

Российский физиологический журнал им. И.М. Сеченова, 2020, T. 106, № 12, стр. 1455-1478
Роль NMDA-рецепторов в эпилептогенезе
Ю. Л. Ергина 1, А. А. Коваленко 1, А. В. Зайцев 1, *
1 Институт эволюционной физиологии и биохимии им. И.М. Сеченова РАН
Санкт-Петербург, Россия
* E-mail: aleksey_zaitsev@mail.ru
Поступила в редакцию 01.09.2020
После доработки 26.09.2020
Принята к публикации 28.09.2020
Аннотация
Эпилепсия характеризуется повторяющимися, возникающими внезапно эпилептическими приступами. При эпилепсии почти в 30% случаев наблюдается фармакорезистентность, в результате чего болезнь может прогрессировать и приводить к ухудшению когнитивных способностей и возникновению сопутствующих психоневрологических заболеваний. Раннее терапевтическое вмешательство способно снизить тяжесть протекания заболевания, а подавление эпилептогенеза считается наиболее перспективной стратегией предотвращения развития эпилепсии после провоцирующих это заболевание событий. NMDA-рецепторы рассматриваются как одна из перспективных мишеней для подавления эпилептогенеза. Нарушение функций NMDA-рецепторов фиксируется на всех этапах развития эпилепсии. Изменения в их экспрессии наблюдаются уже в первые часы после острых судорог, а сами NMDA-рецепторы активно вовлечены в генерацию эпилептической активности. Кроме того, антагонисты NMDA-рецепторов эффективно подавляют эпилептиформную активность в различных моделях судорожных состояний и эпилептического статуса. В этом обзоре мы рассматриваем имеющиеся данные о том, какую роль NMDA-рецепторы играют в развитии эпилепсии, как меняется их экспрессия в разные периоды эпилептогенеза, а также обсуждаем перспективы применения антагонистов и модуляторов NMDA-рецепторов для предотвращения эпилептогенеза.
Эпилепсия – распространенное неврологическое заболевание, характеризующееся повторяющимися неспровоцированными приступами. По оценкам ВОЗ почти 1% населения страдает от эпилепсии. Несмотря на успехи в создании новых противоэпилептических препаратов, полного избавления от судорожных припадков не удается достичь почти у трети больных. Хотя эпилепсия может носить наследственный характер, что часто является следствием генетических аномалий, в большинстве случаев эпилепсия представляет собой приобретенное заболевание. Этиология приобретенной эпилепсии разнообразна, в качестве возможных триггеров могут выступать: эпилептический статус, инсульт, черепно-мозговая травма, пренатальные или перинатальные травмы, инфекции, фебрильные судороги, аутоиммунные заболевания, опухоли головного мозга [1–4]. После воздействия проэпилептических факторов наблюдается так называемый латентный период, предшествующий появлению спонтанной рецидивирующей эпилептической активности. В течение латентного периода наблюдается как гибель нейронов, так и нейрогенез, разрастание аксонов и реорганизация связей между нейронами, изменение баланса возбуждающей и тормозной синаптической передачи, нейровоспалительные процессы и глиоз, нарушения проницаемости гематоэнцефалического барьера [5]. Являясь прогрессирующим заболеванием, эпилепсия сопровождается снижением когнитивных способностей, возникновением сопутствующих психоневрологических нарушений и развитием фармакорезистентности. Раннее терапевтическое вмешательство способно снизить тяжесть протекания заболевания, но именно предотвращение эпилептогенеза считается наиболее перспективной стратегией [6, 7]. К сожалению, до настоящего времени эффективной терапии, предотвращающей развитие эпилепсии, не разработано.
NMDA-рецепторы, ответственные за длительную фазу глутаматергической передачи, рассматриваются в качестве возможной терапевтической мишени для подавления судорожных состояний и эпилептогенеза. Изменения экспрессии этих рецепторов неоднократно показаны в тканях мозга пациентов с эпилепсией [8, 9], а антагонисты NMDA-рецепторов эффективно подавляют эпилептиформную активность в ряде экспериментальных моделей эпилепсии [10, 11]. Однако наличие выраженных побочных эффектов при длительном применении позволяет использовать известные на сегодняшний день антагонисты NMDA-рецепторов исключительно в качестве экстренного средства, например, для лечения эпилептического статуса, устойчивого к терапии первой линии. Тем не менее, продолжается активный поиск фармакологических агентов, способных регулировать работу NMDA-рецепторов с минимальными побочными эффектами. В этом обзоре мы анализируем новые экспериментальные данные об изменениях экспрессии NMDA-рецепторов в различных моделях эпилепсии, их участии в эпилептогенезе, а также рассматриваем перспективы воздействия на NMDA-рецепторы при терапии эпилептогенеза.
ОБЩИЕ СВЕДЕНИЯ ОБ NMDA-РЕЦЕПТОРАХ
NMDA-рецептор – один из трех типов ионотропных глутаматных рецепторов. Ионотропные глутаматные рецепторы представляют собой интегральные мембранные белки, состоящие из четырех крупных субъединиц (более 900 аминокислотных остатков), которые образуют центральную пору ионного канала [12–14]. Функциональные рецепторы образуются исключительно путем сборки субъединиц в пределах одного функционального класса рецепторов [15]. К настоящему моменту известны различные субъединицы NMDA-рецепторов (GluN1, GluN2A-D, GluN3A и GluN3B), отличающихся по своим физиологическим и фармакологическим свойствам [16]. Для сборки функционального NMDA-рецептора требуются две GluN1-субъединицы, являющиеся облигатными, а также присутствие двух других субъединиц – это могут быть две GluN2-субъединицы или же комбинация субъединиц GluN2 и GluN3 [17, 18]. Гомозиготные мутантные мыши, лишенные GluN1-субъединицы, погибают спустя несколько часов после рождения [19].
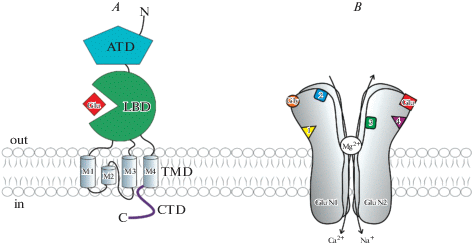
Субъединицы NMDA-рецепторов имеют идентичную мембранную топологию (рис. 1 А). Все субъединицы содержат большой внеклеточный N-концевой домен, три трансмембранных домена (M1, M3, M4), порообразующий (M2) и внутриклеточный С-концевой домены. Функционально у субъединиц можно выделить амино-концевой (ATD), лиганд-связывающий (LBD), трансмембранный (TMD) и цитозольный домены (CTD) [20]. Показано, что ATD участвует в регуляции открытия канала и его инактивации, а также отвечает за аллостерическую регуляцию активности ионного канала путем связывания с модуляторными соединениями. Роль регуляции через ATD для NMDA-рецепторов выражена сильнее, чем для других ионотропных рецепторов глутамата [21]. LBD тремя короткими линкерами связан с TMD. Трансмембранные спирали M1, M3 и M4 от каждой из четырех субъединиц способствуют формированию ионного канала [20]. CTD является наиболее вариабельным доменом и имеет важное значение при регуляции работы канала. Цитозольные домены содержат различные сайты фосфорилирования и сайты связывания внутриклеточных белков, что определяет их роль в регуляции функции рецептора [20]. В частности, CTD GluN1-субъединицы участвует в инактивации рецептора. Цитозольные домены GluN2 необходимы для нормального функционирования NMDA-рецептора, что было продемонстрировано в экспериментах по их удалению или укорочению [22, 23].
Рис. 1.
A – схематическое изображение мембранной топологии субъединицы NMDA-рецептора. ATD – амино-концевой домен, LBD – лиганд-связывающий домен, TMD – трансмембранный домен, CTD – цитозольный домен, Glu – глутамат. B – схематическое изображение регуляторных сайтов NMDA-рецептора. Gly, Glu – сайты специфического связывания медиатора (глутамат) и коагониста (глицин). Модуляторные сайты: 1 – полиаминовый сайт, 2 – сайт связывания неконкурентных антагонистов, 3 – сайт связывания двухвалентных катионов, 4 – сайт связывания нейростероидов. На схеме также показан транспорт ионов Na+, K+ и Ca2+ через ионный канал NMDA-рецептора, в покое пора рецептора заблокирована ионом Mg2+. Воспроизведено с модификациями по [20, 61].
Fig. 1 . A – schematic illustration of the NMDA receptor subunit topology. ATD – amino-terminal domain, LBD – ligand binding domain, TMD – transmembrane domain, CTD – carboxyl-terminal domain, Glu – glutamate. B – schematic illustration of the NMDA receptor regulatory sites. Gly, Glu – a mediator (glutamate) and a coagonist (glycine) specific binding sites. Modulatory sites: 1 – polyamine site, 2 – non-competitive antagonist binding site, 3 – bivalent cation binding site, 4 – neurosteroid binding site. The figure also shows the transport of Na+, K+ and Ca2+ through the NMDA receptor ion channel; at rest, the receptor pore is blocked by the Mg2+. Reproduced with modifications from [20, 61].
Существуют как дигетеромерные, так и тригетеромерные NMDA-рецепторы. В переднем мозге грызунов специфические антитела к GluN2A эффективно иммунопреципитируют к GluN2B и наоборот, что указывает на образование GluN1/GluN2A/GluN2B-тригетеромеров [24]. GluN1/GluN2A/GluN2D- и GluN1/GluN2B/GluN2D-тригетеромеры были обнаружены в таламусе и среднем мозге крысы [25]. Согласно данным иммуноблотного анализа, GluN1/GluN2A/GluN2B-тригетеромеры могут составлять от 10 до 50% и более от общего числа NMDA-рецепторов [26]. Данные последних электрофизиологических исследований указывают на значительный вклад GluN1/GluN2A/GluN2B-тригетеромеров в NMDA-рецептор-опосредованную активность в СА3-СА1 синапсах гиппокампа взрослых животных [27, 28]. Для активации NMDA-рецепторов необходимо одновременное связывание с ними глутамата и глицина, что отличает их от других типов глутаматных рецепторов [29]. Сайты связывания глицина расположены на GluN1- и GluN3-субъединицах [30, 31], в то время как GluN2-субъединицы выступают в качестве сайтов связывания глутамата [30]. Помимо глицина, D- и L-изомеры серина и аланина также служат агонистами субъединицы GluN1 [32, 33]. По сравнению с L-серином, D-серин мощнее и может выступать в качестве основного лиганда к GluN1 в таких областях, как супраоптическое ядро [34]. D-серин синтезируется из L-серина серин-рацемазой в астроцитах [35] и нейронах [36, 37]. К эндогенным агонистам GluN2-субъединицы, помимо глутамата, относятся D- и L-аспартат [38, 39], гомоцистеин и цистеин сульфинат [40, 41].
Четыре глутамат-связывающих GluN2A-D-субъединицы ответственны за биофизические свойства NMDA-рецепторов, а их преобладание зависит от области мозга и периода развития [42–45]. GluN2-субъединицы влияют на эффективность действия глутамата. Например, полумаксимальная эффективная концентрация (ЕС50) для NMDA-рецепторов, содержащих две субъединицы GluN1 и две субъединицы GluN2D, в пять раз ниже, чем для рецепторов, содержащих GluN1/2A, тогда как рецепторы GluN1/2B и GluN1/2C имеют промежуточные значения EC50 [46–48]. Скорость деактивации после отсоединения глутамата, влияющая на продолжительность ВПСТ [49], варьирует более чем в 100 раз между различными GluN2-субъединицами [50]. GluN1/2A- и GluN1/2B-содержащие NMDA-рецепторы имеют более высокую проводимость одиночных каналов, чем GluN1/2C и GluN1/2D [51–53].
Канал NMDA-рецептора проницаем для ионов Ca2+, Na+ и K+. Разные подтипы NMDA-рецепторов демонстрируют сходную проницаемость для ионов Na+ и K+ (PK/PNa = 1.14), при этом они более проницаемы для ионов Ca2+ по сравнению с одновалентными ионами (PCa/PX = 1.8–4.5). Степень проницаемости для Ca2+ зависит от типа GluN2-субъединицы [54, 55]. GluN1/2A и GluN1/2B обладают более высокой проницаемостью для Ca2+ и более чувствительны к Mg2+-блоку, чем GluN1/2C и GluN1/2D [42, 56]. Важной функциональной особенностью NMDA-рецепторов является Mg2+-блок, который сильно зависит от мембранного потенциала и возрастает при его сдвиге к негативным значениям. Величина IC50 (концентрации, обеспечивающей половину от максимально возможного блока) составляет 2, 2, 14 и 10 мкМ для GluN1/2A, GluN1/2B, GluN1/2C и GluN1/2D соответственно при потенциале фиксации равном –100 мВ [57]. Эффективность Mg2+-блока субъединицы GluN2 зависит от множества структурных особенностей, но главным критерием, по-видимому, является единственный аминокислотный остаток в сайте S/L, расположенном в трансмембранной петле M3 [56]. Эти биофизические различия важны тем, что чувствительность к потенциал-зависимому Mg2+-блоку может влиять на временное окно для выработки пластичности, зависящей от времени спайков (spike-timing-dependent plasticity) [58–60].
ФАРМАКОЛОГИЯ NMDA-РЕЦЕПТОРОВ
NMDA-рецепторный комплекс имеет несколько сайтов регуляции: сайты специфического связывания медиатора и коагониста, модуляторные сайты: полиаминовый, сайты связывания двухвалентных катионов и протонов, участки связывания неконкурентных модуляторов [61] (рис. 1B ). Функционирование NMDA-рецепторов модулируется рядом эндогенных и экзогенных соединений, которые могут оказывать противоположный эффект, зависящий от природы лиганда. Соединения, которые взаимодействуют с сайтами связывания основного медиатора или коагониста и блокируют работу NMDA-рецептора, являются конкурентными антагонистами. В то же время существует ряд соединений, которые наряду с глутаматом и глицином являются агонистами данного рецептора. К неконкурентным модуляторам относится широкий спектр лигандов, способных аллостерически взаимодействовать с NMDA-рецептором. Важной особенностью данного типа глутаматных рецепторов является различная чувствительность к неконкурентным антагонистам, которая зависит от субъединичного состава рецептора. Кроме того, существует ряд соединений, блокирующих пору канала NMDA-рецептора [16, 20].
Конкурентные антагонисты
Конкурентными антагонистами NMDA-рецепторов являются производные фосфоновой кислоты, 2-амино-5-фосфо-валериановая кислота (AP5) и 2-амино-7-фосфо-гептановая кислота (AP7). Вскоре после того, как AP7 и AP5 были идентифицированы в качестве сильных селективных антагонистов, был продемонстрирован противосудорожный эффект этих соединений при внутрибрюшинном и внутрижелудочковом введении мышам, тонико-клонические судороги у которых вызывались введением пороговых доз пентилентетразола (PTZ) [62]. Конкурентные антагонисты NMDA-рецепторов показали высокую эффективность в рамках моделей рефлекторной эпилепсии. Например, одна пероральная доза D(-)E-4-(3-фосфонопроп-2-енил)-пиперазин-2-карбоновой кислоты (D-CPPene) предотвращает возникновение фотоиндуцированной эпилептической реакции в течение 24–48 ч у светочувствительного бабуина Papio papio без каких-либо заметных моторных или седативных побочных эффектов [63].
Неконкурентные модуляторы
Аллостерические модуляторы. Поскольку биофизические свойства NMDA-рецепторов сильно зависят от входящих в их состав GluN2-субъединиц, поиск фармакологических агентов, способных контролировать отдельные подтипы этих рецепторов, был сосредоточен на молекулах, избирательно действующих на отдельные GluN2-субъединицы [64, 65].
Кристаллографические исследования продемонстрировали, что GluN2B-селективные негативные аллостерические модуляторы, такие как ифенпродил и Ro 25–6981, связываются с модуляторным сайтом, расположенным между ATD-доменами GluN1- и GluN2B-субъединиц [66–68]. Только один аминокислотный остаток в этом модулирующем сайте отличается у GluN2A- и GluN2B-субъединиц, однако, преобразования этого или других остатков GluN2A в остатки GluN2B недостаточны для изменения чувствительности рецептора к ифенпродилу [66, 69]. Это объясняется особенностями взаимного расположения ATD-доменов, в частности, “карман”, формируемый GluN1/2B-субъединицами, имеет идеальный размер для аналогов ифенпродила, тогда как субъединицы смещены на ~10° в случае GluN1/2A [70].
Каналоблокаторы. Пора ионного канала NMDA-рецептора может быть потенциал-зависимо заблокирована широким спектром органических катионов с разнообразной химической структурой [71]. Подобные соединения, как правило, блокируют открытые каналы активированных рецепторов и являются положительно заряженными соединениями при физиологических значениях pH, такой механизм блока канала рецептора является проявлением неконкурентного антагонизма. В целом, блокаторы открытых каналов можно разделить на три категории в зависимости от способа их взаимодействия с каналом: (1) поэтапные блокаторы (например, производные аминоакридина и тетрапентиламмоний) могут связываться только с открытым каналом, после чего предотвращают его закрытие [72–74]; (2) частичные блокаторы ловушечного типа (например, амантадин и мемантин), входя в канал, препятствуют закрытию каналов, но не могут полностью предотвратить его [75–77]; и (3) полные блокаторы ловушечного типа (например, Mg2+, кетамин, фенциклидин и MK-801), оставаясь внутри поры канала после его закрытия, не препятствуют диссоциации молекул агонистов от сайтов связывания на рецепторе [78, 79]. Было также показано, что некоторые каналоблокаторы способствуют закрытию канала, предположительно воздействуя на воротный механизм [77]. Считается, что часть каналоблокаторов обладает бифункциональностью – например, производные нитромемантина, которые связываются с порой канала и тем самым облегчают взаимодействие нитрогруппы с окислительно-восстановительным регуляторным сайтом рецептора [80].
NMDA-РЕЦЕПТОРЫ И ФЕНОМЕН ЭКСАЙТОТОКСИЧНОСТИ
При эпилептогенезе одним из патогенетических механизмов, приводящих к гибели нейронов, является эксайтотоксичность. Чрезмерная концентрация внеклеточного глутамата может активировать избыточное число постсинаптических NMDA-рецепторов, как синаптических, так и экстрасинаптических, что ведет к поступлению в нейроны большого числа ионов и вызывает их гибель. Кроме того, причиной избыточного накопления глутамата в межклеточном пространстве, помимо высвобождения из пресинаптических терминалей, может быть увеличенный выход глутамата из астроцитов [81, 82]. Определено шесть возможных путей этого процесса: (1) нарушение поглощения глутамата, (2) открытие анионных каналов на астроцитах под воздействием гипотонической среды, (3) Са2+-зависимый экзоцитоз, (4) выход глутамата через цистин-глутаматный антитранспортер, (5) высвобождение глутамата, опосредованное ионотропными пуринергическими рецепторами и (6) через функциональные непарные коннексоны на поверхности клетки [83]. Тем не менее, остается неясным, какой вклад вносит каждый из этих путей. Экспериментальные данные подтверждают роль астроцитов в повышении внеклеточных концентраций глутамата – например, при чрезмерной деполяризации нейронов в ходе эпилептической активности, приводящей к повышению внеклеточной концентрации K+, наблюдалось нарушение поглощения глутамата астроцитами [84]. В норме одна молекула глутамата транспортируется в астроциты и пресинаптические окончания вместе с тремя катионами Na+, при этом один катион K+ удаляется [85]. Повышение внеклеточной концентрации K+ приводит к обратному течению этого процесса – K+ поглощается астроцитами и пресинаптическими окончаниями, а глутамат и Na+ начинают высвобождаться [85].
В 2002 г. Hardingham с соавт. показали, что эксайтотоксическая гибель нейронов связана с активностью экстрасинаптических NMDA-рецепторов, в то время как синаптические NMDA-рецепторы выполняют нейропротекторную функцию [86]. На наличие связи между экстрасинаптическими рецепторами и эксайтотоксичностью указывает и тот факт, что в случае ишемии экстрасинаптические NMDA-рецепторы взаимодействуют с апоптотической киназой DAPK1 [87]. В то же время, некоторые исследования противоречат гипотезе о четком разделении роли синаптических и экстрасинаптических рецепторов в эксайтотоксичности. Так, эксайтотоксическая гибель нейронов была опосредована именно синаптическими NMDA-рецепторами в культуре нейронов гиппокампа [88]. Кроме того, подавление экспрессии скэффолд-белка постсинаптической плотности PSD-95, ассоциированного с NMDA-рецепторами, избирательно ослабляет эксайтотоксичность в культуре нейронов коры [89].
ИЗМЕНЕНИЯ ЭКСПРЕССИИ СУБЪЕДИНИЦ NMDA-РЕЦЕПТОРОВ ВО ВРЕМЯ ЭПИЛЕПТОГЕНЕЗА
При эпилепсии часто выявляется нарушение свойств NMDA-рецепторов, обусловленное изменением их субъединичного состава, числа NMDA-рецепторов или их локализации [90]. Эти изменения со стороны NMDA-рецепторов могут возникать в результате гиперактивности в ЦНС, наблюдаемой при судорожных состояниях, и также могут поддерживать патологическую гиперактивность. Роль NMDA-рецепторов в эпилептогенезе в настоящее время довольно активно исследуется. Ряд экспериментальных работ посвящен изучению изменений экспрессии генов субъединиц NMDA-рецепторов после судорожных состояний (табл. 1) [91–94]. Анализ изменений субъединичного состава NMDA-рецепторов на ранних сроках после первого судорожного приступа позволяет выявить потенциальный вклад данных рецепторов в эпилептогенез.
Таблица 1.
Изменения экспрессии NMDA-рецепторов в различных моделях судорожных состояний и эпилепсии Table 1. Changes in the expression of NMDA receptors in various seizure models and models of epilepsy
| Модель Model |
Срок анализа Measurement time |
Область мозга Brain region |
Изменения
экспрессии Changes in the expression |
Ссылки Links |
|---|---|---|---|---|
| Пилокарпиновая (in vitro)Pilocarpine (in vitro) |
24 ч 24 h |
Культура гиппокампа | GluN1↑ GluN2A↑ |
[95], [96] |
| Hippocampal culture | GluN2B↑ | |||
| Пилокарпиновая (in vivo) Pilocarpine (in vivo) |
24 ч 24 h |
Гиппокамп Hippocampus |
GluN2A↑ GluN2B↑ |
[97] [98] |
| 4–10 недель 4–10 weeks |
Гиппокамп Hippocampus |
GluN2B↑ | ||
| Пентилентетразоловая Pentylenetetrazole |
24 ч 24 h |
Гиппокамп Hippocampus |
GluN2B↑ GluN1↓GluN1↓ | [99]
[100] [101] |
| Киндлинг Конвульсанты: Kindling Convulsants: |
||||
| Пентилентетразол Pentylenetetrazole |
24 ч 24 h |
Гиппокамп Hippocampus |
GluN2B↑GluN1↓ | [102][103] |
| 3-меркаптопропионат 3-mercaptopropionate | 24 ч 24 h |
Гиппокамп Hippocampus |
GluN2A↑ |
[104] |
| Мозжечок Cerebellum |
GluN2B↓ | [105] | ||
| Гиппокамп Hippocampus |
GluN2B↓ | [106] | ||
| Пентилентетразол Pentylenetetrazole |
3 дня 3 days |
Кора, гиппокамп Cortex, hippocampus |
GluN1↑ GluN2A↑ |
[107] |
| Литий-пилокарпиновая Lithium-Pilocarpine |
7 дней 7 days 21 день 21 days |
кора cortex гиппокамп hippocampus |
GluN1↓ GluN2A↓ GluN1↑ GluN2B↑ |
[94] [108] |
| Каинатная Kainate |
24 и 72 ч 24 and 72 hours |
гиппокамп hippocampus |
GluN1↓ | [109] |
| Гипоксическая Hypoxic |
2–4 дня 2–4 days |
гиппокамп hippocampus |
GluN2A↑ GluN3A↑ |
[110] |
Ранние изменения после судорожных состояний
При различных моделях судорожных состояний были выявлены быстрые изменения экспрессии субъединиц NMDA-рецепторов после эпилептической активности. Так, кратковременная гиперэкспрессия GluN1- и GluN2B-субъединиц наблюдается in vitro и in vivo при пилокарпиновой модели [95, 97]. Di Maio с соавт. предположили, что такая гиперэкспрессия является результатом активации двух независимых сигнальных путей: кальций-зависимого НАДФ-оксидазного пути и пути, запускаемого фосфорилированием киназ ERK1/2 [95, 96]. Работая с культурой нейронов гиппокампа, они показали, что кратковременное воздействие пилокарпина вызывает быструю гиперэкспрессию GluN2B-субъединицы, наблюдаемую через 24 ч после воздействия [95], и впоследствии приводит к усилению экспрессии GluN2A-субъединицы [96]. Аналогичные данные были получены в острую фазу пилокарпиновой модели: было выявлено повышение экспрессии как GluN2A-, так и GluN2B-субъединиц [97]. Ранняя активация продукции GluN2B-субъединицы была выявлена в гиппокампе после судорог, вызванных пентилентетразолом [99], и в модели пентилентетразол-индуцированного киндлинга [102]. В то же время в модели киндлинга с использованием 3-меркаптопропионата различными исследователями выявлено снижение продукции GluN2B-субъединицы в гиппокампе и мозжечке [105, 106]. Данное противоречие, возможно, связано с тем, что был использован другой конвульсант. Также в гиппокампе взрослых крыс на следующий день после последнего введения 3-меркаптопропионовой кислоты, которую вводили 1 раз в течение 4 дней в дозе 45 мг/кг, было показано повышение экспрессии гена, кодирующего GluN2A-субъединицу [104]. При использовании модели острого эпилептического статуса через 1 ч после индукции также обнаруживается увеличение экспрессии гена GluN2A-субъединицы в гиппокампальной области мышей [91]. Противоречия в полученных результатах могут быть обусловлены тем, что были использованы различные экспериментальные модели, модельные животные и подходы к анализу экспрессии.
Латентный период эпилептогенеза
Большой интерес вызывают функциональные изменения в работе NMDA-рецепторов во время эпилептогенеза, так как эти изменения могут быть одним из патофизиологических механизмов эпилепсии. Для изучения таких изменений часто используют эпилептические модели на животных. В этих моделях после первичного воздействия, вызвавшего судороги, наблюдается латентный период, после которого формируются спонтанные рецидивирующие судорожные приступы. В частности, такими моделями являются литий-пилокарпиновая [111] и каинатная [112]. Считается, что латентный период сопровождается патофизиологическими изменениями, которые приводят к эпилептизации мозга и развитию спонтанных судорог [113, 114]. В связи с этим особое внимание уделяется изучению изменений, происходящих с NMDA-рецепторами, именно в этот период.
Снижение экспрессии облигатной GluN1-субъединицы обнаруживается в различных областях коры в латентную фазу литий-пилокарпиновой модели [93, 115]. Данное изменение может свидетельствовать об уменьшении общего числа NMDA-рецепторов. Однако в более ранние сроки в гиппокампе, напротив, наблюдается повышение продукции мРНК данной субъединицы [95], это может свидетельствовать о том, что наблюдаемые изменения экспрессии GluN1-субъединицы являются регион-специфичными. Кроме того, показано, что во время латентного периода литий-пилокарпиновой модели эпилепсии наблюдается повышенная иммунореактивность к pGluN1-Ser896-фосфорилированному в положении Ser896 участку С-концевого хвоста GluN1-субъединицы NMDA-рецептора. Фосфорилирование этого сайта регулирует экспрессию NMDA-рецепторов и их кластеризацию [116–119], а также предотвращает кальмодулин-опосредованную инактивацию NMDA-рецепторов [120, 121]. Наибольшая pGluN1-Ser896-иммунореактивность наблюдалась в пирамидных нейронах области CA1 гиппокампа и гранулярных клетках зубчатой извилины на третьи сутки после эпилептического статуса. Спустя неделю после эпилептического статуса, pGluN1-Ser896-иммунореактивность в нейронах областей CA2–СА3 гиппокампа и гранулярных клетках зубчатой извилины снижалась, хотя и оставалась выше, чем у контрольных животных. В пирамидных клетках области CA1 pGluN1-Ser896-иммунореактивность спустя неделю оказалась выше, чем на третьи сутки после эпилептического статуса [122].
В латентный период происходят изменения экспрессии GluN2A- и GluN2B-субъединиц аналогичные изменениям, обнаруженным на ранних сроках анализа. Гипоксические судороги у молодых крыс значительно повышали экспрессию субъединиц GluN2A и GluN3A в гиппокампе через 2–4 дня после судорог [110]. Пилокарпин-индуцированные судороги вызывают значительное увеличение плотности NMDA-рецепторов в гранулярных клетках зубчатой извилины и пирамидных нейронах поля CA3 гиппокампа. Фармакологические эксперименты показали, что GluN2B-субъединица была ответственна за увеличение возбуждающей активности у этих животных [123]. В модели эпилептогенеза, в которой крысы подвергаются периоду гипертермии и претерпевают дисгенез (холодовое повреждение коры), в латентный период (16–20-е постнатальные дни) наблюдалось усиление NMDA-рецептор-опосредованных вызванных ВПСТ в области СА1 гиппокампа. В двух экспериментальных группах – у крыс, перенесших и дисгенез, и гипертермию, а также у крыс, перенесших только дисгенез, в гиппокампе наблюдалось увеличение экспрессии GluN2B-субъединицы [124].
Несмотря на то, что в различных исследованиях обнаруживается повышение доли GluN2B-содержащих NMDA-рецепторов после эпилептического статуса, для эпилептизации мозга необходима именно GluN2A-субъединица. Селективная блокада GluN2A-содержащих NMDA-рецепторов нарушала эпилептогенез и разрастание мшистых волокон в пилокарпиновой- и киндлинг-модели лимбической эпилепсии [125]. Снижение экспрессии GluN2A-субъединицы, выявленное в различных областях коры и гиппокампе через неделю после пилокарпин-индуцированных судорог [115], вероятно, является компенсаторным механизмом, направленным на предотвращение эпилептогенеза.
Кроме того, Eyo с соавт., используя двухфотонную визуализацию с высоким разрешением, исследовали NMDA-рецептор-опосредованную регуляцию физических взаимодействий микроглии и нейронов, которые формируются при повышенной глутаматергической нейротрансмиссии. Было обнаружено, что селективный ингибитор GluN2A NVPAAM007, но не ингибитор GluN2B ифенпродил, блокировал возникновение этих взаимодействий. Согласно этим данным, именно GluN2A-субъединица ответственна за обеспечение физических взаимодействий микроглии и нейронов, причем дифференциальная экспрессия данной субъединицы в различных областях мозга обуславливает возможность формирования этих взаимодействий только в определенных структурах мозга. Авторами предполагается, что такие взаимодействия могут иметь важное значение при различных заболеваниях, в частности, при эпилепсии [126].
Таким образом, GluN2A- и GluN2B-содержащие NMDA-рецепторы вносят вклад в патофизиологические изменения, наблюдаемые при эпилептизации мозга. Повышение доли NMDA-рецепторов, включающих GluN2B-субъединицу, может приводить к большему притоку ионов Ca2+ в нейроны по сравнению с рецепторами, содержащими GluN2A. За счет этого данный подтип NMDA-рецепторов, вероятно, играет важную роль в запуске гипервозбудимости нейронов [96] и эксайтотоксичности [127]. В то же время для эпилептогенеза необходима GluN2A-субъедница [125]. Использование различных экспериментальных моделей эпилепсии при анализе роли NMDA-рецепторов в развитии эпилепсии и получение противоречивых результатов различными исследователями затрудняет понимание механизма эпилептогенеза. Однако не вызывает сомнений, что модуляция функционирования NMDA-рецепторов на этапе эпилептизации мозга является перспективным направлением в лечении эпилепсии.
ПРИМЕНЕНИЕ БЛОКАТОРОВ NMDA-РЕЦЕПТОРОВ ДЛЯ КОНТРОЛЯ ЭПИЛЕПТИЧЕСКИХ ПРИСТУПОВ И ПРЕДОТВРАЩЕНИЯ ЭПИЛЕПТОГЕНЕЗА
Блокаторы NMDA-рецепторов рассматривались в качестве перспективного направления терапии эпилептических приступов [128, 129], однако, из-за сильных побочных эффектов [130–132] их применение в клинике, как правило, ограничено экстренными случаями [133], например, когда пациент демонстрирует фармакорезистентность к препаратам первой линии терапии. Тем не менее, продолжается поиск фармакологических агентов, обладающих меньшими побочными эффектами, равно как и изучаются конкретные механизмы и процессы, запускаемые ранним применением блокаторов NMDA-рецепторов для ослабления или предотвращения эпилептогенеза.
Противосудорожное действие антагонистов NMDA-рецепторов
В настоящее время накоплена большая база экспериментальных исследований, в которых показана высокая противосудорожная эффективность антагонистов NMDA-рецепторов на животных (табл. 2).
Таблица 2.
Экспериментальные исследования противосудорожного действия антагонистов NMDA-рецепторов Table 2. Experimental studies of the anticonvulsant effects of NMDA receptor antagonists
| Модель Model |
Фармакологический агент Pharmacological agent |
Временной интервал Time window |
Эффект Effect |
Ссылки Links |
|---|---|---|---|---|
| Пентилентет разоловая Pentylenetetra zole |
Кетамин, MK-801 Ketamine, MK-801 Мемантин, МК-801 Memantine, MK-801 |
Вводились заранее Administered in advance Вводились заранее Administered in advance |
Повышение порога генерации судорог Increase of seizure threshold Предотвращение клонических и тонических судорог Prevention of clonic and tonic seizures |
[134] [135] |
| Каинатная Kainate |
Кетамин, диазепам Ketamine, diazepam |
Спустя 90 мин после индукции 90 min after induction |
Диазепам отдельно: ослабляет эпилептическую активность, регистируемую на ЭЭГ Кетамин с диазепамом: полностью подавляют эпилептическую активность Diazepam alone: attenuates the EEG registered epileptic activity Ketamine with diazepam: completely suppress epileptic activity |
[136] |
| Зоман-индуцированные судороги Zoman-induced seisures |
Кетамин, атропин Ketamine, atropine |
Спустя 30 мин после индукции. Эффективность падает, если лечение откладывалось более чем на 1 ч 30 min after induction. The efficiency drops if the treatment was postponed for more than 1 h |
Останавливает эпилептическую активность, снижает степень повреждений мозга, обеспечивает
выживание Stops epileptic activity, reduces brain damage, ensures survival |
[137] |
| Киндлинг (электрическая
стимуляция амигдалы) Kindling (electrical stimulation of amygdala) |
L-701,324, фенобарбитал L-701,324, phenobarbital |
Субхроническое введение Subchronic administration |
Повышение порога генерации судорог Increase of seizure threshold |
[138] |
| Пилокарпиновая Pilocarpine |
CI-1041, кетамин CI-1041, ketamine |
Через 15 мин после введения пилокарпина 15 min after administration of pilocarpine |
CI-1041 и кетамин оказывали нейропротекторное воздействие, кетамин также демонстроровал
противосудорожный эффект CI-1041 and ketamine had neuroprotective effects, besides, ketamine demonstrated anticonvulsant effect |
[139] |
| Пикротоксин и понижение
внеклеточной концентрации
Mg2+ (in vitro) Picrotoxin and lowered Mg2+ extracellular concentration (in vitro) |
AP5 | Добавлялся предварительно Administered in advance |
Отсроченное наступление эпилептиформной активности, снижение частоты иктальной активности Delayed onset of epileptiform activity, decreased frequency of ictal activity |
[140] |
| Пилокарпиновая (in vivo) Pilocarpine (in vivo) |
AP5 | Спустя 5 дней в течение 2 недель 5 days later within 2 weeks |
Отсроченное наступление первых спонтанных судорог, снижение их частоты, уменьшение
числа судорог, соответствующих стадиям 3–4 Delayed onset of the first spontaneous seizures, decrease in their frequency, decrease in the number of stage 3–4 seizures |
[140] |
| 4-аминопиридин и изменение внеклеточных ионных концентраций (in vitro) 4-aminopyridine and changes in ionic extracellular concentrations (in vitro) |
AP5 | – | Подавление иктальных разрядов Suppression of ictal discharges |
[141] |
Ионы магния являются естественными каналоблокаторами NMDA-рецепторов. Клинические наблюдения и экспериментальные исследования на животных показали, что истощение запасов Mg2+ ведет к гипервозбудимости нервной системы, что, в конечном итоге, приводит к появлению эпилептической активности или повышенной восприимчивости к эпилептогенным стимулам [142–144]. Изучение метаболизма Mg2+ при эпилепсии позволило установить, что концентрация Mg2+ в сыворотке и спинномозговой жидкости пациентов с тонико-клоническими судорогами были значительно ниже, чем в контрольной группе [145, 146]. Концентрация Mg2+ в сыворотке и спинномозговой жидкости зависела от продолжительности и частоты эпилептической активности. Искусственное повышение концентрации Mg2+ долгое время рассматривалось как эффективное направление терапии. Системное введение Mg2+ оказывало противосудорожное действие в моделях эпилептического статуса [147] в случае аудиогенной и пентилентетразол-индуцированной [148–150], а также NMDA-индуцированной активности [151] у грызунов. Недавно было оценено влияние MgSO4 на судороги, вызванные асфиксией у недоношенных плодов овцы. MgSO4 вводился внутривенно в течение 48 ч на сроке гестации 104 дня (полный срок – 147 дней). Применение MgSO4 не предотвращало судороги, но значительно снижало общее число судорог, их тяжесть и пиковую амплитуду эпилептической активности [152]. Дальнейшие исследования необходимы, чтобы установить оптимальную терапевтическую дозу магния для предотвращения судорог, а также чтобы оценить целесообразность его использования в качестве инструмента для подавления эпилептической активности.
Каналоблокаторы являются перспективным инструментом для подавления судорог, поскольку они снижают чрезмерную активность NMDA-рецепторов, не нарушая их нормальной активности. Подобный эффект объясняется тем, что блокаторы этого типа предпочтительно воздействуют на чрезмерно открытые ионные каналы, а значит будут эффективнее блокировать патологически активированные рецепторы в отличие от нормально работающих рецепторов. Более того, каналоблокаторы почти не нарушают нормальную синаптической передачу благодаря тому, что скорость их выхода из канала относительно высока [153].
ADCI – селективный низкоафинный каналоблокатор NMDA-рецепторов, совмещающий в себе свойства карбамазепина и MK-801 [154]. Sun и Lin [155] сообщили, что ADCI может также блокировать потенциал-управляемые натриевые каналы в нейронах верхнего шейного ганглия и гиппокампа крыс, а также в культивируемых нейронах NT2 человека. В отличие от других каналоблокаторов, ADCI менее токсичен, это может быть связано с тем, что он сильнее блокирует GluN1A/GluN2B-субъединицы (IC50 = 6.6 мкМ), чем GluN1A/GluN2A-субъединицы (IC50 = 14.3 мкМ) [154]. ADCI не вызывает гиперлокомоции или стереотипных движений у крыс; при дозах, в 10 раз превышающих ED50 в тесте максимального электрошока, ADCI не вызывал нарушений поведения в тесте пассивного избегания и в водном лабиринте Морриса, что указывает на отсутствие нарушений памяти. Препарат также не вызывает вакуолизацию нейронов, наблюдаемую при использовании многих других антагонистов NMDA-рецепторов. ADCI демонстрировал противосудорожный эффект в тесте максимального электрошока [156], а также в отношении эпилептической активности, вызванной 4-аминопиридином, NMDA, пентилентетразолом и кокаином [156–158]; клонических судорог, вызванных дендротоксинами [159]; эпилептической активности, наблюдаемой после отмены этанола у мышей [160].
Низкоаффинный блокатор NMDA-рецепторов кетамин показал свою эффективность в качестве противосудорожного препарата в различных моделях эпилепсии – кетамин влияет на эпилептоподобную активность, вызванную NMDA, гуанидиносукцинатом, п-толуидино-3-пропиламино-2-пропанолом, меркаптопропионатом, N-(3,5-диметокси-4-пропоксифенилэтил)-азиридином, лидокаином, пикротоксином, бикукуллином, стрихнином и пентилентетразолом у грызунов [11, 134, 161–165]. В киндлинговой модели эпилепсии кетамин дозозависимо замедлял наступление 3-й и 4-й стадии по шкале Racine y 15-дневных крыс. Кетамин в дозе 20 мг/кг полностью устранял судорожную активность и почти полностью устранял следовые разряды у ювенильных крыс, подвергшихся киндлингу, что намного эффективнее действия более высоких доз кетамина на взрослых крысах. Эти данные подтверждают гипотезу о том, что NMDA-рецепторы играют важную роль в повышенной склонности незрелой нервной системы к эпилептической активности [166]. Кетамин способен действовать синергетически с такими антиэпилептическими препаратами, как вальпроат и карбамазепин, ослабляя MES-индуцированные судороги у мышей [167]. Другие исследования продемонстрировали синергетическое действие диазепама и кетамина на эпилептическую активность, вызванную литий-пилокарпином [168] и каинатом [136]. Субанестезирующие дозы кетамина (10 мг/кг) остановили судороги и предотвратили гибель животных, отравленных зоманом; более того, кетамин ослабил повреждающее действие зоман-индуцированной эпилептической активности на ткани мозга и способствовал выживанию [137].
Мемантин и МК-801 успешно предотвращали клонические и тонические судороги, вызванные пентилентетразолом у мышей [135]. В экспериментах на крысах линии Крушинского–Молодкиной оценивался эффект мемантина и ИЭМ-1921, селективных блокаторов NMDA-рецепторов, на аудиогенные судороги. Блокаторы NMDA-рецепторов вводились до подачи звуковых раздражителей. Спустя 30 мин после введения ИЭМ-1921 (5 мг/кг) у 33% крыс обнаружилось полное отсутствие судорог в ответ на звук, а у 59% животных наблюдалось только двигательное возбуждение, не сопровождавшееся генерализированными клонико-тоническими судорогами. Мемантин (5 мг/кг) не полностью подавлял судороги, но у 50% крыс через 1 ч и у 70% крыс через 2 ч после введения наблюдалось ослабление аудиогенных судорог до уровня двигательного возбуждения [169].
Конкурентный антагонист NMDA-рецепторов AP5 (50 мкM) снижал количество эпилептиформных событий как в зубчатой извилине, так и в области CA1 гиппокампа в переживающих срезах, помещенных в безмагниевый раствор с повышенной концентрацией калия [10]. Эксперименты на изолированном стриатуме, полученном от крысят линии Sprague–Dawley на 7–15-й дни жизни, показали, что стриатум также способен генерировать эпилептиформную активность in vitro без участия коры головного мозга и таламуса. AP5 полностью блокировал иктальные разряды и соответствующие им деполяризационные сдвиги, наблюдаемые при регистрации активности срединных шипиковых нейронов стриатума, в то время как агонист AMPA-рецепторов CNQX не влиял ни на амплитуду, ни на длительность эпилептиформной активности, что указывало на NMDA-рецептор-зависимую природу наблюдаемой активности [141]. Антагонист TCN 201 (3 мкМ), специфически блокирующий GluN2A, и аллостерический антагонист Ro 25-6981 (3 мкМ), блокирующий GluN2B, значительно снижали количество эпилептиформных событий при их использовании в концентрациях гораздо более низких, чем концентрация AP5, что дает надежду на клиническое применение антагонистов, специфичных в отношении субъединичного состава рецептора [10]. Антагонист ифенпродил, блокирующий GluN2B, продемонстрировал широкое терапевтическое временное окно (3 дня), снижая восприимчивость к судорогам в тесте максимального электрошока у крыс, перенесших длительные фебрильные судороги [170].
Для подавления эпилептиформной активности также могут быть использованы антагонисты глицин-связывающего сайта NMDA-рецептора, демонстрирующие более благоприятный профиль переносимости по сравнению с антагонистами сайта связывания глутамата [171]. Хотя субхроническое введение антагониста глицин-связывающего сайта L-701,324 не приводило к противосудорожным или модифицирующим эффектам в отношении судорог у киндлированных мышей, L-701,324 улучшил реакцию на фенобарбитал, что выражалось в повышении порога генерации судорог [138].
Основная проблема применения NMDA-антагонистов заключается в том, что именно высокие дозировки, требуемые для эффективного подавления эпилептической активности, приводят к тяжелым побочным эффектам [172, 173]. Применение антагонистов NMDA-рецепторов снижает NMDA-рецептор-опосредованную синаптическую активность, что, даже в случае использования относительно низких доз антагонистов, может приводить к нарушениям памяти и обучаемости. Более тяжелая гипофункция NMDA-рецепторов может быть причиной психоза [130], характеризующегося галлюцинациями, параноидальным бредом, замешательством, снижением концентрации внимания, изменениями настроения, возбуждением, кошмарами, кататонией, атаксией и потерей чувствительности [131, 132]. Применение антагонистов NMDA-рецепторов может запускать нейродегенеративные процессы в цингулярной и ретросплениальной коре головного мозга [174].
Наконец, немаловажной клинической значимостью обладают данные о преимуществе комбинированной терапии (включающей как блокаторы ГАМКа, так и NMDA-рецепторов) над монотерапией в случае бензодеазепам-резистентной эпилептической активности [175]. Применение комбинации блокаторов ГАМКа и NMDA-рецепторов позволило прекратить эпилептическую активность, устойчивую к монотерапии бензодиазепинами или кетамином. Кроме того, одновременное введение мидазолама и кетамина оказалось эффективнее их последовательного применения в той же дозе, это согласуется с представлениями о том, что фармакорезистентность усиливается с течением времени. Комбинированная терапия блокаторами ГАМКа и NMDA-рецепторов была эффективной спустя 40 мин после начала эпилептической активности как в случае зоман-индуцированной активности, так и в литий-пилокарпиновой модели эпилепсии. Также комбинированная терапия повлияла на долгосрочные последствия эпилептической активности, снизив гибель нейронов и ослабив нарушения пространственной памяти [175].
АНТИЭПИЛЕПТОГЕННОЕ ДЕЙСТВИЕ АНТАГОНИСТОВ NMDA-РЕЦЕПТОРОВ
Эффект D-AP5 на процессы эпилептогенеза был показан in vitro и in vivo. В переживающих срезах предварительное добавление 1 мкМ D-AP5 отсрочило начало эпилептиформной активности и снизило частоту иктальной активности, вызванных пикротоксином и пониженной концентрацией Mg2+. В экспериментах in vivo D-AP5 вводился интрацеребровентрикулярно при помощи осмотического мининасоса спустя 5 дней после индукции эпилептического статуса на протяжении 2 нед., регистрация ЭЭГ продолжалась в течение 2 мес. D-AP5 значительно отсрочил наступление первых спонтанных судорог, вызванных пилокарпином, снизил их частоту, уменьшил число судорог, соответствующих стадиям 3–4. В то же время длительность отдельно взятых периодов спонтанной эпилептической активности не изменилась [140].
Антагонисты NMDA-рецепторов не во всех экспериментах действовали однозначно. Антагонист GluN2B-содержащих NMDA-рецепторов CI-1041 (10 мг/кг), CP-101,606 – антагонист NMDA-рецепторов, обладающий высокой селективностью в отношении дигетеромеров GluN1/GluN2B (10 мг/кг), а также кетамин были использованы для воздействия на этап индукции эпилептического статуса и последующую нейродегенерацию в пилокарпиновой модели эпилепсии. Растворы антагонистов вводились внутрибрюшинно через 15 мин после введения пилокарпина. Кетамин снижал интенсивность устойчивого орофасциального автоматизма, жевания и клонуса передних конечностей, но не восстанавливал выпрямительный рефлекс. Однако применение CI-1041 или CP-101,606 не повлияло на поведенческий паттерн эпилептического статуса. Введение CP-101,606 также не повлияло на интенсивность нейродегенерации в области СА1 гиппокампа, в зубчатой извилине и амигдале на 7-е сутки после введения пилокарпина. В то же время CI-1041 и кетамин снижали нейродегенерацию, наблюдаемую в гиппокампе и миндалине, что указывает на нейропротекторный эффект CI-1041 при отсутствии эффекта на судорожную активность [139]. NBQX, антагонист AMPA-рецепторов, и антагонист NMDA-рецепторов ифенпродил были использованы для предотвращения эпилептогенеза у мышей [176]. Комбинация двух антагонистов вводилась в течение 5 сут, спустя 6 ч после внутригиппокампальной инъекции каината. Через две недели после введения каината у мышей, получающих терапию, наблюдалось меньшее число спонтанных судорог по сравнению с контрольной группой, однако, этот эффект был утрачен в последующие недели. Применение NBQX и ифенпродила временно предотвращало рассеивание гранулярных клеток и сопровождалось гибелью меньшего числа нейронов в зубчатой извилине. Также сообщалось, что введение MK-801 предотвращало повреждение мозговых структур вне зависимости от его противосудорожного действия. Однократное введение низкой дозы MK-801 после того, как эпилептический статус, индуцированный каинатом, уже был остановлен введением диазепама, позволило предотвратить большую часть повреждений головного мозга. Наиболее выраженный нейропротекторный эффект МК-801 наблюдался в передней части дорсального гиппокампа, а областями, на которые МК-801 не оказал нейропротекторного воздействия, были ворота зубчатой извилины и сетчатая часть черной субстанции. Несмотря на это, однократное введение MK-801 не препятствовало развитию спонтанных судорог [177]. Отчасти нейропротекторный эффект MK-801 может объясняться тем, что MK-801 способен блокировать перекисное окисление липидов, индуцированное хинолиновой кислотой в крысином стриатуме. Поскольку основной механизм нейротоксического действия хинолиновой кислоты связан с чрезмерной активацией NMDA-рецепторов, нейропротекторный эффект МК-801 в этом случае, скорее всего, опосредован NMDA-рецепторами [178]. Более длительное применение антагонистов требуется, чтобы уточнить взаимосвязь между дегенеративными изменениями и развитием спонтанных судорог.
Исследования эпилептогенеза на животных позволили выявить новые мишени для терапии, связанные с активностью NMDA-рецепторов. Нокдаун белка Nwd1 оказывает противосудорожное действие, аналогичное действию противоэпилептических препаратов. Подавление Nwd1 in vitro с помощью аденоассоциированных вирусов ведет к ослаблению возбуждающих постсинаптических токов, опосредованных GluN2B-содержащими NMDA-рецепторами, в переживающих срезах гиппокампа. Подавление Nwd1 снижает гипервозбудимость нейронов и уменьшает вероятность возникновения эпилептической активности в ходе каинат-индуцированного эпилептического статуса. Более того, повышенный уровень экспрессии Nwd1 в тканях мозга наблюдался не только у мышей с каинат-индуцированными судорогами, но и у пациентов с эпилепсией височной доли [179].
Регулировать работу NMDA-рецепторов способен также белок σ1R. Кальций-зависимое связывание σ1-рецептора с GluN1-субъединицей NMDA-рецепторов, несущей цитозольный сегмент C1, защищает активность NMDA-рецепторов, а именно вход кальция, от ингибирующего действия кальций-активированного кальмодулина. В то время как агонисты σ1-рецептора способствуют его связыванию с субъединицей GluN1, его антагонисты разрушают эти комплексы, способствуя ингибированию работы NMDA-рецепторов. Каннабидиол, являющийся антагонистом σ1-рецептора, снижал частоту эпилептической активности, вызванной интрацеребровентрикулярным введением NMDA [180]. Также введение каннабидиола приводило к тому, что меньшее количество мышей демонстрировало гипермобильность и клонические судороги. Тонические судороги наблюдались только у 20% мышей, и ни одна мышь не погибла. В присутствии PPCC, агониста σ1-рецептора, каннабидиол прекращал оказывать модулирующее действие на NMDA-индуцированную активность, что свидетельствует о существовании фунциональных связей между σ1-рецепторами и NMDA-рецепторами [180].
Недавно было обнаружено, что у пациентов с эпилепсией и у животных, у которых эпилептическая активность вызывалась экспериментально, повышена экспрессия рецептора GPR40, принадлежащего к семейству рецепторов, сопряженных с G-белком. С использованием двух моделей эпилепсии удалось установить, что активация GPR40 облегчает судороги, вызванные каинатом, а в отношении животных, получавших инъекции пентилентетразола в ходе киндлинга, активация GPR40 снижала подверженность животных к судорогам. Регистрация синаптической активности методом пэтч-кламп показала, что GPR40 влияет на NMDA-рецептор-опосредованную синаптическую передачу. Амплитуда NMDA-рецептор-опосредованных ВПСТ снижалась в присутствии GW9508, селективного агониста GPR40, и увеличивалась в присутствии GW1100 – селективного антагониста GPR40. Напротив, AMPA-рецептор-опосредованные ВПСТ не изменились в присутствии GW9508 и GW1100. Кроме того, GPR40 регулировал поверхностную экспрессию GluN2A и GluN2B [181]. Таким образом, GPR40 способен модулировать эпилептическую активность и является потенциальной мишенью для терапевтического вмешательства.
ЗАКЛЮЧЕНИЕ
Эпилепсия зачастую сопровождается дисрегуляцией NMDA-рецепторов, что делает эти рецепторы перспективной терапевтической мишенью. Несмотря на кажущуюся привлекательность применения антагонистов NMDA-рецепторов для подавления судорог, результаты проведенных исследований на животных оказались противоречивыми. Применение антагонистов NMDA-рецепторов сопровождалось рядом побочных эффектов, таких как: нейродегенеративные изменения в цингулярной и ретросплениальной коре [174], психотомиметические эффекты [182], ухудшение обучаемости [183]. Минимум побочных эффектов вызывало применение антагонистов глицинового сайта и конкурентных антагонистов, в то время как использование высокоаффинных антагонистов открытого состояния канала приводило к обширному спектру нарушений.
Снизить смертность и вероятность развития эпилепсии можно, обеспечив более эффективное лечение эпилептического статуса. Уточнение рекомендаций по терапии эпилептического статуса, основанное на накопленных данных о наблюдаемых в ходе эпилептического статуса изменениях синаптической передачи, сокращение временного промежутка между манифестацией заболевания и началом терапевтического вмешательства, а также раннее использование политерапии потенциально могут быть использованы для предотвращения эпилептогегеза и являются перспективными направлениями лечения эпилепсии.
Список литературы
Berg A.T., Berkovic S.F., Brodie M.J., Buchhalter J., Cross J.H., van Emde Boas W., Engel J., French J., Glauser T.A., Mathern G.W., Moshé S.L., Nordli D., Plouin P., Scheffer I.E. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 51(4): 676–685. 2010.
Sharm A.K., Reams R.Y., Jordan W.H., Miller M.A., Thacker H.L., Snyder P.W. Mesial temporal lobe epilepsy: pathogenesis, induced rodent models and lesions. Toxicol. Pathol. 35(7): 984–999. 2007.
Herman S.T. Epilepsy after brain insult: targeting epileptogenesis. Neurology. 59(9) Suppl 5: S21–S26. 2002.
Anderson G.D., Temkin N.R., Chandler W.L.,Winn H.R. Effect of valproate on hemostatic function in patients with traumatic brain injury. Epilepsy Res. 57(2–3): 111–119. 2003.
Chang B.S., Lowenstein D.H. Epilepsy. N. Engl. J. Med. 349(13): 1257–1266. 2003.
Altındağ E., Erdoğan F.F., Tezer İ., Özkara Ç. Management and Early Treatment of Status Epilepticus in Adults and Children. Turkish J. Neurol. 23(4): 155–161. 2017.
Betjemann J.P., Lowenstein D.H. Status epilepticus in adults. Lancet Neurol. 14(6): 615–624. 2015.
Bayer T.A., Wiestler O.D., Wolf H.K. Hippocampal loss of N-methyl-D-aspartate receptor subunit 1 mRNA in chronic temporal lobe epilepsy. Acta Neuropathol. 89(5): 446–450. 1995.
Mathern G.W., Leite J.P., Babb T.L., Pretorius J.K., Kuhlman P.A., Mendoza D., Fried I., Sakamoto A.C., Assirati J.A., Adelson P.D., Peacock W.J. Aberrant hippocampal mossy fiber sprouting correlates with greater NMDAR2 receptor staining. Neuroreport. 7(5): 1029–1035. 1996.
Punnakkal P., Dominic D. NMDA Receptor GluN2 Subtypes Control Epileptiform Events in the Hippocampus. Neuromolec. Med. 20(1): 90–96. 2018.
Kubová H., Mares P. Effects of MK-801 (dizocilpine) and ketamine on strychnine-induced convulsions in rats: comparison with benzodiazepines and standard anticonvulsants. Physiol. Res. 43(5): 313–320. 1994.
Laube B., Kuhse J., Betz H. Evidence for a tetrameric structure of recombinant NMDA receptors. J. Neurosci. 18(8): 2954–2961. 1998.
Mano I., Teichberg V.I. A tetrameric subunit stoichiometry for a glutamate receptor-channel complex. Neuroreport. 9(2): 327–331. 1998.
Sobolevsky A.I., Rosconi M.P., Gouaux E. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature. 462(7274): 745–756. 2009.
Ayalon G., Segev E., Elgavish S., Stern-Bach Y. Two regions in the N-terminal domain of ionotropic glutamate receptor 3 form the subunit oligomerization interfaces that control subtype-specific receptor assembly. J. Biol. Chem. 280(15): 15053–15060. 2005.
Hansen K.B., Yi F., Perszyk R.E., Furukawa H., Wollmuth L.P., Gibb A.J., Traynelis S.F. Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol. 150(8): 1081–1105. 2018.
Ulbrich M.H., Isacoff E.Y. Subunit counting in membrane-bound proteins. Nat. Methods. 4(4): 319–321. 2007.
Ulbrich M.H., Isacoff E.Y. Rules of engagement for NMDA receptor subunits. Proc. Natl. Acad. Sci. USA. 105(37): 14163–14168. 2008.
Forrest D., Yuzaki M., Soares H.D., Ng L., Luk D.C., Sheng M., Stewart C.L., Morgan J.I., Connor J.A., Curran T. Targeted disruption of NMDA receptor 1 gene abolishes NMDA response and results in neonatal death. Neuron. 13(2): 325–338. 1994.
Traynelis S.F., Wollmuth L.P., McBain C.J., Menniti F.S., Vance K.M., Ogden K.K., Hansen K.B., Yuan H., Myers S.J., Dingledine R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 62(3): 405–496. 2010.
Regan M.C., Romero-Hernandez A., Furukawa H. A structural biology perspective on NMDA receptor pharmacology and function. Current Opin. Struct. Biol. 33: 68–75. 2015.
Punnakkal P., Jendritza P., Köhr G. Influence of the intracellular GluN2 C-terminal domain on NMDA receptor function. Neuropharmacology. 62(5–6): 1985–1992. 2012.
Maki B.A., Aman T.K., Amico-Ruvio S.A., Kussius C.L., Popescu G.K. C-terminal domains of N-methyl-D-aspartic acid receptor modulate unitary channel conductance and gating. J. Biol. Chem. 287(43): 36071–36080. 2012.
Sheng M., Cummings J., Roldan L.A., Jan Y.N., Jan L.Y. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature. 368(6467): 144–147. 1994.
Dunah A.W., Luo J., Wang Y.H., Yasuda R.P., Wolfe B.B. Subunit composition of N-methyl-D-aspartate receptors in the central nervous system that contain the NR2D subunit. Mol. Pharmacol. 53(3): 429–437. 1998.
Dunah A.W., Standaert D.G. Subcellular segregation of distinct heteromeric NMDA glutamate receptors in the striatum. J. Neurochem. 85(4): 935–943. 2003.
Rauner C., Köhr G. Triheteromeric NR1/NR2A/NR2B receptors constitute the major N-methyl-D-aspartate receptor population in adult hippocampal synapses. J. Biol. Chem. 286(9): 7558–7566. 2011.
Tovar K.R., McGinley M.J., Westbrook G.L. Triheteromeric NMDA receptors at hippocampal synapses. J. Neurosci. 33(21): 9150–9160. 2013.
Johnson J.W., Ascher P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature. 325(6104): 529–531. 1987.
Furukawa H., Singh S.K., Mancusso R., Gouaux E. Subunit arrangement and function in NMDA receptors. Nature. 438(7065): 185–192. 2005.
Yao Y., Harrison C.B., Freddolino P.L., Schulten K., Mayer M.L. Molecular mechanism of ligand recognition by NR3 subtype glutamate receptors. EMBO J. 27(15): 2158–2170. 2008.
Pullan L.M., Olney J.W., Price M.T., Compton R.P., Hood W.F., Michel J., Monahan J.B. Excitatory amino acid receptor potency and subclass specificity of sulfur-containing amino acids. J. Neurochem. 49(4): 1301–1307. 1987.
McBain C.J., Kleckner N.W., Wyrick S., Dingledine R. Structural requirements for activation of the glycine coagonist site of N-methyl-D-aspartate receptors expressed in Xenopus oocytes. Mol. Pharmacol. 36(4): 556–565. 1989.
Panatier A., Theodosis D.T., Mothet J., Touquet B., Pollegioni L., Poulain D.A., Oliet S.H.R. Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell. 125(4): 775–784. 2006.
Wolosker H., Blackshaw S., Snyder S.H. Serine racemase: A glial enzyme synthesizing D-serine to regulate glutamate-N-methyl-D-aspartate neurotransmission. Proc. Natl. Acad. Sci. USA. 96(23): 13409–13414. 1999.
Balu D.T., Takagi S., Puhl M.D., Benneyworth M.A., Coyle J.T. D-serine and serine racemase are localized to neurons in the adult mouse and human forebrain. Cell. Mol. Neurobiol. 34(3): 419–435. 2014.
Miya K., Inoue R., Takata Y., Abe M., Natsume R., Sakimura K., Hongou K., Miyawaki T., Mori H. Serine racemase is predominantly localized in neurons in mouse brain. J. Compar. Neurol. 510(6): 641–654. 2008.
Benveniste H. Brain microdialysis. J. Neurochem. 52(6): 1667–1679. 1989.
Zhang X., Nadler J.V. Postsynaptic response to stimulation of the Schaffer collaterals with properties similar to those of synaptosomal aspartate release. Brain Res. 1295: 13–20. 2009.
Do K.Q., Herrling P.L., Streit P., Turski W.A., Cuenod M. In vitro release and electrophysiological effects in situ of homocysteic acid, an endogenous N-methyl-(D)-aspartic acid agonist, in the mammalian striatum. J. Neurosci. 6(8): 2226–2234. 1986.
Yuzaki M., Connor J.A. Characterization of L-homocysteate-induced currents in Purkinje cells from wild-type and NMDA receptor knockout mice. J. Neurophysiol. 82(5): 2820–2826. 1999.
Monyer H., Sprengel R., Schoepfer R., Herb A., Higuchi M., Lomeli H., Burnashev N., Sakmann B., Seeburg P.H. Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science. 256(5060): 1217–1221. 1992.
Monyer H., Burnashev N., Laurie D.J., Sakmann B., Seeburg P.H. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 12(3): 529–540. 1994.
Akazawa C., Shigemoto R., Bessho Y., Nakanishi S., Mizuno N. Differential expression of five N-methyl-D-aspartate receptor subunit mRNAs in the cerebellum of developing and adult rats. J. Compar. Neurol. 347(1): 150–160. 1994.
Zhong J., Carrozza D.P., Williams K., Pritchett D.B., Molinoff P.B. Expression of mRNAs encoding subunits of the NMDA receptor in developing rat brain. J. Neurochem. 64(2): 531–539. 1995.
Erreger K., Geballe M.T., Kristensen A., Chen P.E., Hansen K.B., Lee C.J., Yuan H., Le P., Lyuboslavsky P.N., Micale N., Jørgensen L., Clausen R.P., Wyllie D.J.A., Snyder J.P., Traynelis S.F. Subunit-specific agonist activity at NR2A-, NR2B-, NR2C-, and NR2D-containing N-methyl-D-aspartate glutamate receptors. Mol. Pharmacol. 72(4): 907–920. 2007.
Chen P.E., Geballe M.T., Katz E., Erreger K., Livesey M.R., O’Toole K.K., Le P., Lee C.J., Snyder J.P., Traynelis S.F., Wyllie D.J.A. Modulation of glycine potency in rat recombinant NMDA receptors containing chimeric NR2A/2D subunits expressed in Xenopus laevis oocytes. J. Physiol. 586(1): 227–245. 2008.
Hansen K.B., Bräuner-Osborne H., Egebjerg J. Pharmacological characterization of ligands at recombinant NMDA receptor subtypes by electrophysiological recordings and intracellular calcium measurements. Comb. Chem. High Throughput Screen. 11(4): 304–315. 2008.
Lester R.A.J., Clements J.D., Westbrook G.L., Jahr C.E. Channel kinetics determine the time course of NMDA receptor-mediated synaptic currents. Nature. 346(6284): 565–567. 1990.
Vicini S., Wang J.F., Li J.H., Zhu W.J., Wang Y.H., Luo J.H., Wolfe B.B., Grayson D.R. Functional and pharmacological differences between recombinant N-methyl-D-aspartate receptors. J. Neurophysiol. 79(2): 555–566. 1998.
Erreger K., Chen P.E., Wyllie D.J.A., Traynelis S.F. Glutamate receptor gating. Crit. Rev. Neurobiol. 16(3): 187–224. 2004.
Glasgow N.G., Siegler- Retchless B., Johnson J.W. Molecular bases of NMDA receptor subtype-dependent properties. J. Physiol. 593(1): 83–95. 2015.
Yuan H., Geballe M.T., Hansen K.B, Traynelis S.F. Structure and Function of the NMDA Receptor. In: Structural And Functional Organization of the Synapse. Eds. J.W. Hell, M.D. Ehlers. Boston. MA. Springer. 289–316. 2008.
Burnashev N., Zhou Z., Neher E., Sakmann B. Fractional calcium currents through recombinant GluR channels of the NMDA, AMPA and kainate receptor subtypes. J. Physiol. 485(2): 403–418. Jun. 1995.
Jatzke C., Watanabe J., Wollmuth L.P. Voltage and concentration dependence of Ca(2+) permeability in recombinant glutamate receptor subtypes. J. Physiol. 538(1): 25–39. 2002.
Siegler Retchless B., Gao W., Johnson J.W. A single GluN2 subunit residue controls NMDA receptor channel properties via intersubunit interaction. Nat. Neurosci. 15(3): 406–413. S1-2. 2012.
Kuner T., Schoepfer R. Multiple structural elements determine subunit specificity of Mg2+ block in NMDA receptor channels. J. Neurosci. 16(11): 3549–3558. 1996.
Nevian T., Sakmann B. Single spine Ca2+ signals evoked by coincident EPSPs and backpropagating action potentials in spiny stellate cells of layer 4 in the juvenile rat somatosensory barrel cortex. J. Neurosci. 24(7): 1689–1699. 2004.
Nevian T., Sakmann B. Spine Ca2+ signaling in spike-timing-dependent plasticity. J. Neurosci. 26(43): 11001–11013. 2006.
Carter B.C., Jahr C.E. Postsynaptic, not presynaptic NMDA receptors are required for spike-timing-dependent LTD induction. Nat. Neurosci. 19(9): 1218–1224. 2016.
Tomek S.E., Lacrosse A.L., Nemirovsky N.E., Olivev M.F. NMDA Receptor Modulators in the Treatment of Drug Addiction. Pharmaceuticals (Basel). 6(2): 251–268. 2013.
Croucher M.J., Collins J.F., Meldrum B.S. Anticonvulsant action of excitatory amino acid antagonists. Science. 216(4548): 899–901. 1982.
Patel S., Chapman A.G., Graham J.L., Meldrum B.S., Frey P. Anticonvulsant activity of the NMDA antagonists, D(-)4-(3-phosphonopropyl)-piperazine-2-carboxylic acid (D-CPP) and D(-)(E)-4-(3-phosphonoprop-2-enyl)-piperazine-2-carboxylic acid (D-CPPene) in a rodent and a primate model of reflex epilepsy. Epilepsy Res. 7(1): 3–10. 1990.
Burnell E.S., Irvine M., Fang G., Sapkota K., Jane D.E., Monaghan D.T. Positive and Negative Allosteric Modulators of N-Methyl-d-aspartate (NMDA) Receptors: Structure-Activity Relationships and Mechanisms of Action. J. Med. Chem. 62(1): 3–23. 2019.
Ogden K.K., Traynelis S.F. New advances in NMDA receptor pharmacology. Trends Pharmacol. Sci. 32(12): 726–733. 2011.
Karakas E., Simorowski N., Furukawa H. Subunit arrangement and phenylethanolamine binding in GluN1/GluN2B NMDA receptors. Nature. 475(7355): 249–253. 2011.
Karakas E., Furukawa H. Crystal structure of a heterotetrameric NMDA receptor ion channel. Science. 344(6187): 992–997. 2014.
Stroebel D., Buhl D.L., Knafels J.D., Chanda P.K., Green M., Sciabola S., Mony L., Paoletti P., Pandit J. A Novel Binding Mode Reveals Two Distinct Classes of NMDA Receptor GluN2B-selective Antagonists. Mol. Pharmacol. 89(5): 541–551. 2016.
Burger P.B., Yuan H., Karakas E., Geballe M., Furukawa H., Liotta D.C., Snyder J.P., Traynelis S.F. Mapping the binding of GluN2B-selective N-methyl-D-aspartate receptor negative allosteric modulators. Mol. Pharmacol. 82(2): 344–359. 2012.
Romero-Hernandez A., Simorowski N., Karakas E., Furukawa H. Molecular Basis for Subtype Specificity and High-Affinity Zinc Inhibition in the GluN1-GluN2A NMDA Receptor Amino-Terminal Domain. Neuron. 92(6): 1324–1336. 2016.
Parsons C.G., Quack G., Bresink I., Baran L., Przegalinski E., Kostowski W., Krzascik P., Hartmann S., Danysz W. Comparison of the potency, kinetics and voltage-dependency of a series of uncompetitive NMDA receptor antagonists in vitro with anticonvulsive and motor impairment activity in vivo. Neuropharmacology. 34(10): 1239–1258. 1995.
Benveniste M., Mayer M.L. Trapping of glutamate and glycine during open channel block of rat hippocampal neuron NMDA receptors by 9-aminoacridine. J. Physiol. 483(2): 367–384. 1995.
Bolshakov K.V., Gmiro V.E., Tikhonov D.B., Magazanik L.G. Determinants of trapping block of N-methyl-d-aspartate receptor channels. J. Neurochem. 87(1): 56–65. 2003.
Barygin O.I., Gmiro V.E., Kim K.K., Magazanik L.G., Tikhonov D.B. Blockade of NMDA receptor channels by 9-aminoacridine and its derivatives. Neurosci. Lett. 451(1): 29–33. 2009.
Blanpied T.A., Boeckman F.A., Aizenman E., Johnson J.W. Trapping channel block of NMDA-activated responses by amantadine and memantine. J. Neurophysiol. 77(1): 309–323. 1997.
Kotermanski S.E., Wood J.T., Johnson J.W. Memantine binding to a superficial site on NMDA receptors contributes to partial trapping. J. Physiol. 587(19): 4589–4604. 2009.
Johnson J.W., Glasgow N.G., Povysheva N.V. Recent insights into the mode of action of memantine and ketamine. Curr. Opin. Pharmacol. 20: 54–63. 2015.
Sobolevsky A.I., Yelshansky M.V. The trapping block of NMDA receptor channels in acutely isolated rat hippocampal neurones. J. Physiol. 526(3): 493–506. 2000.
Poulsen M.H., Andersen J., Christensen R., Hansen K.B., Traynelis S.F., Strømgaard K., Kristensen A.S. Binding of ArgTX-636 in the NMDA receptor ion channel. J. Mol. Biol. 427 (1): 176–189. 2015.
Takahashi H., Xia P., Cui J., Talantova M., Bodhinathan K., Li W., Saleem S., Holland E.A., Tong G., Piña-Crespo J., Zhang D., Nakanishi N., Larrick J.W., McKercher S.R., Nakamura T., Wang Y., Lipton S.A. Pharmacologically targeted NMDA receptor antagonism by NitroMemantine for cerebrovascular disease. Sci. Rep. 5: 14781. 2015.
Hamilton N.B., Attwell D. Do astrocytes really exocytose neurotransmitters? Nat. Rev. Neurosci. 11(4): 227–238. 2010.
Duan S., Anderson C.M., Keung E.C., Chen Y., Chen Y., Swanson R.A. P2X7 receptor-mediated release of excitatory amino acids from astrocytes. J. Neurosci. 23(4): 1320–1328. 2003.
Malarkey E.B., Parpura V. Mechanisms of glutamate release from astrocytes. Neurochem. Int. 52(1–2): 142–154. 2008.
Szatkowski M., Barbour B., Attwell D. Non-vesicular release of glutamate from glial cells by reversed electrogenic glutamate uptake. Nature. 348(6300): 443–446. 1990.
Nicholls D., Attwell D. The release and uptake of excitatory amino acids. Trends Pharmacol. Sci. 11(11): 462–468. 1990.
Hardingham G.E., Bading H. Coupling of extrasynaptic NMDA receptors to a CREB shut-off pathway is developmentally regulated. Biochim. Biophys. Acta. 1600(1–2): 148–153. 2002.
Tu W., Xu X., Peng L., Zhong X., Zhang W., Soundarapandian M.M., Balel C., Wang M., Jia N., Zhang W., Lew F., Chan S.L., Chen Y., Lu Y. DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell. 140(2): 222–234. 2010.
Wroge C.M., Hogins J., Eisenman L., Mennerick S. Synaptic NMDA receptors mediate hypoxic excitotoxic death. J. Neurosci. 32(19): 6732–6742. 2012.
Sattler R., Xiong Z., Lu W.Y., Hafner M., MacDonald J.F., Tymianski M. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science. 284(5421): 1845–1848. 1999.
Tajima N., Karakas E., Grant T., Simorowski N., Diaz-Avalos R., Grigorieff N., Furukawa H. Activation of NMDA receptors and the mechanism of inhibition by ifenprodil. Nature. 534(7605): 63–68. 2016.
Ahmadirad N., Shojaei A., Javan M., Pourgholami M.H., Mirnajafi-Zadeh J. Effect of minocycline on pentylenetetrazol-induced chemical kindled seizures in mice. Neurol. Sci. 35(4): 571–576. 2014.
Davoudi M., Shojaei A., Palizvan M.R., Javan M., Mirnajafi-Zadeh J. Comparison between standard protocol and a novel window protocol for induction of pentylenetetrazol kindled seizures in the rat. Epilepsy Res. 106(1–2): 54–63. 2013.
Lopes M.W., Soares F.M.S., de Mello N., Nunes J.C., Cajado A.G., de Brito D., de Cordova F.M., da Cunha R.M.S., Walz R., Leal R.B. Time-dependent modulation of AMPA receptor phosphorylation and mRNA expression of NMDA receptors and glial glutamate transporters in the rat hippocampus and cerebral cortex in a pilocarpine model of epilepsy. Exp. Brain Res. 226(2): 153–163. 2013.
Zubareva O.E., Kovalenko A.A., Karyakin V.B., Kalemenev S.V., Lavrent’eva V.V., Magazanik L.G., Zaitsev A.V. Changes in the Expression of Genes of the Glutamate Transporter and Subunits of the NMDA and AMPA Receptors in the Rat Amygdala in the Lithium–Pilocarpine Model of Epilepsy. Neurochem. J. 12(3): 222–227. 2018.
di Maio R., Mastroberardino P.G., Hu X., Montero L.M., Greenamyre J.T. Pilocapine alters NMDA receptor expression and function in hippocampal neurons: NADPH oxidase and ERK1/2 mechanisms. Neurobiol. Dis. 42(3): 482–95. 2011.
di Maio R., Mastroberardino P.G., Hu X., Montero L.M., Greenamyre J.T. Thiol oxidation and altered NR2B/NMDA receptor functions in in vitro and in vivo pilocarpine models: implications for epileptogenesis. Neurobiol. Dis. 49: 87–98. 2013.
Alsharafi W.A., Xiao B., Li J. MicroRNA-139-5p negatively regulates NR2A-containing NMDA receptor in the rat pilocarpine model and patients with temporal lobe epilepsy. Epilepsia. 57(11): 1931–1940. 2016.
Müller L., Tokay T., Porath K., Köhling R., Kirschstein T. Enhanced NMDA receptor-dependent LTP in the epileptic CA1 area via upregulation of NR2B. Neurobiol. Dis. 54: 183–193. 2013.
Postnikova T.Y., Zubareva O.E., Kovalenko A.A., Kim K.K., Magazanik L.G., Zaitsev A.V. Status epilepticus impairs synaptic plasticity in rat hippocampus and is followed by changes in expression of NMDA receptors. Biochemistry. 82(3): 282-290. 2017.
Alinaghipour A., Mazoochi T., Ardjmand A. Low-dose ethanol ameliorates amnesia induced by a brief seizure model: the role of NMDA signaling. Neurol. Res. 41(7): 624–632. 2019.
Wang W.-P., Lou Y., Li Z.-Z., Li P., Duan R.-S. Change of hippocampal NMDA receptor and emotional behavior and spatial learning and memory in status epilepticus rat model. Chinese J. Appl. Physiol. 23(1): 51–55. 2007.
Zhu X., Dong J., Shen K., Bai Y., Zhang Y., Xuan L., Chao J., Yao H. NMDA receptor NR2B subunits contribute to PTZ-kindling-induced hippocampal astrocytosis and oxidative stress. Brain Res. Bull. 114: 70–78. 2015.
Lasoń W., Turchan J., Przewłocka B., Łabuz D., Machelska H., Przewłocki R. Effects of pentylenetetrazol kindling on glutamate receptor genes expression in the rat hippocampus. Brain Res. 785(2): 355–358. 1998.
Gori M.B., Girardi E. 3-Mercaptopropionic acid-induced repetitive seizures increase glun2a expression in rat hippocampus: A potential neuroprotective role of cyclopentyladenosine. Cell. Mol. Neurobiol. 33(6): 803–813. 2013.
Girardi E., Auzmendi J., Charó N., Gori M.B., Castro M. 3-Mercaptopropionic Acid-Induced Seizures Decrease NR2B Expression in Purkinje Cells: Cyclopentyladenosine Effect. Cell. Mol. Neurobiol. 30(7): 985–990. 2010.
Auzmendi J., González N., Girardi E. The NMDAR subunit NR2B expression is modified in hippocampus after repetitive seizures. Neurochem. Res. 34(5): 819–826. 2009.
Zhu L.-J., Chen Z., Zhang L.-S., Xu S.-J., Xu A.-J., Luo J.-H. Spatiotemporal changes of the N-methyl-d-aspartate receptor subunit levels in rats with pentylenetetrazole-induced seizures. Neurosci. Lett. 356(1): 53–56. 2004.
Peng W.-F., Ding J., Li X., Fan F., Zhang Q.-Q.,Wang X. N-methyl-d-aspartate receptor NR2B subunit involved in depression-like behaviours in lithium chloride-pilocarpine chronic rat epilepsy model. Epilepsy Res. 119: 77–85. 2016.
Lasón W., Turchan J., Przewłocki R., Machelska H., Labuz D., Przewłocka B. Effects of pilocarpine and kainate-induced seizures on N-methyl-D-aspartate receptor gene expression in the rat hippocampus. Neuroscience. 78(4): 997–1004. 1997.
Zhou C., Sun H., Klein P.M., Jensen F.E. Neonatal seizures alter NMDA glutamate receptor GluN2A and 3A subunit expression and function in hippocampal CA1 neurons. Front. Cell. Neurosci. 9: 362. 2015.
Curia G., Longo D., Biagini G., Jones R.S.G., Avoli M. The pilocarpine model of temporal lobe epilepsy. J. Neurosci. Methods. 172(2): 143–157. 2008.
Lévesque M., Avoli M. The kainic acid model of temporal lobe epilepsy. Neurosci. Biobehav. Rev. 37(10): 2887–2899. 2013.
Dalby N.O., Mody I. The process of epileptogenesis: A pathophysiological approach. Current Opin. Neurol. 14(2): 187–192. 2001.
Pitkänen A., Sutula T.P. Is epilepsy a progressive disorder? Prospects for new therapeutic approaches in temporal-lobe epilepsy. Lancet Neurol. 1(3): 173–181. 2002.
Zubareva O.E., Kovalenko A.A., Kalemenev S.V., Schwarz A.P., Karyakin V.B., Zaitsev A. V. Alterations in mRNA expression of glutamate receptor subunits and excitatory amino acid transporters following pilocarpine-induced seizures in rats. Neurosci. Lett. 686: 94–100. 2018.
Ehlers M.D., Tingley W.G., Huganir R.L. Regulated subcellular distribution of the NR1 subunit of the NMDA receptor. Science. 269(5231): 1734–1737. 1995.
Tingley W.G., Ehlers M.D., Kameyama K., Doherty C., Ptak J.B., Riley C.T., Huganir R.L. Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-D-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J. Biol. Chem. 272(8): 5157–5166. 1997.
Crump F.T., Dillman K.S.,Craig A.M. cAMP-dependent protein kinase mediates activity-regulated synaptic targeting of NMDA receptors. J. Neurosci. 21(14): 5079–5088. 2001.
Fong D.K., Rao A., Crump F.T., Craig A.M. Rapid synaptic remodeling by protein kinase C: Reciprocal translocation of NMDA receptors and calcium/calmodulin-dependent kinase II. J. Neurosci. 22(6): 2153–2164. 2002.
Ehlers M.D., Zhang S., Bernhardt J.P., Huganir R.L. Inactivation of NMDA receptors by direct interaction of calmodulin with the NR1 subunit. Cell. 84(5): 745–755. 1996.
Hisatsune C., Umemori H., Inoue T., Michikawa T., Kohda K., Mikoshiba K., Yamamoto T. Phosphorylation-dependent regulation of N-methyl-D-aspartate receptors by calmodulin. J. Biol. Chem. 272(33): 20805–20810. 1997.
Ryu H.J., Kim J.E., Yeo S.I., Kim D.S., Kwon O.S., Choi S.Y. Potential roles of D-serine and serine racemase in experimental temporal lobe epilepsy. J. Neurosci. Res. 88(11): 2469–2482. 2010.
Naylor D.E., Liu H., Niquet J., Wasterlain C.G. Rapid surface accumulation of NMDA receptors increases glutamatergic excitation during status epilepticus. Neurobiol. Dis. 54: 225–238. 2013.
Ouardouz M., Lema P., Awad P.N., di Cristo G., Carmant L. N-methyl-d-aspartate, hyperpolarization-activated cation current (I h) and -aminobutyric acid conductances govern the risk of epileptogenesis following febrile seizures in rat hippocampus. Eur. J. Neurosci. 31(7): 1252–1260. 2010.
Chen Q., He S., Hu X.L., Yu J., Zhou Y., Zheng J., Zhang S., Zhang C., Duan W.H., Xiong Z.Q. Differential Roles of NR2A- and NR2B-Containing NMDA Receptors in Activity-Dependent Brain-Derived Neurotrophic Factor Gene Regulation and Limbic Epileptogenesis. J. Neurosci. 27(3): 542–552. 2007.
Eyo U.B., Bispo A., Liu J., Sabu S., Wu R., Dibona V.L., Zheng J., Murugan M., Zhang H., Tang Y., Wu L.J. The GluN2A Subunit Regulates Neuronal NMDA receptor-Induced Microglia-Neuron Physical Interactions. Sci. Rep. 8(1): 828. 2018.
Wu Y., Chen C., Yang Q., Jiao M., Qiu S. Endocytosis of GluN2B-containing NMDA receptor mediates NMDA-induced excitotoxicity. Mol. Pain. 13: 1744806917701921. 2017.
Bertram E.H., Lothman E.W. NMDA receptor antagonists and limbic status epilepticus: a comparison with standard anticonvulsants. Epilepsy Res. 5(3): 177–184. 1990.
Rogawski M.A. The NMDA receptors. NMDA antagonists and epilepsy therapy. Drugs. 44(3): 279–292. 1992.
Farber N.B. The NMDA receptor hypofunction model of psychosis. Ann. N. Y. Acad. Sci. 1003: 119–130. 2003.
Muir K.W., Lees K.R. Clinical experience with excitatory amino acid antagonist drugs. Stroke. 26(3): 503–513. 1995.
Gouzoulis-Mayfrank E., Heekeren K., Neukirch A., Stoll M., Stock C., Obradovic M., Kovar K.A. Psychological effects of (S)-ketamine and N,N-dimethyltryptamine (DMT): a double-blind, cross-over study in healthy volunteers. Pharmacopsychiatry. 38(6): 301–311. 2005.
Zeiler F.A. Early Use of the NMDA Receptor Antagonist Ketamine in Refractory and Superrefractory Status Epilepticus. Crit. Care Res. Pract. 2015: 831260. 2015.
Ghasemi M., Shafaroodi H., Nazarbeiki S., Meskar H., Heydarpour P., Ghasemi A., Talab S.S., Ziai P., Bahremand A., Dehpour A.R. Voltage-dependent calcium channel and NMDA receptor antagonists augment anticonvulsant effects of lithium chloride on pentylenetetrazole-induced clonic seizures in mice. Epilepsy Behav. 18(3): 171–178. 2010.
Lukomskaya N.Y., Rukoyatkina N.I., Gorbunova L.V., Magazanik L.G., Gmiro V.E. Studies of the roles of NMDA and AMPA glutamate receptors in the mechanism of corasole convulsions in mice. Neurosci. Behav. Physiol. 34(8): 783–789. 2004.
Vermoesen K., Smolders I., Massie A., Michotte Y., Clinckers R. The control of kainic acid-induced status epilepticus. Epilepsy Res. 90(1–2): 164–166. 2010.
Dorandeu F., Carpentier P., Baubichon D., Four E., Bernabé D., Burckhart M.F., Lallement G. Efficacy of the ketamine-atropine combination in the delayed treatment of soman-induced status epilepticus. Brain Res. 1051(1–2): 164–175. 2005.
Zellinger C., Salvamoser J.D., Soerensen J., van Vliet E.A., Aronica E., Gorter J., Potschka H. Pre-treatment with the NMDA receptor glycine-binding site antagonist L-701,324 improves pharmacosensitivity in a mouse kindling model. Epilepsy Res. 108(4): 634–643. 2014.
Loss C.M., da Rosa N.S., Mestriner R.G., Xavier L.L.,Oliveira D.L. Blockade of GluN2B-containing NMDA receptors reduces short-term brain damage induced by early-life status epilepticus. Neurotoxicology. 71: 138–149. 2019.
Clasadonte J., Dong J., Hines D.J., Haydon P.G. Astrocyte control of synaptic NMDA receptors contributes to the progressive development of temporal lobe epilepsy. Proc. Natl. Acad. Sci. USA. 110(43): 17540–17545. 2013.
Yu W., Calos M., Pilitsis J., Shin D.S.H. Deconstructing the neural and ionic involvement of seizure-like events in the striatal network. Neurobiol. Dis. 52: 128–136. 2013.
Hanna S., Harrison M., Macintyre I., Fraser R. The syndrome of magnesium deficiency in man. Lancet. 276(7143): 172–176. 1960.
Buck D.R., Mahoney A.W., Hendricks D.G. Effect of magnesium deficiency on nonspecific excitability level (NEL) and audiogenic seizure susceptibility. Pharmacol. Biochem. Behav. 5(5): 529–534. 1976.
Randall R.E.J., Rossmeisl E.C., Bleifer K.H. Magnesium depletion in man. Ann. Intern. Med. 50(2): 257–287. 1959.
Govil M.K., Mangal B.D., Alam S.M., Mahendru R.K., Srivastava D.K., Mudgal J.C. Serum and cerebrospinal fluid calcium and magnesium levels in cases of idiopathic grand mal epilepsy and induced convulsions. J. Assoc. Physic. India. 29(9): 695–699. 1981.
Sinert R., Zehtabchi S., Desai S., Peacock P., Altura B.T., Altura B.M. Serum ionized magnesium and calcium levels in adult patients with seizures. Scand. J. Clin. Lab. Invest. 67(3): 317–326. 2007.
Mikati M.A., Injibar H., Kurdi R.M., El H.J., Abou R.S., Lteif L., Abdul J.M., Francis E., Geha G., Farhat F. Effects of magnesium sulfate in kainic acid-induced status epilepticus. J. Med. Liban. 54(4): 200–204. 2006.
Ghasemi A., Saberi M., Ghasemi M., Shafaroodi H., Moezi L., Bahremand A., Montaser-Kouhsari L., Ziai P., Dehpour A.R. Administration of lithium and magnesium chloride inhibited tolerance to the anticonvulsant effect of morphine on pentylenetetrazole-induced seizures in mice. Epilepsy Behav. 19(4): 568–574. 2010.
Bac P., Herrenknecht C., Binet P., Durlach J. Audiogenic seizures in magnesium-deficient mice: effects of magnesium pyrrolidone-2-carboxylate, magnesium acetyltaurinate. magnesium chloride and vitamin B-6. Magnes. Res. 6(1): 11–19. 1993.
Safar M.M., Abdallah D.M., Arafa N.M., Abdel-Aziz M.T. Magnesium supplementation enhances the anticonvulsant potential of valproate in pentylenetetrazol-treated rats. Brain Res. 1334: 58–64. 2010.
Cotton D.B., Hallak M., Janusz C., Irtenkauf S.M., Berman R.F. Central anticonvulsant effects of magnesium sulfate on N-methyl-D-aspartate-induced seizures. Am. J. Obstet. Gynecol. 168(3): 974–978. 1993.
Bennet L., Galinsky R., Draghi V., Lear C.A., Davidson J.O., Unsworth C.P., Gunn A.J. Time and sex dependent effects of magnesium sulphate on post-asphyxial seizures in preterm fetal sheep. J. Physiol. 596(23): 6079–6092. 2018.
Lipton S.A. Failures and successes of NMDA receptor antagonists: molecular basis for the use of open-channel blockers like memantine in the treatment of acute and chronic neurologic insults. NeuroRx. 1(1): 101–110. 2004.
Bialer M., Johannessen S.I., Kupferberg H.J., Levy R.H., Loiseau P., Perucca E. Progress report on new antiepileptic drugs: a summary of the fourth Eilat conference (EILAT IV). Epilepsy Res. 34(1): 1–41. 1999.
Sun L., Lin S.S. The anticonvulsant SGB-017 (ADCI) blocks voltage-gated sodium channels in rat and human neurons: comparison with carbamazepine. Epilepsia. 41(3): 263–270. 2000.
Rogawski M.A., Yamaguchi S., Jones S.M., Rice K.C., Thurkauf A., Monn J.A. Anticonvulsant activity of the low-affinity uncompetitive N-methyl-D-aspartate antagonist (+-)-5-aminocarbonyl-10,11-dihydro-5H-dibenzoad-cyclohepten-5,10-imine (ADCI): comparison with the structural analogs dizocilpine (MK-801) and carbamazepine. J. Pharmacol. Exp. Ther. 259(1): 30–37. 1991.
Seidleck B.K., Thurkauf A., Witkin J.M. Evaluation of ADCI against convulsant and locomotor stimulant effects of cocaine: comparison with the structural analogs dizocilpine and carbamazepine. Pharmacol. Biochem. Behav. 47(4): 839–844. 1994.
Geter-Douglass B., Witkin J.M. Behavioral effects and anticonvulsant efficacies of low-affinity, uncompetitive NMDA antagonists in mice. Psychopharmacology. 146(3): 280–289. 1999.
Coleman M.H., Yamaguchi S., Rogawski M.A. Protection against dendrotoxin-induced clonic seizures in mice by anticonvulsant drugs. Brain Res. 575(1): 138–142. 1992.
Grant K.A., Snell L.D., Rogawski M.A., Thurkauf A., Tabakoff B. Comparison of the effects of the uncompetitive N-methyl-D-aspartate antagonist (+-)-5-aminocarbonyl-10,11-dihydro-5H-dibenzoad-cyclohepten-5,10-imine (ADCI) with its structural analogs dizocilpine (MK-801) and carbamazepine on ethanol withdrawal seizures. J. Pharmacol. Exp. Ther. 260(3): 1017–1022. 1992.
Ghasemi M., Shafaroodi H., Nazarbeiki S., Meskar H., Ghasemi A., Bahremand A., Ziai P., Dehpour A.R. Inhibition of NMDA receptor/NO signaling blocked tolerance to the anticonvulsant effect of morphine on pentylenetetrazole-induced seizures in mice. Epilepsy Res. 91(1): 39–48. 2010.
Taberner P.V. The anticonvulsant activity of ketamine against seizures induced by pentylenetetrazol and mercaptopropionic acid. Eur. J. Pharmacol. 39(2): 305–311. 1976.
Velísková J., Velísek L., Mares P., Rokyta R. Ketamine suppresses both bicuculline- and picrotoxin-induced generalized tonic-clonic seizures during ontogenesis. Pharmacol. Biochem. Behav. 37(4): 667–674. 1990.
D’Hooge R., Pei Y.Q., de Deyn P.P. N-methyl-D-aspartate receptors contribute to guanidinosuccinate-induced convulsions in mice. Neurosci. Lett. 157(2): 123–126. 1993.
Stafstrom C.E., Sasaki-Adams D.M. NMDA-induced seizures in developing rats cause long-term learning impairment and increased seizure susceptibility. Epilepsy Res. 53(1–2): 129–137. 2003.
Trommer B.L., Pasternak J.F. NMDA receptor antagonists inhibit kindling epileptogenesis and seizure expression in developing rats. Brain Res. Dev. 53(2): 248–252. 1990.
Borowicz K.K., Łuszczki J., Czuczwar S.J. Interactions between non-barbiturate injectable anesthetics and conventional antiepileptic drugs in the maximal electroshock test in mice – an isobolographic analysis. Eur. Neuropsychopharmacol. 14(2): 163–172. 2004.
Martin B.S., Kapur J. A combination of ketamine and diazepam synergistically controls refractory status epilepticus induced by cholinergic stimulation. Epilepsia. 49(2): 248–255. 2008.
Kim K.K., Zaitsev A.V., Lavrent’eva V.V., Zhabko E.P., Vataev S.I., Lukomskaya N.Y., Magazanik L.G. Effects of Ionotropic Glutamate Receptor Blockers on Pentylenetetrazole-Induced Seizures in Krushinskii–Molodkina Rats. Neurosci. Behav. Physiol. 44(8): 945–950. 2014.
Chen B., Feng B., Tang Y., You Y., Wang Y., Hou W., Hu W., Chen Z. Blocking GluN2B subunits reverses the enhanced seizure susceptibility after prolonged febrile seizures with a wide therapeutic time-window. Exp. Neurol. 283: 29–38. 2016.
Jansen M., Dannhardt G. Antagonists and agonists at the glycine site of the NMDA receptor for therapeutic interventions. Eur. J. Med. Chem. 38(7–8): 661–670. 2003.
Chen H.-S.V., Lipton S.A. The chemical biology of clinically tolerated NMDA receptor antagonists. J. Neurochem. 97(6): 1611–1626. 2006.
Kalia L.V., Kalia S.K., Salter M.W. NMDA receptors in clinical neurology: excitatory times ahead. Lancet Neurol. 7(8): 742–755. 2008.
Berger P., Farrel K., Sharp F., Skolnick P. Drugs acting at the strychnine-insensitive glycine receptor do not induce HSP-70 protein in the cingulate cortex. Neurosci. Lett. 168(1–2): 147–150. 1994.
Niquet J., Lumley L., Baldwin R., Rossetti F., Suchomelova L., Naylor D., Estrada I.B.F., Schultz M., Furtado M. de A., Wasterlain C.G. Rational polytherapy in the treatment of cholinergic seizures. Neurobiol. Dis. 133: 104537. 2020.
Schidlitzki A., Twele F., Klee R., Waltl I., Römermann K., Bröer S., Meller S., Gerhauser I., Rankovic V., Li D., Brandt C., Bankstahl M., Töllner K., Löscher W. A combination of NMDA and AMPA receptor antagonists retards granule cell dispersion and epileptogenesis in a model of acquired epilepsy. Sci. Rep. 7(1): 1–19. 2017.
Brandt C., Potschka H., Loscher W., Ebert U. N-methyl-D-aspartate receptor blockade after status epilepticus protects against limbic brain damage but not against epilepsy in the kainate model of temporal lobe epilepsy. Neuroscience. 118(3): 727–740. 2003.
Santamaría A., Ríos C. MK-801, an N-methyl-d-aspartate receptor antagonist, blocks quinolinic acid-induced lipid peroxidation in rat corpus striatum. Neurosci. Lett. 159(1): 51–54. 1993.
Yang Q., Huang Z., Luo Y., Zheng F., Hu Y., Liu H., Zhu S., He M., Xu D., Li Y., Yang M., Yang Y., Wei X., Gao X., Wang W., Ma J., Ma Y., Wang X., Wang Q. Inhibition of Nwd1 activity attenuates neuronal hyperexcitability and GluN2B phosphorylation in the hippocampus. EBioMedicine. 47: 470–483. 2019.
Rodríguez-Muñoz M., Onetti Y., Cortés-Montero E., Garzón J., Sánchez-Blázquez P. Cannabidiol enhances morphine antinociception, diminishes NMDA-mediated seizures and reduces stroke damage via the sigma 1 receptor. Mol. Brain. 11(1): 1–12. 2018.
Yang Y., Tian X., Xu D., Zheng F., Lu X., Zhang Y., Ma Y., Li Y., Xu X., Zhu B., Wang X. GPR40 modulates epileptic seizure and NMDA receptor function. Sci. Adv. 4(1): 1–12. 2018.
Koek W., Colpaert F.C. Selective blockade of N-methyl-D-aspartate (NMDA)-induced convulsions by NMDA antagonists and putative glycine antagonists: relationship with phencyclidine-like behavioral effects. J. Pharmacol. Exp. Ther. 252(1): 349–357. 1990.
Chiamulera C., Costa S., Reggiani A. Effect of NMDA- and strychnine-insensitive glycine site antagonists on NMDA-mediated convulsions and learning. Psychopharmacology (Berl). 102(4): 551–552. 1990.
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова