производства АТФ c цикла трикарбоновых кислот (ЦТК) и электрон-транспортной цепи. Так, например, инозитолтрифосфат (IP3), в качестве вторичного мессенджера, может активировать ЦТК путем увеличения концентрации Ca2+ в матриксе митохондрий [96]. И одна из основных функций сигма-1-рецепторов как раз заключается в регуляции передачи Ca2+-сигналов между ЭР и митохондриями [95] (рис. 2). При нормальных концентрациях Ca2+ в ЭР (0.5–1.0 мМ) сигма-1-рецепторы находятся на MAM в комплексе с ЭР-шапероном, глюкозо-регулируемым белком GRP78, также известным как BiP (binding immunoglobulin protein). При снижении концентрации Ca2+ сигма-1-рецептор диссоциирует от BiP и действует, как свободный шаперон, стабилизируя IP3-рецепторы (IP3R), усиливая передачу Ca2+-сигналов от ЭР в митохондрии для увеличения производства АТФ [97]. В другом случае сигма-1 рецепторы могут ингибировать поступление Ca2+ в депо путем ослабления связывания Ca2+-чувствительного белка STIM1 c каналом Orai1 (депо-зависимый вход кальция 1), снижая уровень Ca2+ в ЭР, возможно для предотвращения чрезмерной Ca2+-трансмиссии в митохондрии и/или ингибирования апоптотических механизмов [99]. Сигма-1-рецепторы также способны регулировать фосфорилирование тау-белка, рост аксонов [100] и образование дендритных шипиков в гиппокампе [98]. Снижение количества сигма-1-рецепторов наблюдается в мозге пациентов с БА [101]. Было показано, что введение агонистов этих рецепторов защищает культуру нейронов от токсического воздействия Aβ, предотвращая нейродегенерацию [102]. В целом, многофункциональность сигма-1-рецепторов представляет большой терапевтический интерес, так как с их помощью можно смягчить симптомы нейродегенерации, но пока ни один селективный агонист сигма-1-рецепторов для лечения не используется [101].
Рис. 2.
Многообразие Ca2+-механизмов в трехчастном синапсе. Высвобождение и захват глиотрансмиттеров, регуляция содержания внеклеточного калия находится под контролем астроцитарных Ca2+-событий: астроциты обеспечивают обратный захват глутамата из синаптической щели через глутаматные транспортеры; высвобождение глутамата и D-серина из астроцитов является необходимым условием для инициации LTP в синапсах гиппокампа. Основные события, приводящие к увеличению уровня [Ca2+]i, включают активацию G-белковых рецепторов и PLC для продуцирование IP3, который связывается с IP3R, приводя к высвобождению Ca2+ из ЭР, а также поступление Ca2+ в клетку через nAChR, TRP и Orai. Увеличения [Ca2+]i могут быть ограничены одним астроцитом или передаваться в соседние. Sig-1 (σ) рецепторы на МАМ регулируют передачу Ca2+-сигналов между ЭР и митохондриями, стабилизируя IP3-рецепторы, для согласования производства АТФ. Дисрегуляция Ca2+-механизмов в астроцитах может являться причиной астроцитарной атрофии, утраты подконтрольных синапсов и других патологических событиях БА.
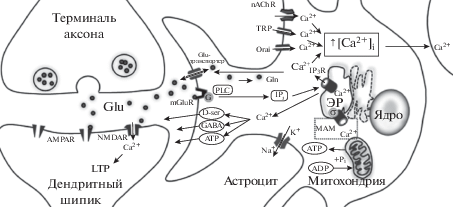
При БА, помимо отклонений в кальциевом и липидном обмене, наблюдаются нарушения функционирования митохондрий, которые включают снижение активности дыхательной цепи, окислительного фосфорилирования и увеличение продукции активных форм кислорода [103–105]. Современные гипотезы, касающиеся митохондриальной дисфункции, предполагают, что этот дефект является следствием накопления Aβ в митохондриях [106]. Но митохондриальные фенотипы с энергетическим дефицитом могут наблюдаться задолго до появления бляшек, а также у пациентов без диагностированной БА. Были проведены исследования, в которых обнаружилось, что фрагмент C99 способен перемещаться путем еще неизвестного механизма на МАМ, где расщепляется γ-секретазой, c образованием Aβ [107]. Значительное увеличение C99 в МАМ было зафиксировано в культуре, в клетках модельных животных и в клетках, полученных от пациентов с БА [48]. Поэтому C99, а не Aβ, предлагается как ранний критический компонент патогенеза БА с токсическим воздействием на MAM и митохондрии. Накопление С99 в МАМ приводит к усилению регуляции гидролиза сфингомиелина сфингомиелиназами (SMases), в результате снижается содержание сфингомиелина и увеличивается выход продукта гидролиза сфингомиелина – церамида [105]. Церамид является проапоптотической молекулой и ингибитором митохондриального дыхания. Действительно, в PS1-мутантных (mPS1) клетках и у пациентов с БА функционирование дыхательной цепи снижено, как и функционирование дыхательного суперкомплекса [105]. Как патогенные мутации в PSEN1, так и снижение активности γ-секретазы вызывают повышение уровня МАМ-C99, который способствует развитию различных признаков нейродегенерации, включая дерегуляцию баланса кальция и липидов, митохондриальные нарушения, которые позднее могут инициировать образование амилоидных бляшек и тау-клубков [48]. Известно, что в нормальных условиях PS1 способен усиливать экспрессию белка PGC-1α (peroxisome proliferator-activated receptor gamma coactivator 1α), главного регулятора митохондриального биогенеза и энергетического метаболизма [108]. В случае семейной БА, мутации в PSEN1 аннулируют его способность регулировать экспрессию PGC-1α, что приводит к уменьшению производства АТФ и скорости потребления кислорода. Возможно, этот же эффект имеет место и в спорадической форме БА [108]. Кроме того, патологическое воздействие mPS1 может проявляться в нарушении кальциевого обмена между митохондриями и цитозолем. Увеличение выработки активных форм кислорода дисфункциональными митохондриями активирует патологический каскад событий, вызывающий метаболические изменения, которые могут повлиять на множество важных нейрональных процессов, включая экспрессию АРР и образование Аβ, и в конечном счете, эти изменения приводят к потере когнитивных функций. Однако даже при таком объяснении дисфункция митохондрий вряд ли может претендовать на фундаментальную причину заболевания, так как несмотря на раннее проявление, она не является специфичной для БА, проявляясь при ряде других нейродегенеративных заболеваниях и, вероятно, является следствием других первичных событий в прогрессировании БА [105].
После многочисленных исследований стало ясно, что БА не может объясняться моноказуально. Но несмотря на то, что каждая гипотеза включает в себя довольно сложный набор субмеханизмов (например, эксайтотоксичность, воспаление, глиоз, избыток активных форм кислорода и оксида азота) и эти механизмы частично перекрываются друг с другом, ни одна из вышеперечисленных гипотез полностью не учитывает широкий спектр патологических изменений у пациентов с БА. Стандартный нейроцентрический подход к БА с привлечением глии только в качестве суб-участника изжил себя и не дает увидеть полной картины патофизиологии ранней стадии заболевания.
РОЛЬ ГЛИАЛЬНЫХ КЛЕТОК В БОЛЕЗНИ АЛЬЦГЕЙМЕРА
Несмотря на выраженную гетерогенность глиальных клеток, их объединяет одна фундаментальная задача: поддержание баланса в ЦНС. Глиальные клетки обеспечивают соответствующий субстрат и среду для функционирования нейронов. Любые изменения глиальных регуляторных функций могут в значительной степени способствовать прогрессированию нейродегенеративных заболеваний или даже стать одним из центральных элементов начала болезни. В настоящее время выделяют четыре типа глиальных клеток в ЦНС, а именно: астроциты, олигодендроциты, полидендроциты (также называемые NG2-глия) и микроглию [109]. Данные последних исследований указывают на то, что астроциты и микроглия могут играют далеко не последнюю роль в патогенезе БА [14].
Микроглия
Микроглиальные клетки составляют врожденную иммунную систему мозга (около 10% всех клеток нервной системы) и являются первой линией защиты от инвазивных патогенов [110]. В ЦНС микроглия представляет собой фенотипически разнообразную группу клеток, которые легко реагируют на сигналы из внеклеточной среды, изменяя свой статус активации и экспрессию фенотипических маркеров [111]. В зрелом мозге микроглиальные клетки встречаются в трех различных состояниях: в виде “покоящейся” микроглии, “активированной” микроглии и “фагоцитарной” микроглии. В нормальном мозге микроглиальные клетки присутствуют в состоянии “покоя”, или точнее “мониторинга”, характеризуясь небольшой сомой и множеством очень тонких, сильно разветвленных отростков, с помощью расширения и втягивания которых они непрерывно анализируют внеклеточную среду [111]. Наличие в микроглиальных отростках многочисленных рецепторов делает их идеальными датчиками статуса тканей ЦНС [112]. В патологических ситуациях, таких как нейродегенеративные заболевания, инсульт, травма или опухоль, эти клетки “активируются”, становясь подвижными и мигрируют, окружая поврежденные или мертвые клетки, очищая область от “клеточного мусора”, что полезно, так как создает пространство для новых клеточных соединений, тем самым помогая системе регенерировать [110].
В ходе нейродегенерации меняется не только морфологический фенотип микроглии, но и ее общее количество. Микроглиальные клетки высвобождают нейротрофические факторы и множество провоспалительных медиаторов, включая цитокины, активные формы кислорода, комплементарные факторы, нейротоксические секреторные продукты и свободные радикалы. При БА микроглии отведена ключевая роль в удалении бляшек Aβ [113]. Она снижает аккумуляцию Aβ за счет его активного фагоцитоза и последующей деградации [114]. Активация происходит в ответ на образование амилоидных бляшек, так как Aβ и APP могут являться мощными активаторами микроглии [115]. Воспалительный микроглиальный ответ может привести к дисфункциям и гибели нейронов [116].
Согласно поляризационной концепции в зависимости от среды, в которой происходит активация, или стимулирующих факторов, микроглия обладает состояниями “классической активации”, “альтернативной активации” и “приобретенной дезактивации” [117]. Классическая активация связана с продуцированием провоспалительных цитокинов фактора некроза опухоли α (TNFα) и интерлейкина-1β (IL-1β), оксида азота (NO), активных форм кислорода и различных протеаз [118]. Микроглиальный фенотип в этом состоянии называют “M1 микроглия”. Термин “M2 микроглия” используют для обозначения состояний альтернативной активации и приобретенной дезактивации. Альтернативная активация связана с IL-4 или IL-13 и способствует восстановлению тканей и внеклеточного матрикса при воспалении [117, 119]. Приобретенная дезактивация – это еще одно состояние для снятия симптомов воспаления, которое характеризуется поглощением апоптотических клеток или воздействием противовоспалительных цитокинов, таких как IL-10 и трансформирующий фактор роста 1 β (TGFβ) [120]. IL-10 сам по себе или в дополнение к TGFβ инициирует клеточные сигнальные пути, которые приводят к ингибированию продуцирования провоспалительных цитокинов, увеличению экспрессии рецепторов - поглотителей и дальнейшему увеличению продукции IL-10 [117]. Микроглия, окружающая и фагоцитирующая амилоидные бляшки при БА, обычно проявляет фенотип активации M2 [121]. Однако c возрастом, увеличение как количества, так и размера Aβ бляшек при БА коррелирует с уменьшением фагоцитарной способности микроглии [50, 121]. Фагоцитарная активность микроглии ослабляется провоспалительными цитокинами, такими как интерферон γ (IFNγ), IL-1β и TNFα, что, скорее всего, переводит микроглию в агрессивное провоспалительное состояние M1 [110], вызывающее нейротоксичность и необратимую утрату нейронов [120] (рис. 1Б).
В стареющем мозге микроглиальные клетки могут подвергаться различным молекулярным и клеточным изменениям, в результате чего они теряют способность защищать мозг, что, вероятно, может предшествовать нейродегенерации. Нормальное старение сопровождается увеличением числа провоспалительных медиаторов IL-1β и IL-6 при одновременном снижении уровня IL-10 в головном мозге [120]. При нейродегенеративных заболеваниях, в том числе и при БА, баланс между микроглиальными фенотипами M1 и M2 нарушен во время прогрессирования хронического воспаления [120] (рис. 1Б). Разработка методов манипулирования статусом реактивации микроглии – переключение между микроглиальными фенотипами M1/M2 (от цитотоксического к нейропротективному) позволит снизить воспаление и сдерживать утрату нейронов. Однако некоторые исследователи предполагают, что парадигма о фенотипах M1 и M2 неверна и требует пересмотра [122, 123], так как эти фенотипы не обязательно исключают друг друга и часто сосуществуют; такой смешанный фенотип зависит от баланса активирующей и ингибирующей активности и тканевой среды [122].
Физиологическая роль астроцитов
Астроциты – самый распространенный и разнообразный тип глиальных клеток в ЦНС. Они сильно различаются по своей морфологии и функции и в различных областях мозга могут проявлять разные физиологические свойства, поэтому часто используется собирательный термин “астроглия”. Основными типами астроглиальных белков промежуточных филаментов (глиальных фибрилл) являются глиальный фибриллярный кислый белок (GFAP), виментин и нестин [112]. Но далеко не всегда, например, экспрессию GFAP можно использовать как маркер идентификации астроцитов. Во-первых, уровни экспрессии GFAP значительно различаются в зависимости от астроцитарного подтипа и расположения клеток; во-вторых, некоторые астроциты в норме вообще не экспрессируют GFAP, а только в патологических условиях, как, например, клетки Мюллера (глиальные клетки сетчатки). Наиболее общим признаком астроцитарной клетки является наличие двух типов контактных участков: с нейронами (в области синапсов в сером веществе и с аксоном в белом веществе) и кровеносной системой или стенками желудочков мозга [124].
Четыре класса GFAP+ (положительных) астроцитов наблюдаются в мозге человека: интерламинарные (межслойные) астроциты I и II слоя коры головного мозга (специфичны для приматов), протоплазматические астроциты в слоях II-VI, варикозные проекционные астроциты в слоях V и VI (у высших приматов) и фиброзные (волокнистые) астроциты в белом веществе [125, 126]. Самый распространенный тип в мозге приматов и грызунов – протоплазматические астроциты. У человека они наиболее крупные (длина отростков примерно в 2.6 раза больше, чем у грызунов, в среднем около 100 мкм), имеют характерную “кустообразную” или “губчатую”, заполняющую пространство морфологию и анатомически организованы в отдельные домены (диаметр домена до 400 мкм), т.е., распределяются таким образом, что каждый астроцит занимает собственное пространство с небольшим перекрытием (но намного большим, чем у грызунов) с соседними клетками, охватывая соматы нейронов, синапсы и кровеносные сосуды в своей окрестности [125, 127–130]. Было подсчитано, что один астроцитарный домен может охватить до 120 000 синапсов в мозге грызунов и до 2 млн синапсов в мозге человека [125, 127]. В начале развития, на стадии роста, соседние астроциты значительно перекрываются между собой, но уже к 21-му дню постнатального периода у грызунов эти перекрытия устраняются [131]. Пересечение астроцитарных доменов в мозге молодых здоровых животных минимально, но увеличивается с возрастом или при некоторых неврологических заболеваниях, например, эпилепсии [130].
Астроциты способствуют образованию и поддержанию гематоэнцефалического барьера, регулируют церебральную микроциркуляцию, обеспечивают нейрональный обмен веществ и структурную поддержку синапсов, регулируют баланс ЦНС в отношении внеклеточной концентрации нейротрансмиттеров и ионов [132]. Поглощение ионов калия астроцитами имеет решающее значение для поддержания баланса ионов во внеклеточной среде. Калий распределяется по глиальному синцитию и, в случае его избыточного выделения нейронами, может быть удален через концевые ножки (“endfeet”) астроцитов в кровоток. Астроциты играют модуляторную и защитную роль в трехчастном синапсе с пре- и постсинаптическими нейронами, удаляя из синаптической щели избыток γ-аминомасляной кислоты, глутамата и других нейротрансмиттеров [133] (рис. 2). Примерно 80% глутамата, выделяемого синапсами во внеклеточное пространство, захватывается перисинаптическими отростками астроцитов через глутаматные транспортеры. Посредством глутаминсинтетазы астроциты превращают глутамат в глутамин, который затем передают нейронам для повторного синтеза глутамата [134]. Такой механизм удовлетворяет метаболическую потребность и предохраняет нейроны от глутаматной эксайтотоксичности [132]. Астроциты транспортируют глюкозу из крови через глюкозные транспортеры (GLUT1) и хранят ее в виде гликогена или превращают в лактат, по необходимости доставляя нейронам [135]. Астроциты также способствуют превращению филоподий в зрелые шипики и выделяют нейротрофические факторы для формирования, поддержания и стабилизации синапсов [132].
Астроциты могут влиять на синаптическую передачу посредством Ca2+-зависимого высвобождения глиотрансмиттеров, таких как глутамат и D-серин [136, 137] (рис. 2). Например, участие астроцитов необходимо для генерации LTP, индуцированной холинергической активностью, в синапсах гиппокампа: высвобождение глутамата, стимулированное увеличением внутриклеточного Ca2+ в астроцитах и одновременная постсинаптическая деполяризация пирамидальных нейронов области СА1 вызывает LTP в синапсах CA3–CA1 посредством пресинаптической активации метаботропных глутаматных рецепторов (mGluR) [137]. Высвобождение из астроцитов D-серина, коагониста NMDAR, под контролем астроцитарных кальциевых сигнальных путей, позволяет индуцировать LTP в культуре нейронов и в переживающих срезах гиппокампа [133, 138] (рис. 2). Имеются данные об активном участии астроцитов в формировании памяти [139]. Кроме того, астроциты экспрессируют рецепторы врожденного иммунитета, включая толл-подобные рецепторы, NOD-подобные рецепторы, маннозный рецептор и компоненты системы комплемента [140]. Совместно с микроглией астроциты продуцируют широкий спектр хемокинов и цитокинов, влияя на иммунную систему мозга.
Астроцитарная атрофия и реактивный глиоз
Астроцитарная атрофия, характеризующаяся значительным уменьшением размера астроцитарных сомат и отростков, снижением ветвления, общей поверхности и объема GFAP, показана на очень ранних стадиях БА, еще до появления классических признаков патологии. Исследования на мышиной модели БА 3×TG выявили начало астроцитарной атрофии в энторинальной коре уже в месячном возрасте, в префронтальной коре в возрасте 3-х месяцев и значительно позднее в гиппокампе (9–12 месяцев), что говорит о большей уязвимости первых двух зон. Снижения плотности астроцитов при этом не было зафиксировано [141, 142]. Подчеркнем, что астроцитарная атрофия обнаруживается в вышеуказанных областях мозга до появления внеклеточных отложений Аβ. Возможно, именно это событие, при котором происходит снижение охвата синапсов астроцитарными отростками из-за общего сокращения астроцитарных доменов, порождает начальную стадию патологии. Отсутствие астроцитарной поддержки приводит к нарушениям в синаптической передаче и связности нейронной сети, затем утрате синапсов и нейродегенерации, являющихся основной причиной ухудшения когнитивных способностей и памяти на продромальных стадиях БА [142].
На более поздних стадиях заболевания атрофированные астроциты, которые локализуются, в основном, дистально (>50 мкм) от границ образовавшихся амилоидных бляшек, сосуществуют с популяцией так называемых реактивных астроцитов, напротив, находящихся вблизи бляшек амилоида [141, 142] (рис. 1В, Д). В подтверждение существования двух различных популяций астроцитов в мозге трансгенных животных свидетельствуют эксперименты с визуализацией Сa2+in vivo, которые продемонстрировали увеличение Сa2+-возбудимости в астроцитах, контактирующих с бляшками Аβ [8]. Реактивный астроцитоз выражается в гипертрофии сомат и отростков, общего увеличение объема и площади поверхности GFAP+-астроцитов [141, 144]. Согласно исследованиям при травмах головного мозга IP3-зависимый сигнальный Ca2+-каскад в самих астроцитах является необходимым для запуска реактивного астроглиоза [145] – полезного механизма для осуществления регенерации: реактивные астроциты усиливают трофическую поддержку нейронов и способствуют образованию глиального рубца, который изолирует область повреждения от остальной ткани ЦНС [146]. При БА реактивные астроциты группируются вокруг бляшек Aβ, становясь частью воспалительного процесса: совместно с активированной микроглией они начинают продуцировать различные провоспалительные цитокины, такие как IL-1 и TNFα, вызывая иммунный ответ ЦНС (рис. 1Б–Г). Однако эти изменения имеют не только положительные последствия. Например, повышение уровня экспрессии IL-1 астроцитами неоднозначно влияет на патофизиологию БА: множество экспериментальных данных подтвердило участие IL-1 в механизмах нейродегенерации, таупатии и гиппокамп-зависимом дефиците пространственной памяти [147–149]; другие исследования определили роль IL-1 как адаптивную, способствующую увеличению клиренса Аβ микроглиальными клетками без апоптотического или нейродегенеративного эффектов [150, 151].
Переход астроцитов на воспалительный фенотип сопровождается утратой их регуляторных функций и приводит к дегенерации клеток, зависящих от этих функций [144, 152]. Нарастающее при БА доминирование фенотипов реактивных астроцитов, с их пониженной способностью к поглощению глутамата через глутаматные транспортеры, ведет к развитию глутаматной эксайтотоксичности и дальнейшей нейродегенерации. Реактивные астроциты также считаются ответственными за сосудистые дисфункции, периваскулярный амилоидоз и нарушение функционирования гематоэнцефалического барьера [155] (рис. 1В). И хотя реактивный астроцитоз ингибирует образование и рост амилоидных бляшек [146], этого механизма недостаточно для предотвращения токсического эффекта, вызванного чрезмерным накоплением амилоида. Реактивные астроциты фагоцитируют протофибриллы Aβ, но не разрушают их полностью, образуя крупные астроцитарные эндосомы [156]. Известно, что синаптическая дисфункция и когнитивный дефицит при БА коррелирует с ростом числа реактивных астроцитов [157]. Предотвращение астроцитарной гипертрофии путем двойной генетической абляции генов глиальных фибриллярных кислых белков и виментина (GFAP–/–Vim–/–) улучшает посттравматическую регенерацию синапсов и аксонов, но способствует образованию бляшек Aβ [153]. В эксперименте отростки GFAP–/–Vim–/–-астроцитов, прилегающих к амилоидным бляшкам, не становились гипертрофированными, а сами астроциты показали лишь очень ограниченное физическое взаимодействие с амилоидными бляшками, однако впоследствие это привело к тому, что амилоидные отложения и признаки нейродегенерации были более заметными, чем у контрольной группы мышей [153]. Эти данные указывают на то, что делеции GFAP и Vim не влияют на первоначальное осаждение бляшек, но усугубляют их последующее накопление, поскольку активированные астроциты, по-видимому, ингибируют рост бляшек за счет прямого взаимодействия с ними [146, 153]. Таким образом, хотя астроцитарная активация играет защитную роль в головном мозге при травмах и нейродегенеративных заболеваниях, последствия деятельности реактивных астроцитов и воспаление могут ускорять прогрессирование БА [154]. Интересно, что реактивный астроглиоз в мозге, пораженном БА, проявляется не повсеместно. У мышей 3×TG отложения Aβ вызывали появление реактивных астроцитов в гиппокампе, но не в энторинальной коре, где в любом возрасте астроциты демонстрировали только “атрофическую” морфологию [141–143].
ДИСРЕГУЛЯЦИЯ КАЛЬЦИЯ В АСТРОЦИТАХ ПРИ БОЛЕЗНИ АЛЬЦГЕЙМЕРА
Хотя астроциты долгое время принято было характеризовать как “пассивные” или “электрически невозбудимые” (в отличие от нейронов), сейчас субстратом клеточной возбудимости в них считаются Ca2+-сигналы, поскольку стимуляция астроглии различными нейромедиаторами, нейромодуляторами и нейрогормонами почти всегда вызывает цитоплазматические Ca2+-реакции. Собственно говоря, ионы кальция являются универсальными внутриклеточными мессенджерами и участвуют в регуляции большинства известных клеточных реакций. Кальций вовлечен в такие процессы, как экзоцитоз, встраивание рецепторов в мембрану, экспрессию генов и может иметь опосредованное влияние на процессы обучения и памяти [112]. Молекулярные каскады, контролирующие гомеостаз и сигналы Сa2+, включают Сa2+-каналы, обменники, АТФ-зависимые Сa2+-транспортеры, а также Сa2+-связывающие белки и Сa2+-зависимые ферменты (например, киназы, фосфатазы и т.д.) [158]. Универсальная роль Сa2+-сигналов в клеточном метаболизме является основой регуляторных (и не только) функций астроцитов. Так, повышение астроцитарного [Сa2+]i в ответ на активность нейронов приводит к Сa2+-зависимому высвобождению глиотрансмиттеров, включая глутамат, D-серин, АТФ и метаболиты арахидоновой кислоты [159]. Глутамат-зависимая генерация астроцитарных кальциевых сигналов является общим механизмом, посредством которого информация о локальном уровне активности нейронов передается в сосудистую сеть головного мозга [160]. В свою очередь уровень кальциевой активности в астроцитах определяет степень охвата синапсов астроцитарными процессами и влияет на поглощение перисинаптического глутамата [161] (рис. 2). Астроциты способны не только реагировать на внешнюю стимуляцию увеличением уровня внутриклеточного кальция, но и могут передавать эти сигналы соседним (не стимулированным) астроцитам через щелевые контакты или с помощью внеклеточных сигнальных молекул [162]. Это явление называется “межклеточные волны Ca2+” (ICWs, “intercellular Ca2+ waves”) [163]. Кальциевая волна представляет собой локализованное увеличение цитозольного Ca2+, за которым следует последовательность подобных событий, которые могут быть ограничены одной клеткой (внутриклеточные волны) или передаваться в соседние (межклеточные волны). Межклеточные волны распространяются со скоростью 15–27 мкм/с и могут затрагивать клетки в радиусе 200–350 мкм [164]. В зависимости от состояния внутриклеточных биофизических параметров (например, скорости утечки кальция из внутриклеточных хранилищ) динамика этих волн может кодировать информацию о внешних стимулах в амплитудной, частотной и пространственно-временной модуляции [165, 166]. Некоторые авторы полагают, что Ca2+-активность астроцитарной сети представляет собой форму межклеточной коммуникации, которая оказывает физиологически значимый эффект на активность нейронов [163].
Механизмы пространственно-временной интеграции активности Ca2+ в астроцитах играют важную роль в управлении функциями мозга [167]. Основные события, приводящие к внутриклеточным волнам Са2+ в астроцитах, обычно включают активацию рецепторов, связанных с G-белком, активацию фосфолипазы C (PLC) и продуцирование IP3, который связывается с IP3R, приводя к высвобождению Са2+ из ЭР [163] (рис. 2). Внутриклеточные волны Са2+ следуют определенному пространственному пути внутри клеток, который зависит от близости IP3 -рецепторов ЭР. Если ближайший IP3R слишком удален, Ca2+ буферизуется до базального уровня и внутриклеточная Ca2+-волна заканчивается. Таким образом, пространственное распределение IP3R обеспечивает траекторию для внутриклеточных волн Са2+. Подобные события, происходящие внутри отдельных астроцитарных микродоменов, не синхронизированные со смежными астроцитами, могут регулировать локальный уровень внеклеточного калия, глутамата, ГАМК, аденозина, NO и др., а также, возможно, влияют на образование дендритных шипиков путем выделения трофических факторов [168].
Считается, что аномальные кальциевые осцилляции имеют патологическое значение для развития БА: способствуют синаптической дисфункции и утрате синаптических контактов, увеличению производства Aβ, когнитивному дефициту и в конечном итоге приводят к гибели нейронов [169]. Неуловимые изменения Ca2+-гомеостаза на ранних стадиях БА могут развиваться и накапливать последствия в течение десятилетий до обнаружения следов дисбаланса, и в конечном счете способствовать возникновению специфических симптомов [11]. Аргументом в пользу данной гипотезы является связь между генетическим аномальным фоном БА и нарушениями баланса кальция в клетках, которая обеспечивается пресенилинами: нейроны, выделенные из трансгенных животных, экспрессирующих мутантный PS1, демонстрируют повышенную восприимчивость к эксайтотоксичности, что инициируется чрезмерным высвобождением Ca2+ из ЭР с помощью IP3R [170]. Непосредственное взаимодействие между мутантным PS и IP3-рецепторами может приводить к увеличению вероятности открытия последних и увеличению чувствительности канала к IP3 [169].
Влияние Aβ на кальциевые сигналы в астроцитах
Большое количество исследований, посвященных БА, сообщают о влиянии отложений Aβ на Ca2+ баланс в соседствующих нейронах и синаптическую передачу [171–177]. Эффекты Аβ-олигомеров на глиальные клетки были исследованы в значительно меньшей степени и, в основном, in vitro в культивируемых первичных астроцитах. Однако недавние эксперименты in situ оценивают Аβ-индуцированные Ca2+-изменения в астроцитах как даже более значительные, чем таковые в нейронах [10]. Поскольку симптомы БА наиболее тесно связаны с синаптической дисфункцией (на ранних стадиях), за которой следует утрата нейронов (на более поздних и конечных стадиях заболевания), наблюдаемые во многих трансгенных моделях мышей аберрантные изменения кальция в астроцитах, способные влиять на передачу сигналов в синапсах, представляют особый интерес. Например, исследование астроцитов в смешанной астроглиально-нейронной культуре выявили спорадические аберрантные Сa2+-сигналы, вызванные добавлением токсичных фрагментов амилоида Aβ, которые приводили к гибели нейронов, но не астроцитов [178]. Следует отметить, что нейроны проявляют выраженную кальциевую гиперактивность вблизи бляшек, тогда как астроциты усиливают активность как вблизи, так и проксимально от бляшек, т.е., отложения Aβ могут инициировать нарушения кальциевых внутри- и межклеточных сигнальных путей в астроцитах, не находящихся в области поражения [8, 179]. Эксперименты с использованием двухфотонной микроскопии через отверстие (окно) черепной коробки показали, что базальный уровень [Ca2+]i в реактивных астроцитах, связанных с бляшками Aβ был почти удвоен по сравнению с контрольными мышами дикого типа (WT, “wild type”) в клетках у мышиной модели APP/PS1. Это [Ca2+]i-увеличение проявлялось параллельно со спонтанной Ca2+-активностью, синхронной гиперактивностью и распространяющимися по астроглиальному синцитию аберрантных Ca2+-волн. Такая аберрантная кальциевая активность проявлялась на поздних стадиях БА, когда бляшки уже присутствовали, и не зависела от нейронов, поскольку не блокировалась тетродотоксином [8]. На более ранних стадиях патологии, изучавшихся у другой модели мышей APP Swe в возрасте 2–4 месяца (т.е. до накопления внеклеточных амилоидных отложений) также наблюдалось повышение частоты спонтанных кальциевых колебаний [180].
Транскрипционные эффекты Аβ на Ca2+-регулирующие гены в астроцитах включают увеличение экспрессии mGluR типа 5, IP3R, CaSR, ионного канала с транзиторным рецепторным потенциалом (TRP) и Orai [134, 181–184]. Механизмы Аβ-индуцированных [Ca2+]i колебаний могут затрагивать и другие пути поступления Ca2+ в клетки, а также выброс Ca2+ из внутриклеточных хранилищ [57, 178]. Небольшое увеличение базального уровня [Ca2+]i в присутствии Аβ может быть достаточным для запуска сигнальных каскадов, вызывающих долгосрочные изменения функционирования астроцитов. Например, низкие (200 пМ) концентрации Аβ способны модифицировать высокопроницаемые для кальция α7-никотиновые рецепторы (α7nAChRs), увеличивая частоту и амплитуду спонтанных внутриклеточных Ca2+-сигналов в астроцитах [186]. Длительное (24–72 ч) воздействие на культивируемые астроциты коры и гиппокампа низкими концентрациями (0.1–100 нМ) Aβ42-олигомеров положительно регулирует транскрипцию генов никотиновых ацетилхолиновых рецепторов типов α7, α4 и β2 [168]. Кроме того, Aβ увеличивает уровни астроцитарного [Ca2+]i путем его высвобождения из ЭР через рианодиновые рецепторы (RyR) [186]. В контексте подобных исследований БА были обнаружены различия между Ca2+-динамикой астроцитов гиппокампа и энторинальной коры, которые могут быть весьма существенными, так как астроциты из разных областей мозга демонстрируют совершенно различную физиологию и экспрессируют разные наборы рецепторов к нейротрансмиттерам. Астроциты, выделенные из энторинальной коры мышей с моделью БА 3×TG, после воздействия на них Aβ не демонстрировали различия Ca2+-сигналов с таковыми в контрольных клетках (в отличие от вышеописанных экспериментов с гиппокампальными астроцитами) [182, 184]. Отсутствие модификаций Ca2+-динамики в астроцитах энторинальной коры может быть связано с отсутствием проявления реактивного астроглиоза в ответ на отложения Aβ [142].
Явление кальциевой дисрегуляции в глиальных клетках проявляется в большинстве, если не во всех моделях животных, в которых обнаруживаются гистопатологические признаки и клинические симптомы БА, хотя они и не воспроизводят патологию в точности, особенно когда речь идет о спорадической форме заболевания. Имеющиеся в настоящее время данные исследований свидетельствуют о том, что связующим звеном между нарушениями кальциевого баланса, индуцируемыми Aβ и гибелью нейронов может являться кальцинейрин (CaN), Ca2+/кальмодулин-зависимая фосфатаза, которая активируется небольшими длительными увеличениями [Ca2+]i [187]. Активация CaN приводит к транслокации в ядро фактора транскрипции NF-kB, что усиливает экспрессию как mGluR5, так и IP3R типа 2 (доминирующая изоформа в астроцитах); аналогично, но без участия NF-kB – для IP3R типа 1 [183]. Отметим, что сверхэкспрессия CaN была зафиксирована в реактивных астроцитах, взаимодействующих с бляшками амилоида, полученных из гиппокампов мышей с моделью БА [188] и у пациентов с БА [183]. С.М. Norris с соавт. считают, что сверхэкспрессия CaN может быть достаточной причиной для запуска активации астроцитов при БА [188]. Связь между Aβ и CaN предполагается и другими исследователями, как в нейронах [187], так и в астроцитах [189]. Необходимы дальнейшие эксперименты для установления точной роли этой фосфатазы при БА.
Принято считать, что накопление Aβ исходит из нейронов, но есть экспериментальные доказательства того, что Aβ может продуцироваться и астроглией [181, 190]. Астроциты экспрессируют все компоненты для амилоидогенной и неамилоидогенной обработки APP, включая его самого, BACE1, γ-секретазу и другие ферменты [181]. Этот факт, как предполагают некоторые авторы [142, 143], также свидетельствует в пользу предположения о первостепенной роли астроглии в патогенезе БА. Реактивные астроциты могут являться дополнительным источником Aβ [190] и тем самым способствовать “порочному кругу”, при котором Aβ индуцирует нарушения передачи Ca2+-сигналов в астроцитах и нейронах, в свою очередь приводящих к накоплению Aβ и другим патологическим процессам при БА (рис. 1).
ЗАКЛЮЧЕНИЕ
Большинство исследований, посвященных БА, сфокусированы на функционировании и патологии нейронов. На сегодняшний день по-прежнему доминирующей является амилоидная гипотеза развития БА, с тем уточнением, что именно внутриклеточные растворимые формы, а позднее – внеклеточные агрегаты Aβ оказывают токсическое воздействие на нейроны, индуцируя глиальную активацию и последующую нейродегенерацию. Однако многочисленные эксперименты, нацеленные на удаление Aβ, пока так и не привели к успеху. Применение препаратов для подавления воспалительного процесса, который считается одной из основных причин поздней нейродегенерации, также не дало положительных результатов. Сейчас в литературе появляется все больше доказательств, указывающих на то, что именно изменения в работе глиальных клеток приводят к патологическим изменениям при БА и некоторых других заболеваниях ЦНС. Результаты вышеописанных экспериментов позволяют предположить, что мишенью Aβ являются в первую очередь астроциты (рис. 3). Нам известно, что Aβ способен к дисрегуляции уровня астроцитарного кальция путем изменения мембранной Са2+-проницаемости и/или усилению выделения Са2+ из хранилищ. Возможно, что тонкие Ca2+-механизмы в астроцитах подвергаются патологическим изменениям на самых ранних стадиях БА и влекут за собой астроцитарную атрофию. Домены атрофированных астроцитов сокращаются, в результате уменьшается или вовсе исчезает астроцитарная поддержка нейронов и синапсов, изначально находящихся под протекцией астроцитов в данной области (рис. 3). Это приводит к нарушению передачи сигналов в синапсах, глутаматной эксайтотоксичности, а затем к полной утрате синаптических контактов. Aβ-зависимая Ca2+-дисрегуляция в астроцитах может накапливаться в течение длительного времени, “обрастая” все новыми последствиями, такими как: реактивный астроглиоз, воспаление, снижение фагоцитарной функции микроглии, нарушение поддержания гемотоэнцефалического барьера, ухудшение мозгового кровообращения, эксайтотоксичность, гиперактивация GSK3β, перепроизводство гиперфосфорилированного белка тау, накопление внеклеточных олигомерных и префибриллярных форм Aβ, дисфункции митохондрий и МАМ. Апофеозом становится нейродегенерация, начинающаяся с наиболее уязвимых зон головного мозга.
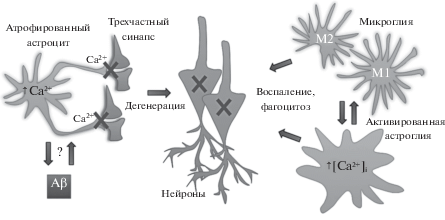
Рис. 3.
Глиально-кальциевая гипотеза болезни Альцгеймера. Ранняя стадия заболевания характеризуется наличием атрофированных астроцитов, которые перестают оказывать достаточную поддержку синапсам в своих доменах, возможно, в результате нарушения локальных кальциевых событий в соме и отростках. Это событие вызывает нарушение передачи сигналов в синапсах, приводит к экситотоксичности, синаптической деградации и гибели нейронов. Причиной этих изменений может служить увеличение внутриклеточных растворимых, а позже, и внеклеточных нерастворимых форм Aβ. Дисрегулированные кальциевые сигналы в атрофированных астроцитах, накопление Aβ и деградация нейронов активируют сигнальные каскады, приводящие к реактивному астроцитозу и активации микроглии. Все последующие патологические события являются следствием кальциевых нарушений в астроцитах и глиальных воспалительных реакций.
Вклад глиальных клеток, в особенности астроцитов, в нейродегенеративные расстройства сложен и многогранен. Нейроглиальные реакции являются основополагающими для определения прогрессирования и исхода патологий ЦНС. Возможно ли использовать модуляцию передачи кальциевых сигналов в астроцитах посредством генетических или фармакологических манипуляций для предотвращения накопления Aβ и других характерных нарушений? На этот вопрос можно попытаться ответить, отойдя от нейроцентризма и приступив к изучению роли глиальных кальциевых сигналов в функционировании ЦНС. Рассмотрение астроцитов как активных участников обработки информации, использующих кальциевые колебания как инструмент, посредством которого они реагируют, интегрируют и передают информацию в мозге, поможет ускорить прогресс в нахождении новых терапевтических стратегий, направленных на возвращение астроцитам физиологической роли при нейродегенерации, и, следовательно, привести к клиническому улучшению у пациентов.
Список литературы
Alzheimer Alois. About a peculiar disease of the cerebral cortex. Gen. J. Psychiatry Mental-Judicial Med. 64(1–2): 146–148. 1907.
Alzheimer’s Disease International (ADI). World Alzheimer Report 2015, The Global Economic Impact of Dementia. 1–92. 2015.
Alzheimer A. Beitrage zur Kenntnis der pathologischen Neuroglia und ihrer Beziehungenzu den Abbauvorgangen im Nervengewebe. In: Nissl F, Alzheimer A. (eds) Histologische undhistopathologische Arbeiten uber die Grosshirnrinde mit besonderer Berucksichtigung der pathologischen Anatomie der Geisteskrankheiten. 1–3: 401–562. 1910.
Jana M., Palencia C.A., Pahan K. Fibrillar amyloid-beta peptides activate microglia via TLR2: implications for Alzheimer’s disease. J. Immunol. 181(10): 7254–7262. 2008.
Krabbe G., Halle A., Matyash V., Rinnenthal J.L., Eom G.D., Bernhardt U., Miller K.R., Prokop S., Kettenmann H., Heppner F.L. Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS One. 8(4): e60921. 2013.
Dossi E., Vasile F., Rouach N. Human astrocytes in the diseased brain. Brain Res. Bull. 136: 139–156. 2018.
Wyss-Coray T., Rogers J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2(1): a006346. 2012.
Kuchibhotla K.V., Lattarulo C.R., Hyman B.T., Bacskai B.J. Synchronous Hyperactivity and Intercellular Calcium Waves in Astrocytes in Alzheimer Mice. Science. 323(5918): 1211–1215. 2009.
Shigetomi E., Patel S., Khakh B.S. Probing the complexities of astrocyte calcium signaling. Trends in Cell Biology. 26(4): 300–312. 2016.
Tyurikova O., Zheng K., Rings A., Drews A., Klenerman D., Rusakov D.A. Monitoring Ca2+ elevations in individual astrocytes upon local release of amyloid beta in acute brain slices. Brain Res. Bull. 136: 85–90. 2018.
Stutzmann G.E. The Pathogenesis of Alzheimers Disease – Is It a Lifelong “Calciumopathy”? The Neuroscientist. 13(5): 546–559. 2007.
Bekris L.M., Yu C.E., Bird T.D., Tsuang D.W. Genetics of Alzheimer’s disease. J. Geriatr. Psychiatry Neurol. 23(4): 213–227. 2010.
Kang J., Lemaire H.G., Unterbeck A., Salbaum J.M., Masters C.L., Grzeschik K.H., Multhaup G., Beyreuther K., Müller-Hill B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 325(6106): 733–736. 1987.
Cabezas Lopategui I., Batista Herrera A., Rol Penton G. The role of glial cells in Alzheimer disease: Potential therapeutic implications. Neurologia. 29(5): 305–309. 2014.
Rodriguez J.J., Verkhratsky A. Neuroglial roots of neurodegenerative diseases? Mol. Neurobiol. 43(2): 87–96. 2011.
Chung W.S., Welsh C.A., Barres B.A., Stevens B. Do glia drive synaptic and cognitive impairment in disease? Nat. Neurosci. 18(11): 1539–1545. 2015.
Bird T.D. Genetic aspects of Alzheimer disease. Genet. Med. 10(4): 231–239. 2008.
Malashenkova I.K., Krynskiy S.A., Mamoshina M.V., Didkovskiy N.A. APOEgene polymorphism: The impact of APOE4 allele on systemic inflammation and its role in the pathogenesis of Alzheimer’s disease. Med. Immunol. 20(3): 303–312. 2018.
Gunten A.V., Clerc M.T., Tomar R., John-Smith P.S. Evolutionary Considerations on Aging and Alzheimer’s Disease. J. Alzheimers Dis. Parkinsonism. 8(1): 423. 2018.
Xu W., Tan L., Yu, J.T. The Role of PICALM in Alzheimer’s Disease. Mol. Neurobiol. 52(1): 399–413. 2015.
Griciuc A., Serrano-Pozo A., Parrado A.R., Lesinski A.N., Asselin C.N., Mullin K., Hooli B., Choi S.H., Hyman B.T., Tanzi R.E. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 78(4): 631–643. 2013.
Jiang T., Yu J.T., Hu N., Tan M.S., Zhu X.C., Tan L. CD33 in Alzheimer’s disease. Mol. Neurobiol. 49(1): 529–535. 2014.
Tan L., Wang H.F., Tan M.S., Tan C.C., Zhu X.C., Miao D., Yu W.J., Jiang T., Tan L., Yu J.T. and Alzheimer’s Disease Neuroimaging Initiative. Effect of CLU genetic variants on cerebrospinal fluid and neuroimaging markers in healthy, mild cognitive impairment and Alzheimer’s disease cohorts. Sci. Rep. 6: 26027. 2016.
Heslegrave A., Heywood W., Paterson R., Magdalinou N., Svensson J., Johansson P., Öhrfelt A., Blennow K., Hardy J., Schott J., Mills K., Zetterberg H. Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer’s disease. Mol. Neurodegener. 11: 3. 2016.
Guerreiro R., Wojtas A., Bras J., Carrasquillo M., Rogaeva E., Majounie E., Cruchaga C., Sassi C., Kauwe J.S., Younkin S., Hazrati L., Collinge J., Pocock J., Lashley T., Williams J., Lambert J.C., Amouyel P., Goate A., Rademakers R., Morgan K., Powell J., St George-Hyslop P., Singleton A., Hardy J.; Alzheimer Genetic Analysis Group. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 368(2): 117–127. 2012.
Celarain N., Sanchez-Ruiz de Gordoa J., Zelaya M.V., Roldán M., Larumbe R., Pulido L., Echavarri C., Mendioroz M. TREM2 upregulation correlates with 5-hydroxymethycytosine enrichment in Alzheimer’s disease hippocampus. Clin. Epigenetics. 8: 37. 2016.
Korvatska O., Leverenz J.B., Jayadev S., McMillan P., Kurtz I., Guo X., Rumbaugh M., Matsushita M., Girirajan S., Dorschner M.O., Kiianitsa K., Yu C.E., Brkanac Z., Garden G.A., Raskind W.H., Bird T.D. R47H Variant of TREM2 Associated With Alzheimer Disease in a Large Late-Onset Family: Clinical, Genetic, and Neuropathological Study. JAMA Neurol. 72(8): 920–927. 2015.
Baranowski J.B., Bott K.N., MacPherson R.E.K. Evaluation of neuropathological effects of a high-fat high-sucrose diet in middle-aged male C57BL6/J mice. Physiol. Rep. 6(11): e13729. 2018.
Solomon A., Kivipelto M., Wolozin B., Zhou J., Whitmer R.A. Midlife serum cholesterol and increased risk of Alzheimer’s and vascular dementia three decades later. Dement. Geriatr. Cogn. Disord. 28(1): 75–80. 2009.
Yaffe K., Lindquist K., Schwartz A.V., Vitartas C., Vittinghoff E., Satterfield S., Simonsick E.M., Launer L., Rosano C., Cauley J.A., Harris T. Advanced glycation end product level, diabetes, and accelerated cognitive aging. Neurology. 77(14): 1351–1356. 2011.
Rusanen M., Kivipelto M., Quesenberry C.P., Zhou J., Whitmer R.A. Heavy Smoking in Midlife and Long-term Risk of Alzheimer Disease and Vascular Dementia. Arch. Intern. Med. 171(4): 333–339. 2011.
Scarmeas N., Luchsinger J.A., Brickman A.M., Cosentino S., Schupf N., Xin-Tang M., Gu Y., Stern Y. Physical activity and Alzheimer disease course. Am. J. Geriatr. Psychiatry. 19(5): 471–481. 2011.
Masters C.L., Bateman R., Blennow K., Rowe C.C., Sperling R.A., Cummings J.L. Alzheimer’s disease. Nature Rev. Disease Primers. 1: 15056. 2015.
Qiu C., Kivipelto M., von Strauss E. Epidemiology of Alzheimer’s disease: occurrence, determinants, and strategies toward intervention. Dialogues Clin. Neurosci. 11(2): 111–128. 2009.
Alzheimer's Association. 2012 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia. J. Alzheimer’s Assoc. 8(2): 131–168. 2012.
Alzheimer's Association. 2016 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia. J. Alzheimer’s Assoc. 12(4): 459–509. 2016.
Chin A.L., Negash S., Hamilton R. Diversity and disparity in dementia: the impact of ethnoracial differences in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 25(3): 187–195. 2011.
Hebert L.E., Weuve J., Scherr P.A., Evans D.A. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology. 80(19): 1778–1783. 2013.
Altmann A., Tian L., Henderson V.W., Greicius M.D. Alzheimer’s Disease Neuroimaging Initiative Investigators. Sex modifies the APOE-related risk of developing Alzheimer disease. Ann. Neurol. 75(4): 563–573. 2014.
Mielke M.M., Vemuri P., Rocca W.A. Clinical epidemiology of Alzheimer’s disease: assessing sex and gender differences. Clin. Epidemiol. 6: 37–48. 2014.
Mosconi L., Rahman A., Diaz I., Wu X., Scheyer O., Hristov H.W., Vallabhajosula S., Isaacson R.S., de Leon M.J., Brinton R.D. Increased Alzheimer’s risk during the menopause transition: A 3-year longitudinal brain imaging study. PLoS One. 13(12): e0207885. 2018.
Nelson P.T., Alafuzoff I., Bigio E.H., Bouras C., Braak H., Cairns N.J., Castellani R.J., Crain B.J., Davies P., Del Tredici K., Duyckaerts C., Frosch M.P., Haroutunian V., Hof P.R., Hulette C.M., Hyman B.T., Iwatsubo T., Jellinger K.A., Jicha G.A., Kövari E., Kukull W.A., Leverenz J.B., Love S., Mackenzie I.R., Mann D.M., Masliah E., McKee A.C., Montine T.J., Morris J.C., Schneider J.A., Sonnen J.A., Thal D.R., Trojanowski J.Q., Troncoso J.C., Wisniewski T., Woltjer R.L., Beach T.G. Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. J. Neuropathol. Exp. Neurol. 71(5): 362–381. 2012.
De Paula V.J., Radanovic M., Diniz B.S., Forlenza O.V. Alzheimer disease. Subcell. Biochem. 65: 329–352. 2012.
Roberts G.W., Gentleman S.M., Lynch A., Murray L., Landon M., Graham, D.I. Beta Amyloid protein deposition in the brain after severe head injury: Implications for the pathogenesis of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry. 57(4): 419–425. 1994.
Rhein V., Eckert A. Effects of Alzheimer’s amyloid-beta and tau protein on mitochondrial function – Role of glucose metabolism and insulin signaling. Arch. Physiol. Biochem. 113(3): 131–141. 2007.
Mokhtar S.H., Bakhuraysah M.M., Cram D.S., Petratos S. The Beta-Amyloid Protein of Alzheimer’s Disease: Communication Breakdown by Modifying the Neuronal Cytoskeleton. Internat. J. Alzheimer’s Dis. 2013: 910502. 2013.
Magalingam K.B., Radhakrishnan A., Ping N.S., Haleagrahara N. Current Concepts of Neurodegenerative Mechanisms in Alzheimer’s Disease. Biomed. Res. Int. 8: 3740461. 2018.
Pera M., Larrea D., Guardia-Laguarta C., Montesinos J., Velasco K.R., Agrawal R.R., Xu Y., Chan R.B., Di Paolo G., Mehler M.F., Perumal G.S., Macaluso F.P., Freyberg Z.Z., Acin-Perez R., Enriquez J.A., Schon E.A., Area-Gomez E. Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. EMBO J. 36(22): 3356–3371. 2017.
Glabe C.G. Conformation-dependent antibodies target diseases of protein misfolding. Trends Biochem. Sci. 29(10): 542–547. 2004.
Kim J., Basak J.M., Holtzman D.M. The role of apolipoprotein E in Alzheimer’s disease. Neuron. 63(3): 287–303. 2009.
Mawuenyega K.G., Sigurdson W., Ovod V., Munsell L., Kasten T., Morris J.C., Yarasheski K.E., Bateman R.J. Decreased Clearance of CNS Amyloid-β in Alzheimer’s Disease. Science. 330(6012): 1774. 2010.
Recuero M., Serrano E., Bullido M.J., Valdivieso F. Aβ production as consequence of cellular death of a human neuroblastoma overexpressing APP. FEBS Letters. 570(1–3): 114–118. 2004.
Gu L., Guo Z. Alzheimer’s Aβ42 and Aβ40 peptides form interlaced amyloid fibrils. J. Neurochem. 126(3): 305–311. 2013.
Hecimovic S., Wang J., Dolios G., Martinez M., Wang R., Goate A.M. Mutations in APP have independent effects on Aβ and CTFγ generation. Neurobiol. Dis. 17(2): 205–218. 2014.
Kumar-Singh S., Theuns J., Van Broeck B., Pirici D., Vennekens K., Corsmit E., Cruts M., Dermaut B., Wang R., Van Broeckhoven C. Mean Age-of-Onset of Familial Alzheimer Disease Caused by Presenilin Mutations Correlates With Both Increased Aβ42 and Decreased Aβ40. Human Mutation. 27(7): 686–696. 2006.
Jonsson T., Atwal J.K., Steinberg S., Snaedal J., Jonsson P.V., Bjornsson S., Stefansson H., Sulem P., Gudbjartsson D., Maloney J., Hoyte K., Gustafson A., Liu Y., Lu Y., Bhangale T., Graham R.R., Huttenlocher J., Bjornsdottir G., Andreassen O.A., Jönsson E.G., Palotie A., Behrens T.W., Magnusson O.T., Kong A., Thorsteinsdottir U., Watts R.J., Stefansson K. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 488(7409): 96–99. 2012.
Abramov A.Y., Canevari L., Duchen M.R. Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J. Neurosci. 24: 565–575. 2004.
Golpich M., Amini E., Mohamed Z., Azman Ali R., Mohamed Ibrahim N., Ahmadiani A. Mitochondrial Dysfunction and Biogenesis in Neurodegenerative diseases: Pathogenesis and Treatment. CNS Neurosci. Therap. 23(1): 5–22. 2017.
Beach T.G., Monsell S.E., Phillips L.E., Kukull W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005-2010. J. Neuropathol. Exp. Neurol. 71(4): 266–273. 2012.
Serrano-Pozo A., Qian J., Monsell S.E., Blacker D., Gómez-Isla T., Betensky R.A., Growdon J.H., Johnson K.A., Frosch M.P., Sperling R.A., Hyman B.T. Mild to moderate Alzheimer dementia with insufficient neuropathological changes. Ann. Neurol. 75(4): 597–601. 2014.
Sonnen J.A., Larson E.B., Crane P.K., Haneuse S., Haneuse S., Li G., Schellenberg G.D., Craft S., Leverenz J.B., Montine T.J. Pathological correlates of dementia in a longitudinal, population-based sample of aging. Ann. Neurol. 62(4): 406–413. 2007.
Larson M.E., Lesne S.E. Soluble Aβ oligomer production and toxicity. J. Neurochem. 120(Suppl 1): 125–139. 2011.
Sengupta U., Nilson A.N., Kayed R. The Role of Amyloid-β Oligomers in Toxicity, Propagation, and Immunotherapy. EBioMedicine. 6: 42–49. 2016.
Tai L.M., Ghura S., Koster K.P., Liakaite V., Maienschein-Cline M., Kanabar P., Collins N., Ben-Aissa M., Lei A.Z., Bahroos N., Green S.J., Hendrickson B., Van Eldik L.J., LaDu M.J. APOE-modulated Aβ-induced neuroinflammation in Alzheimer’s disease: Current landscape, novel data, and future perspective. J. Neurochem. 133(4): 465–488. 2015.
Chiarini A., Armato U., Gardenal E., Gui L., Dal Prà I. Amyloid β-Exposed Human Astrocytes Overproduce Phospho-Tau and Overrelease It within Exosomes, Effects Suppressed by Calcilytic NPS 2143-Further Implications for Alzheimer’s Therapy. Front Neurosci. 11: 217. 2017.
Shahani N., Brandt R. Functions and malfunctions of the tau proteins. Cellular and molecular life sciences. 59(10): 1668–1680. 2002.
Drechsel D.N., Hyman A.A., Cobb M.H., Kirschner M.W. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol. Biol. Cell. 3(10): 1141–1154. 1992.
Mandelkow E., Bergen V.M., Biernat J., Mandelkow E.M. Structural principles of tau and the paired helical filaments of Alzheimer’s disease. Brain Pathology. 17(1): 83–90. 2007.
Ballatore C., Lee V.M., Trojanowski J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 8(9): 663–672. 2007.
Bretteville A., Planel E. Tau aggregates: Toxic, inert, or protective species? J. Alzheimers Dis. 14(4): 431–436. 2008.
Morris M., Maeda S., Vossel K., Mucke L. The many faces of tau. Neuron. 70(3): 410–426. 2011.
Simic G., Babic Leko M., Wray S., Harrington C., Delalle I., Jovanov-Milošević N., Bažadona D., Buée L., de Silva R., Di Giovanni G., Wischik C., Hof P.R. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules. 6(1): 6. 2016.
Schulz K.L., Eckert A., Rhein V., Mai S., Haase W., Reichert A.S., Jendrach M., Müller W.E., Leuner K. A new link to mitochondrial impairment in tauopathies. Mol. Neurobiol. 46(1): 205–216. 2012.
Scheff S.W., Price D.A., Schmitt F.A., DeKosky S.T., Mufson E.J. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 68(18): 1501–1508. 2007.
Minger S.L., Esiri M.M., McDonald B., Keene J., Carter J., Hope T., Francis P.T. Cholinergic deficits contribute to behavioral disturbance in patients with dementia. Neurology. 55(10): 1460–1467. 2000.
Auld D.S., Kornecook T.J., Bastianetto S., Quirion R. Alzheimer’s disease and the basal forebrain cholinergic system: relations to beta-amyloid peptides, cognition, and treatment strategies. Prog. Neurobiol. 68(3): 209–245. 2002.
Mufson E.J., Counts S.E., Perez S.E., Ginsberg S.D. Cholinergic system during the progression of Alzheimer’s disease: therapeutic implications. Expert. Rev. Neurother. 8(11): 1703–1718. 2008.
Steriade M. Acetylcholine systems and rhythmic activities during the waking–sleep cycle. Prog. Brain Res. 145: 179–196. 2004.
Hasselmo M.E. The role of acetylcholine in learning and memory. Curr. Opin. Neurobiol. 16(6): 710–715. 2006.
Platt B., Riedel G. The cholinergic system, EEG and sleep. Behav. Brain Res. 221(2): 499–504. 2011.
Ballinger E.C., Ananth M., Talmage D.A., Role L.W. Basal Forebrain Cholinergic Circuits and Signaling in Cognition and Cognitive Decline. Neuron. 91(6): 1199–1218. 2016.
Maurer S.V., Williams C.L. The Cholinergic System Modulates Memory and Hippocampal Plasticity via Its Interactions with Non-Neuronal Cells. Front. Immunol. 8: 1489. 2017.
Paul S., Jeon W.K., Bizon J.L., Han J.S. Interaction of basal forebrain cholinergic neurons with the glucocorticoid system in stress regulation and cognitive impairment. Front. Aging Neurosci. 7: 43. 2015.
Geula C., Nagykery N., Nicholas A., Wu C.K. Cholinergic neuronal and axonal abnormalities are present early in aging and in Alzheimer disease. J. Neuropathol. Exp. Neurol. 67(4): 309–318. 2008.
Mesulam M., Shaw P., Mash D., Weintraub S. Cholinergic Nucleus Basalis Tauopathy Emerges Early in the Aging-MCI-AD Continuum. Ann. Neurol. 55: 815–828. 2004.
Grothe M., Heinsen H., Teipel S.J. Atrophy of the cholinergic Basal forebrain over the adult age range and in early stages of Alzheimer’s disease. Biol. Psychiatry. 71(9): 805–813. 2011.
Kilimann I., Grothe M., Heinsen H., Alho E.J., Grinberg L., Amaro E.Jr., Dos Santos G.A., da Silva R.E., Mitchell A.J., Frisoni G.B., Bokde A.L., Fellgiebel A., Filippi M., Hampel H., Klöppel S., Teipel S.J. Subregional basal forebrain atrophy in Alzheimer’s disease: A multicenter study. J. Alzheimers Dis. 40(3): 687–700. 2014.
Schmitz T.W., Nathan Spreng R. Alzheimer’s Disease Neuroimaging Initiative. Basal forebrain degeneration precedes and predicts the cortical spread of Alzheimer’s pathology. Nat. Communcat. 7: 13249. 2016.
Ferreira-Vieira T.H., Guimaraes I.M., Silva F.R., Ribeiro F.M. Alzheimer’s Disease: Targeting the Cholinergic System. Current Neuropharmacol. 14(1): 101–115. 2016.
Wu H., Williams J., Nathans J. Complete morphologies of basal forebrain cholinergic neurons in the mouse. Elife. 3: e02444. 2014.
Llorens-Martin M., Jurado J., Hernandez F., Avila J. GSK-3β, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 2014. 7: 46.
Deng Y., Xiong Z., Chen P., Wei J., Chen S., Yan Z. β-amyloid impairs the regulation of N-methyl-D-aspartate receptors by glycogen synthase kinase 3. Neurobiol. Aging. 35(3): 449–459. 2014.
Григорян Г.А. Роль гликогенсинтазы киназы-3 в механизмах обучения и памяти. Ж. высш. нерв. деят. 63(5): 507–519. 2013 [Grigoryan G.A. Role of glycogen synthase kinase-3 in mechanisms of learning and memory. Zh.Vyssh. Nerv. Deiat. Im I. P. Pavlova. 63(5): 507–519. 2013 [In Russ.].
Samadi A., Valderas C., de los Ríos C., Bastida A., Chioua M., González-Lafuente L., Colmena I., Gandía L., Romero A., Del Barrio L., Martín-de-Saavedra M.D., López M.G., Villarroya M., Marco-Contelles J. Cholinergic and neuroprotective drugs for the treatment of Alzheimer and neuronal vascular diseases. II. Synthesis, biological assessment, and molecular modelling of new tacrine analogues from highly substituted 2-aminopyridine-3-carbonitriles. Bioorg. Med. Chem. 19(1): 122–133. 2011.
Hayashi T. Regulating ankyrin dynamics: Roles of sigma-1 receptors. Proc. Natl. Acad. Sci. USA. 98(2): 491–496. 2001.
Hayashi T., Rizzuto R., Hajnoczky G., Su T.P. MAM: More than just a housekeeper. Trends Cell Biol. 19(2): 81–88. 2009.
Hayashi T., Su T.P. Sigma-1 Receptor Chaperones at the ER-Mitochondrion Interface Regulate Ca2+ Signaling and Cell Survival. Cell. 131(3): 596–610. 2007.
Weng T.Y., Shang-Yi Y.A., Su T.P. Roles of sigma-1 receptors on mitochondrial functions relevant to neurodegenerative diseases. J. Biomed. Sci. 24: 74. 2017.
Srivats S., Balasuriya D., Pasche M., Vistal G., Edwardson J.M., Taylor C.W., Murrell-Lagnado R.D. Sigma1 receptors inhibit store-operated Ca2+ entry by attenuating coupling of STIM1 to Orai1. J. Cell. Biol. 213(1): 65–79. 2016.
Tsai S.Y., Pokrass M.J., Klauer N.R., Nohara H., Su T.P. Sigma-1 receptor regulates Tau phosphorylation and axon extension by shaping p35 turnover via myristic acid. Proc. Nat. Acad. Sci. USA. 112(21): 6742–6747. 2015.
Nguyen L., Lucke-Wold B.P., Mookerjee S.A., Cavendish J.Z., Robson M.J., Scandinaro A.L., Matsumoto R.R. Role of sigma-1 receptors in neurodegenerative diseases. J. Pharmacol. Sci. 127(1): 17–29. 2015.
Behensky A.A., Yasny I.E., Shuster A.M., Seredenin S.B., Petrov A.V., Cuevas J. Afobazole activation of σ-1 receptors modulates neuronal responses to amyloid-β25-35. J. Pharmacol. Exp. Ther. 347(2): 468–477. 2013.
Reddy P.H. Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer’s disease. Exp Neurol. 218(2): 286–292. 2009.
Cabezas-Opazo F.A., Vergara-Pulgar K., Perez M.J., Jara C., Osorio-Fuentealba C., Quintanilla R.A. Mitochondrial Dysfunction Contributes to the Pathogenesis of Alzheimer’s Disease. Oxid Med. Cell Longev. 2015: 509654. 2015.
Area-Gomez E., Groof A., Bonilla E., Montesinos J., Tanji K., Boldogh I., Pon L., Schon E.A. A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell Death. Dis. 9(3): 335. 2018.
Manczak M., Calkins M.J., Reddy P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 20(13): 2495–2509. 2011.
Schreiner B., Hedskog L., Wiehager B., Ankarcrona M. Amyloid-β peptides are generated in mitochondria-associated endoplasmic reticulum membranes. J. Alzheimers Dis. 43(2): 369–374. 2015.
Robinson A., Grosgen S., Mett J., Zimmer V.C., Haupenthal V.J., Hundsdörfer B., Stahlmann C.P., Slobodskoy Y., Müller U.C., Hartmann T., Stein R., Grimm M.O. Upregulation of PGC-1α expression by Alzheimer’s disease-associated pathway: presenilin 1/amyloid precursor protein (APP)/intracellular domain of APP. Aging Cell. 13(2): 263–272. 2014.
Dzamba D., Harantova L., Butenko O., Anderova M. Glial Cells – The Key Elements of Alzheimers Disease. Curr. Alzheimer Res. 13(8): 894–911. 2016.
Hanisch U.K., Kettenmann H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10(11): 1387–1394. 2007.
Mandrekar S., Landreth G.E. Microglia and Inflammation in Alzheimer’s Disease. CNS & Neurol. Disord. Drug Targets. 9(2): 156–167. 2010.
Verkhratsky A., Butt A. Glial Neurobiology. John Wiley & Sons, Ltd. England. The Atrium, Southern Gate, Chichester, West Sussex PO19 8SQ. 2007.
Weitz T.M., Town T. Microglia in Alzheimer’s Disease: It’s All About Context. Int. J. Alzheimers Dis. 2012: 314185. 2012.
Bolmont T., Haiss F., Eicke D., Radde R., Mathis C.A., Klunk W.E., Kohsaka S., Jucker M., Calhoun M.E. Dynamics of the Microglial/Amyloid Interaction Indicate a Role in Plaque Maintenance. J. Neurosci. 28(16): 4283–4292. 2008.
Barger S.W., Harmon A.D. Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature. 388(6645): 878–881. 1997.
Solito E., Sastre M. Microglia function in Alzheimer’s disease. Front. Pharmacol. 3: 14. 2012.
Colton C.A. Heterogeneity of microglial activation in the innate immune response in the brain. J. Neuroim. Pharmacol. 4(4): 399–418. 2009.
Block M.L., Zecca L., Hong J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8(1): 57–69. 2007.
Ponomarev E.D., Maresz K., Tan Y., Dittel B.N. CNS-Derived Interleukin-4 Is Essential for the Regulation of Autoimmune Inflammation and Induces a State of Alternative Activation in Microglial Cells. J. Neurosci. 27(40): 10714–10721. 2007.
Tang Y., Le W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 53(2): 1181–1194. 2016.
Jimenez S., Baglietto-Vargas D., Caballero C., Moreno-Gonzalez I., Torres M., Sanchez-Varo R., Ruano D., Vizuete M., Gutierrez A., Vitorica J. Inflammatory Response in the Hippocampus of PS1M146L/APP751SL Mouse Model of Alzheimer’s Disease: Age-Dependent Switch in the Microglial Phenotype from Alternative to Classic. J. Neurosci. 28(45): 11650–11661. 2008.
Martinez F.O., Gordon S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Reports. 6: 13. 2014.
Ransohoff R.M. A polarizing question: Do M1 and M2 microglia exist. Nat. Neurosci. 19(8): 987–991. 2016.
Verkhratsky A., Orkand R.K., Kettenmann H. Glial calcium: Homeostasis and signaling function. Physiol. Rev. 78(1): 99–141. 1998.
Oberheim N.A., Takano T., Han X., He W., Lin J.H., Wang F., Xu Q., Wyatt J.D., Pilcher W., Ojemann J.G., Ransom B.R., Goldman S.A., Nedergaard M. Uniquely hominid features of adult human astrocytes. J. Neurosci. 29(10): 3276–3287. 2009.
Vasile F., Dossi E., Rouach N. Human astrocytes: Structure and functions in the healthy brain. Brain Struct. Funct. 222(5): 2017–2029. 2017.
Bushong E.A., Martone M.E., Jones Y.Z., Ellisman M.H. Stratum radiatum occupy separate anatomical domains. J. Neurosci. 22(1): 183–192. 2002.
Oberheim N.A., Wang X., Goldman S., Nedergaard M. Astrocytic complexity distinguishes the human brain. Trends Neurosci. 29(10): 547–553. 2006.
Halassa M.M., Fellin T., Takano H., Dong J.H., Haydon P.G. Synaptic Islands Defined by the Territory of a Single Astrocyte. J. Neurosci. 27(24): 6473–6477. 2007.
Oberheim N.A., Tian G.F., Han X., Peng W., Takano T., Ransom B., Nedergaard M. Loss of Astrocytic Domain Organization in the Epileptic Brain. J. Neurosci. 28(13): 3264–3276. 2008.
Bushong E.A., Martone M.E., Ellisman M.H. Maturation of astrocyte morphology and the establishment of astrocyte domains during postnatal hippocampal development. Int. J. Dev. Neurosci. 22(2):73–86. 2004.
Wang D.D., Bordey A. The Astrocyte Odyssey. Progr. Neurobiol. 86(4): 342–367. 2008.
Araque A., Carmignoto G., Haydon P.G., Oliet S.H.R., Robitaille R., Volterra A. Gliotransmitters Travel in Time and Space. Neuron. 81(4): 728–739. 2014.
Dal Pra I., Chiarini A., Armato U. Antagonizing amyloid-β/calcium-sensing receptor signaling in human astrocytes and neurons: A key to halt Alzheimer’s disease progression? Neural. Regen. Res. 10: 213–218. 2015.
Magistretti P.J., Allaman I. A Cellular Perspective on Brain Energy Metabolism and Functional Imaging. Neuron. 86(4): 883–901. 2015.
Henneberger C., Bard L., Rusakov D.A. D-Serine: A key to synaptic plasticity? The Internat. J. Biochem. Cell Biol. 44(4): 587–590. 2012.
Navarrete M., Gertrudis P., Fernandez de Sevilla D., Gomez-Gonzalo M., Nunez A., Martín E.D., Araque A. Astrocytes mediate in vivo cholinergic-induced synaptic plasticity. PLoS Biology. 10(2): e1001259. 2012.
Henneberger C., Papouin T., Oliet S.H.R., Rusakov D.A. Long-term potentiation depends on release of d-serine from astrocytes. Nature. 463(7278): 232–236. 2010.
Zorec R., Horvat A., Vardjan N., Verkhratsky A. Memory Formation Shaped by Astroglia. Front. Integr. Neurosci. 9: 56. 2015.
Farina C., Aloisi F., Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 28(3): 138–145. 2007.
Olabarria M., Noristani H.N., Verkhratsky A., Rodríguez J.J. Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia. 58(7): 831–838. 2010.
Yeh C., Vadhwana B., Verkhratsky A., Rodriguez J.J. Early Astrocytic Atrophy in the Entorhinal Cortex of a Triple Transgenic Animal Model of Alzheimer’s Disease. ASN Neuro. 3(5): 271–279. 2011.
Kulijewicz-Nawrot M., Verkhratsky A., Chvatal A., Sykova E., Rodríguez J.J. Astrocytic cytoskeletal atrophy in the medial prefrontal cortex of a triple transgenic mouse model of Alzheimer’s disease. J. Anat. 221(3): 252–262. 2012.
Zamanian J.L., Xu L., Foo L.C., Nouri N., Zhou L., Giffard R.G., Barres B.A. Genomic Analysis of Reactive Astrogliosis. J. Neurosci. 32(18): 6391–6410. 2012.
Kanemaru K., Kubota J., Sekiya H., Hirose K., Okubo Y., Iino M. Calcium-dependent N-cadherin up-regulation mediates reactive astrogliosis and neuroprotection after brain injury. Proc. Natl. Acad. Sci. USA. 110(28): 11612–11617. 2013.
Pekny M., Wilhelmsson U., Pekna M. The dual role of astrocyte activation and reactive gliosis. Neurosci. Lett. 565: 30–38. 2014.
Allan S.M., Tyrrell P.J., Rothwell N.J. Interleukin-1 and neuronal injury. Nat. Rev. Immunol. 5(8): 629–640. 2005.
Kitazawa M., Cheng D., Tsukamoto M.R., Koike MA, Wes PD, Vasilevko V, Cribbs DH, LaFerla FM. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β‑catenin pathway function in an Alzheimer’s disease model. J. Immunol. 187(12): 6539–6549. 2011.
Ghosh S., Wu M.D., Shaftel S.S., Kyrkanides S., LaFerla F.M., Olschowka J.A., O’Banion M.K. Sustained interleukin-1β overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J. Neurosci. 33(11): 5053–5064. 2013.
Matousek S.B., Ghosh S., Shaftel S.S., Kyrkanides S., Olschowka J.A., O’Banion M.K. Chronic IL-1β-mediated neuroinflammation mitigates amyloid pathology in a mouse model of Alzheimer’s disease without inducing overt neurodegeneration. J. Neuroim. Pharmacol. 7(1): 156–164. 2011.
Ben-Menachem-Zidon O., Ben-Menahem Y., Ben-Hur T., Yirmiya R. Intra-hippocampal transplantation of neural precursor cells with transgenic over-expression of IL-1 receptor antagonist rescues memory and neurogenesis impairments in an Alzheimer’s disease model. Neuropsychopharmacology. 39(2): 401–414. 2013.
Orre M., Kamphuis W., Osborn L.M., Melief J., Kooijman L., Huitinga I., Klooster J., Bossers K., Hol E.M. Acute isolation and transcriptome characterization of cortical astrocytes and microglia from young and aged mice. Neurobiol. Aging. 35(1): 1–14. 2014.
Kraft A.W., Hu X., Yoon H., Yan P., Xiao Q., Wang Y., Gil S.C., Brown J., Wilhelmsson U., Restivo J.L., Cirrito J.R., Holtzman D.M., Kim J., Pekny M., Lee J.M. Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. The FASEB Journal. 27(1):187–198. 2013.
Meraz-Rios M.A., Toral-Rios D., Franco-Bocanegra D., Villeda-Hernandez J., Campos-Pena V. Inflammatory process in Alzheimer’s Disease. Front. Integr. Neurosci. 7: 59. 2013.
Bell R.D., Zlokovic B.V. Neurovascular mechanisms and blood–brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 118(1): 103–113. 2009.
Sollvander S., Nikitidou E., Brolin R., Soderberg L., Sehlin D., Lannfelt L., Erlandsson A. Accumulation of amyloid-β by astrocytes result in enlarged endosomes and microvesicle-induced apoptosis of neurons. Mol. Neurodegenerat. 11: 38. 2016.
Singh A., Wickliffe C.A. Astrocytes and synaptic plasticity in health and disease. Exp. Brain Res. 235(6): 1645–1655. 2017.
Verkhratsky A., Rodríguez J.J., Parpura V. Calcium signalling in astroglia. Mol Cell Endocrinol. 353(1–2): 45–56. 2012.
Acosta C., Anderson H.D., Anderson C.M. Astrocyte Dysfunction in Alzheimer Disease. J. Neurosci. Res. 95(12): 2430–2447. 2017.
Petzold G.C., Murthy V.N. Role of astrocytes in neurovascular coupling. Neuron. 71(5): 782–796. 2011.
Tanaka M., Shih P.Y., Gomi H., Yoshida T., Nakai J., Ando R., Furuichi T., Mikoshiba K., Semyanov A., Itohara S. Astrocytic Ca2+ signals are required for the functional integrity of tripartite synapses. Mol. Brain. 6: 6. 2013.
Giaume C. Astroglial Wiring is Adding Complexity to Neuroglial Networking. Front. Neuroenerg. 2: 129. 2010.
Scemes E., Giaume C. Astrocyte Calcium Waves: What They Are and What They Do. Glia. 54(7):716–725. 2006.
Kang M., Othmer H.G. Spatiotemporal characteristics of calcium dynamics in astrocytes. Chaos. 19(3): 037116. 2009.
De Pitta M., Volman V., Levine H., Pioggia G., De Rossi D., Ben-Jacob E. Coexistence of amplitude and frequency modulations in intracellular calcium dynamics. Phys. Rev. E. Stat. Nonlin. Soft. Matter. Phys. 77(3 Pt 1): 030903. 2008.
Guerra-Gomes S., Sousa N., Pinto L., Oliveira J.F. Functional Roles of Astrocyte Calcium Elevations: From Synapses to Behavior. Front. Cell. Neurosci. 11: 427. 2018.
Wu Y., Tang X., Arizono M., Bannai H., Shih P., Semyanov A. Spatiotemporal calcium dynamics in single astrocytes and its modulation by neuronal activity. Cell Calcium. 55(2): 119–129. 2014.
Khakh B.S., McCarthy K.D. Astrocyte Calcium Signaling: From Observations to Functions and the Challenges Therein. Cold Spring Harb. Perspect. Biol. 7(4): a020404. 2015.
Lim D., Ronco V., Grolla A.A., Verkhratsky A., Genazzani A.A. Glial Calcium Signalling in Alzheimer’s Disease. Rev. Physiol. Biochem.Pharmacol. 167: 45–65. 2014.
Nelson O., Tu H., Lei T., Bentahir M., De Strooper B., Bezprozvanny I. Familial Alzheimer disease-linked mutations specifically disrupt Ca2+ leak function of presenilin 1. J. Clin. Invest. 117(5): 1230–1239. 2007.
Mattson M.P., Cheng B., Davis D., Bryant K., Lieberburg I., Rydel R.E. Beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J. Neurosci. 12(2): 376–389. 1992.
Blanchard B.J., Chen A., Rozeboom L.M., Stafford K.A., Weigele P., Ingram V.M. Efficient reversal of Alzheimer’s disease fibril formation and elimination of neurotoxicity by a small molecule. Proc. Natl. Acad. Sci. USA. 101(40): 14326–14332. 2004.
Demuro A., Mina E., Kayed R., Milton S.C., Parker I., Glabe C.G. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 280(17): 17294–17300. 2005.
Snyder E.M., Nong Y., Almeida C.G., Paul S., Moran T., Choi E.Y., Nairn A.C., Salter M.W., Lombroso P.J., Gouras G.K., Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat. Neurosci. 8(8): 1051–1058. 2005.
Celsi F., Svedberg M., Unger C., Cotman C.W., Carrì M.T., Ottersen O.P., Nordberg A., Torp R. Beta-amyloid causes downregulation of calcineurin in neurons through induction of oxidative stress. Neurobiol. Dis. 26(2): 342–352. 2007.
Lacor P.N., Buniel M.C., Furlow P.W., Clemente A.S., Velasco P.T., Wood M., Viola K.L., Klein W.L. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J. Neurosci. 27(4): 796–807. 2007.
Alberdi E., Sanchez-Gomez M.V., Cavaliere F., Perez-Samartín A., Zugaza J.L., Trullas R., Domercq M., Matute C. Amyloid beta oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium. 47(3): 264–272. 2010.
Abramov A.Y., Canevari L., Duchen M.R. Changes in intracellular calcium and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. J. Neurosci. 23(12): 5088–5095. 2003.
Busche M.A., Eichhoff G., Adelsberger H., Abramowski D., Wiederhold K.H., Haass C., Staufenbiel M., Konnerth A., Garaschuk O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science. 321(5896): 1686–1689. 2008.
Takano T., Han X., Deane R., Zlokovic B., Nedergaard M. Two-photon imaging of astrocytic Ca2+ signaling and the microvasculature in experimental mice models of Alzheimer’s disease. Ann. N.Y. Acad. Sci. 1097: 40–50. 2007.
Grolla A.A., Fakhfouri G., Balzaretti G., Marcello E., Gardoni F., Canonico P.L., DiLuca M., Genazzani A.A., Lim D. Aβ leads to Ca2+ signaling alterations and transcriptional changes in glial cells. Neurobiol. Aging. 34(2): 511–22. 2013.
Grolla A.A., Sim J.A., Lim D., Rodriguez J.J., Genazzani A.A., Verkhratsky A. Amyloid-β and Alzheimer’s disease type pathology differentially affects the calcium signalling toolkit in astrocytes from different brain regions. Cell Death & Disease. 4: e623. 2013.
Lim D., Iyer A., Ronco V., Grolla A.A., Canonico P.L., Aronica E., Genazzani A.A. Amyloid beta deregulates astroglial mGluR5-mediated calcium signaling via calcineurin and Nf-kB. Glia. 61(7):1134–1145. 2013.
Ronco V., Grolla A.A., Glasnov T.N., Canonico P.L., Verkhratsky A., Genazzani A.A., Lim D. Differential deregulation of astrocytic calcium signalling by amyloid-β, TNFα, IL-1β and LPS. Cell Calcium. 55(4): 219–229. 2014.
Lee L., Kosurid P., Arancio O. Picomolar Amyloid-β Peptides Enhance Spontaneous Astrocyte Calcium Transients. J. Alzheimer’s Dis. 38(1): 49–62. 2014.
Alberdi E., Wyssenbach A., Alberdi M., Sánchez-Gómez M.V., Cavaliere F., Rodríguez J.J., Verkhratsky A., Matute C. Ca(2+)-dependent endoplasmic reticulum stress correlates with astrogliosis in oligomeric amyloid β-treated astrocytes and in a model of Alzheimer’s disease. Aging Cell. 12(2): 292–302. 2013.
Wu H.Y., Hudry E., Hashimoto T., Kuchibhotla K., Rozkalne A., Fan Z., Spires-Jones T., Xie H., Arbel-Ornath M., Grosskreutz C.L., Bacskai B.J., Hyman B.T. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J. Neurosci. 30(7): 2636–2649. 2010.
Norris C.M., Kadish I., Blalock E.M., Chen K.C., Thibault V., Porter N.M., Landfield P.W., Kraner S.D. Calcineurin Triggers Reactive/Inflammatory Processes in Astrocytes and Is Upregulated in Aging and Alzheimer’s Models. J. Neurosci. 25(18): 4649–4658. 2005.
Abdul H.M., Sama M.A., Furman J.L., Mathis D.M., Beckett T.L., Weidner A.M., Patel E.S., Baig I., Murphy M.P., LeVine H.3rd., Kraner S.D., Norris C.M. Cognitive decline in Alzheimer’s disease is associated with selective changes in calcineurin/NFAT signaling. J. Neurosci. 29(41): 12957–12969. 2009.
Rossner S., Lange-Dohna C., Zeitschel U., Perez-Polo J.R. Alzheimer’s disease beta-secretase BACE1 is not a neuron-specific enzyme. J. Neurochem. 92(2): 226–234. 2005.
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова