

Российский физиологический журнал им. И.М. Сеченова, 2020, T. 106, № 2, стр. 135-156
Роль аутофагии в ишемическом и реперфузионном повреждении сердца
Н. С. Воронков 1, 2, *, Л. Н. Маслов 1, 2
1 Научно-исследовательский институт кардиологии
Томск, Россия
2 Томский национальный исследовательский медицинский центр РАН
Томск, Россия
* E-mail: maslov@cardio-tomsk.ru
Поступила в редакцию 09.09.2019
После доработки 07.11.2019
Принята к публикации 07.11.2019
Аннотация
Ишемия/реперфузия (И/Р) сердца приводит к усилению аутофагического потока. Прекондиционирование стимулирует аутофагический поток за счет активации киназы AMPK и PI3-киназы при ингибировании киназы mTOR. Кардиопротекторный эффект посткондиционирования связан с активацией аутофагии и повышением активности NO-синтазы и AMPK. И/Р стимулирует аутофагию, а гонадэктомия ее подавляет. Адаптация к гипоксии оказывает кардиопротекторный эффект, который, возможно, связан с активацией аутофагии. Возможно, что негативный эффект жировой диеты на толерантность сердца к И/Р связан с ингибированием аутофагии. Голодание стимулирует аутофагию, этот эффект сопровождается повышением толерантности сердца к действию И/Р. Окислительный стресс стимулирует аутофагию при И/Р сердца. Супероксидный радикал, генерируемый НАДФН-оксидазой, выступает в роли триггера аутофагии, по-видимому, за счет активации киназы AMPK. Есть основания полагать, что киназы AMPK, GSK-3β, JNK, MEK и ERK стимулируют аутофагию, а mTOR, Akt- и PI3-киназа ингибируют аутофагию при И/Р сердца. Установлено, что транскрипционные факторы FoxO1, FoxO3, NF-κB и HIF-1α усиливают аутофагию при И/Р сердца. Транскрипционные факторы STAT1 и p53 ингибируют аутофагию в условиях И/Р сердца. miR-325, miR-145 и miR-144 симулируют аутофагию, а miR-30a, miR-221, miR-638 и miR-144 ингибируют аутофагию при И/Р сердца. Представленные данные свидетельствуют, что H2S повышает толерантность сердца к действию И/Р и ингибирует аутофагию. Гемоксигеназа-1 стимулирует аутофагию и предупреждает повреждение митохондрий при гипоксии/реоксигенации кардиомиоцитов. Активация аутофагии во время И/Р может быть протекторной или повреждающей в зависимости от экспериментальной модели. Инфаркт-лимитирующий эффект зависит не только от того, как влияет то или иное соединение на аутофагию, но и от того, как оно влияет на апоптоз, некроптоз и некроз кардиомиоцитов при И/Р сердца.
Аутофагия или аутофагоцитоз – это процесс энзиматической деградации белков, жиров, нуклеиновых кислот и целых органелл в лизосомах. Термин аутофагия происходит от древнегреческого αὐτός ауто – “сам” и φαγεῖν – “есть”. История аутофагии началась с открытия лизосом, которое был сделано в 1955 г. коллективом исследователей, возглавляемых C. de Duve [1]. Он же предложил термин лизосомы от греческого λύσις — “разложение” и σώμα — “тело” [1]. Лизосомы представляют из себя везикулу, окруженную двойной клеточной мембраной и содержащую около 50 кислых гидролаз, катализирующих реакцию [2]:
Кислые гидролазы включают фосфатазы, нуклеазы, гликозидазы, протеазы, пептидазы, сульфатазы, липазы, которые активны при кислой pH 4.7 [3].
В январе 1962 г. K.R. Porter и T. Ashford показали, что после применения глюкагона лизосомы клеток печени начинают поглощать клеточные органеллы, в том числе митохондрии [4]. В исследовании, опубликованном в 1963 г., у крыс моделировали различные патологические состояния и проводили морфологические исследования печени, почек и поджелудочной железы. Было обнаружено секвестирование цитоплазмы с последующей деградацией в лизосомах, то есть аутофагия. Сам термин “аутофагия” впервые прозвучал на the Ciba Foundation Symposium on Lysosomes, который проходил в Лондоне 12–14 февраля 1963 г. [5]. Термин “аутофагоцитоз” (autophagocytosis) впервые использовал J. Radewa [6]. Позднее появились статьи, которые подтвердили существование аутофагии [7, 8].
Известно, что существует три типа аутофагии. Макроаутофагия включает образование везикул с двойной мембраной, называемых аутофагосомами, которые связывают клеточные белки, глюциды, липиды и органеллы, а затем доставляют их в лизосомы для деградации. Микроаутофагия относится к процессу, посредством которого клеточные элементы, которые подвержены деградации, непосредственно поглощаются лизосомами [9]. Опосредованная шапероном аутофагия характеризуется связыванием белков, содержащих последовательность KFERQ, с шапероном Hsc70, который транспортирует целевые белки в лизосомы с участием белка 2А лизосомальной мембраны (Lamp2A) [9]. Макроаутофагия (ее обычно называют аутофагией) имеет решающее значение для деградации органелл и адаптации к клеточному стрессу, тогда как две другие формы аутофагии постоянно участвуют в специализированных клеточных функциях [9, 10]. Мы многим обязаны своими знаниями о природе аутофагии Yoshinori Ohsumi, лауреату Нобелевской премии 2016 г.
На начальном этапе аутофагия сопровождается образованием фрагмента мембраны, который происходит из эндоплазматического ретикулума, его обычно называют фагофором (phagophore). При участии белкового комплекса Beclin–Vps34 и при участии белка LC3 (microtubule-associated protein 1 light chain 3) образуется везикула, называемая аутофагосомой [9, 11]. Около 80 белков Atg, кодируемых генами, связанными с аутофагией (autophagy-related genes, Atg), участвуют в образовании аутофагосом [9, 11, 12]. Белки паркин (parkin) и P62 (sequestosome 1/SQSTM1) играют ключевую роль в митофагии [9, 13]. Обычно при усилении аутофагического потока (an autophagic flow) увеличивается уровень беклина-1, повышается соотношение LC3-II/LC3-I, а уровень P62 снижается [14–18]. Вместе с тем, некоторые авторы сообщают об увеличении уровня P62 в ответ на ишемию/реперфузию (И/Р) сердца [19, 20]. На заключительной стадии аутофагии аутофагосома сливается с лизосомой и образуется аутолизосома, в которой происходит расщепление внутриклеточных структур. В слиянии аутофагосом и лизосом принимает участие белок митофузин [21].
Ишемия/реперфузия и аутофагия. В ряде исследований было показано, что И/Р стимулирует аутофагию кардиомиоцитов [22–28]. Некоторые исследователи полагают, что аутофагия во время ишемии носит протекторный эффект, а во время реперфузии аутофагия вызывает повреждения сердца [24]. Утверждают, что аутофагия усиливается при ишемии сердца, но еще в большей степени она увеличивается во время реперфузии [23]. Коронароокклюзия (30 мин) стимулирует аутофагию особенно сильно в зоне, граничащей с областью инфаркта [22]. В культивируемых неонатальных кардиомиоцитах крысы депривация глюкозы ведет к усилению экспрессии белков, участвующих в аутофагии LC3 и Atg12 [29]. Показано, что у свиней три эпизода ишемии (90 мин), вызванные снижением коронарного кровотока на 30%, вызывают усиление аутофагии, которая, по мнению авторов, способствует снижению интенсивности апоптоза [30]. Авторы считают, что аутофагия – это гомеостатический механизм, который способствует выживанию кардиомиоцитов в условиях И/Р [30]. Как ни парадоксально, но аутофагия может иметь защитное значение при И/Р сердца [13]. Например, у свиней, подвергшихся 45-минутной окклюзии и реперфузии коронарной артерии, индуктор аутофагии хлорамфеникол уменьшал размер инфаркта [31]. Сообщают, что аутофагия активируется в миокарде человека после воздействия И/Р [12]. В исследовании, выполненном в 2014 г., использовали ушко правого предсердия сердца человека, полученное после кардиоплегической остановки сердца [12]. Экспрессию Atg оценивали с помощью ПЦР. Оказалось, что И/Р приводит к усилению экспрессии 11-ти и даунрегуляции 3-х из 84-х белков Atg. Усиливается шаперон-опосредованная аутофагия, о чем свидетельствует увеличение уровня HSPA8, HSP90AA1. Соотношение LC3-II/LC3-I также увеличивается [12]. Представленные данные свидетельствуют, что И/Р сердца человека способствует появлению аутофагии. В исследовании, выполненном на мышах, воспроизводили коронароокклюзию (120 мин) или коронароокклюзию (30 мин) и реперфузию (90 мин) [32]. Ишемия не влияла на уровень маркеров аутофагии LC3-II, P62, беклина-1 и LAMP2 (lysosome-associated membrane protein-2). Во время реперфузии уровень LC3-II, P62, беклина-1 увеличивался, а уровень LAMP2 снижался [32]. Стимуляцию аутофагии в ответ на И/Р сердца отмечают другие исследователи [19, 33–38]. Активация аутофагии наблюдается в ответ на гипоксию/реоксигенацию изолированных кардиомиоцитов [36, 39–42].
Следует отметить, что некоторым исследователям не удалось обнаружить усиление аутофагии кардиомиоцитов в ответ на гипоксию/реоксигенацию [43]. У мышей вызывали коронароокклюзию (1 и 4 ч) и реперфузию (24 ч). Кроме того, использовали непрерывную коронароокклюзию (24 ч). Ни в одном из этих случаев не было обнаружено увеличение соотношения LC3-II/LC3-I [44]. Изолированные неонатальные кардиомиоциты крыс и кардиомиобласты H9c2 подвергали гипоксии/реоксигенации (Г/Р) [45]. Г/Р вызывала увеличение экспрессии беклина-1 и LC3-II у неонатальных кардиомиоцитов, но снижала уровень этих маркеров аутофагии в клетках H9c2. Есть данные о том, что И/Р сердца in vivo вызывает снижение интенсивности аутофагии [46].
Таким образом, большинство данных свидетельствует, что И/Р сердца приводит к усилению аутофагического потока (рис. 1).
Рис. 1.
Процесс аутофагии и его роль в ишемическом/реперфузионном повреждении кардиомиоцитов. Fig. 1. The process of autophagy and its role in ischemic/reperfusion injury of cardiomyocytes.
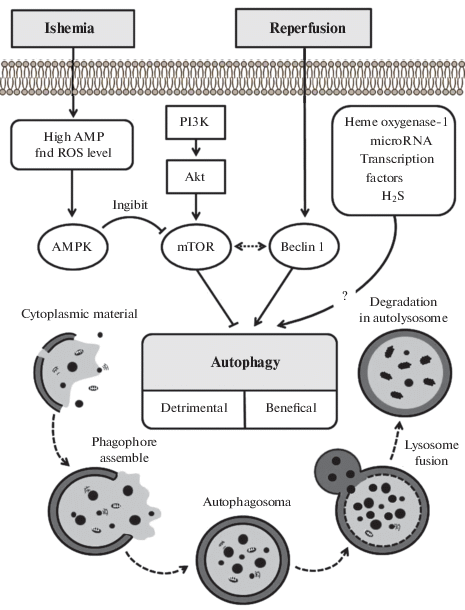
Прекондиционирование и посткондиционирование. Ишемическое прекондиционирование (ИПре) и посткондиционирование (ИПост) – это два феномена, обеспечивающих повышение толерантности сердца к действию длительной И/Р. Прекондиционирование достигается с помощью нескольких сеансов кратковременной И/Р перед длительной ишемией [47]. Посткондиционирование обеспечивает устойчивость сердца к действию реперфузии с помощью нескольких сеансов кратковременной реперфузии/ишемии после длительной ишемии [47]. Дистантное прекондиционирование – это повышение устойчивости сердца к длительной И/Р с помощью кратковременной И/Р дистантного органа [47].
В исследовании, выполненном на изолированном перфузируемом сердце крысы, ИПре воспроизводили с помощью четырех циклов ишемии (5 мин) и реперфузии (10 мин) перед длительной ишемией (30 мин) [48]. Ингибитор PI3-киназы вортманнин вводили внутрибрюшинно за 30 мин до изоляции сердца. Прекондиционирование вызвало усиление экспрессии белков, участвующих в аутофагии LC3-II и беклин-1. Кроме того, усиливается экспрессия белка BAG-1 (Bcl-2-associated athanogene), который увеличивает выживаемость клеток в неблагоприятных условиях и является кошапероном, взаимодействующим с Hsc70/Hsp70 (heat shock proteins). Вортманнин устранял кардиопротекторный эффект ИПре и ингибировал экспрессию LC3-II, беклина-1 и BAG-1. Интрамиокардиальная инъекция BAG-1 siРНК (small interfering RNA) уменьшала индукцию LC3-II и устраняла кардиопротекцию, вызванную ИПре [48]. Исследования проводили на обычных C57BL/6J мышах (wild-type) и животных с нокаутом гена, кодирующего паркин (Parkin–/–) [13]. Прекондиционирование воспроизводили с помощью трех сеансов ишемии (3 мин) и реперфузии (3 мин), для биохимических исследований сердце иссекали. Длительную И/Р воспроизводили с помощью коронароокклюзии (20 мин) и реперфузии (22 ч). Прекондиционирование приводило к увеличению экспрессии паркина и накоплению его в митохондриях. Прекондиционирование вызывало увеличение экспрессии белка P62, участвующего в аутофагии. ИПре уменьшало соотношение размер инфаркта/область риска (РИ/ОР) у обычных мышей, но не влияло на РИ/ОР у мышей Parkin–/– [13]. Областью риска принято называть зону И/Р. Представленные данные свидетельствуют, что ИПре стимулируют аутофагию, а белок паркин участвует в инфаркт-лимитирующем эффекте прекондиционирования.
Исследование выполнялось на пациентах с коронарным шунтированием, которое включало кардиоплегическую остановку (ишемия) сердца [49]. Дистантное ИПре воспроизводили с помощью трех циклов ишемии (5 мин) и реперфузии (5 мин) левой руки перед разрезом кожи. Трансмуральную миокардиальную биопсию (2–5 мг) забирали перед началом искусственного кровообращения и через 5–10 мин после начала реперфузии. Дистантное ИПре способствовало снижению уровня маркера некроза миокарда тропонина I в сыворотке крови после операции, то есть оказывало кардиопротекторный эффект. В качестве маркеров аутофагии определяли беклин-1, фосфорилированный беклин-1, P62, LC3-II. Исследователям не удалось обнаружить разницу по этим маркерам между группой плацебо и группой дистантного посткондиционирования [49]. Следовательно, кардиопротекторный эффект дистантного прекондиционирования при коронарном шунтировании не зависит от аутофагии. Дистантное ИПре воспроизводили у мышей с помощью четырех циклов ишемии (5 мин) и реперфузии (5 мин) конечностей [50]. Сердце изолировали сразу после прекондиционирования (first window) или через 24 ч после прекондиционирования (отсроченное прекондиционирование) и подвергали глобальной ишемии (30 мин) и реперфузии (60 мин). В обоих случаях прекондиционирование сопровождалось уменьшением размера инфаркта и увеличением давления, развиваемого левым желудочком. При раннем прекондиционировании происходило увеличение количества фосфорилированной p-AMPK (AMP-activated protein kinase), снижение количества фосфорилированной киназы p-mTOR (mammalian target of rapamycin) и повышение соотношения LC3-II/LC3-I при неизменном уровне беклина-1, Atg5 и P62. При отсроченном прекондиционировании происходило снижение количества p-AMPK, p-mTOR, наблюдалось снижение уровня беклина-1 при неизменном уровне Atg5, P62 и соотношения LC3-II/LC3-I. Авторы заключили, что только раннее дистантное прекондиционирование стимулирует аутофагию [50]. В исследовании, выполненном на изолированном перфузируемом сердце крысы, было показано, что прекондиционирование повышает толерантность сердца к действию И/Р [14]. Показано, что прекондиционирование способствовало увеличению соотношения LC3-II/LC3-I при одновременном снижении количества P62, что авторы расценивают как увеличение аутофагического потока. Вортманнин, ингибитор PI3-киназы, устранял усиление аутофагического потока и кардиопротекторный эффект прекондиционирования [14]. Представленные данные свидетельствуют, что кардиопротекторный эффект прекондиционирования может быть следствием активации PI3-киназы и усиления аутофагии. Вместе с тем, есть данные, свидетельствующие о том, что кардиопротекторное действие дистанционного ИПре реализуется без участия аутофагии [49].
Культуру неонатальных кардиомиоцитов подвергали аноксии (30 мин) и реоксигенации (60 мин) [51]. Посткондиционирование воспроизводили с помощью трех циклов реоксигенации (1 мин) и аноксии (1 мин). ИПост уменьшало количество клеток в состоянии некроза, апоптоза и аутофагии. У крыс вызывали коронароокклюзию (30 мин) и реперфузию (30 мин) [52]. Посткондиционирование воспроизводили с помощью трех циклов реперфузии (30 с) и реокклюзии (30 с). Рапамицин, ингибитор киназы mTOR и активатор аутофагии, устранял инфаркт-лимитирующий эффект ИПост. Следовательно, есть основания предполагать, что ИПост супрессирует аутофагию. Исследования проводили на крысах, выполняли коронароокклюзию (30 мин) и реперфузию (120 мин) [21]. Хлороквин вводили внутрибрюшинно (10 мг/кг) за 90 мин до коронароокклюзии. Ингаляцию севофлураном начинали во время реперфузии. Севофлуран имитировал феномен ИПост и уменьшал соотношение РИ/ОР. Ингибитор аутофагии хлороквин устранял инфаркт-лимитирующий эффект севофлурана, при этом сам не влиял на размер инфаркта [21]. Сходные данные получили другие исследователи, использовавшие севофлуран [33]. У крыс воспроизводили коронароокклюзию (30 мин) и реперфузию (120 мин) [53]. Посткондиционирование моделировали с помощью трех циклов реперфузии (10 с) и ишемии (10 с). Ингибитор аутофагии 3-метиладенин (3-МА) вводили внутрибрюшинно перед ишемией. ИПост приводило к многократному увеличению количества аутофагических вакуолей, увеличению соотношения LC3-II/LC3-I и повышению количества беклина-1 и белка LAMP-2. Посткондиционирование способствовало уменьшению соотношения РИ/ОР примерно на 50%, а 3-МА устранял этот эффект [53]. Эти факты указывают на то, что ИПост стимулирует аутофагию, которая участвует в инфаркт-лимитирующем эффекте посткондиционирования. У крыс воспроизводили коронароокклюзию (30 мин) и реперфузию (1, 2, 3, 6, 12 и 24 ч) [35]. ИПост вызывали с помощью трех циклов реперфузии (30 с) и ишемии (30 с) после длительной ишемии. Ишемия/реперфузия способствовали увеличению соотношения LC3-II/LC3-I, повышению уровня беклина-1, LC3-II и P62, что говорит об активации аутофагии. Посткондиционирование ингибировало аутофагическую активность в первый час реперфузии, усиливало аутофагию в течение 2–6 ч реперфузии, уменьшало аутофагию через 12–24 ч аутофагии согласно величине соотношения LC3-II/LC3-I. Динамика изменения беклина-1 была иной, ИПост увеличивало этот показатель в первый час реперфузии, снижало через 2–6 ч после возобновления кровотока и увеличивало через 12–24 ч. Ингибитор аутофагии хлороквин устранял инфаркт-лимитирующий эффект ИПост при коронароокклюзии и реперфузии (3 ч) [35]. Представленные данные свидетельствуют о том, что ИПост оказывает неоднозначный эффект на аутофагию, которая участвует в инфаркт-лимитирующем эффекте ИПост. У мышей воспроизводили коронароокклюзию (30 мин) и реперфузию (60 мин) [20]. Посткондиционирование индуцировали с помощью трех циклов реперфузии (30 с) и ишемии (30 с). Посткондиционирование H9c2 клеток включало три цикла гипоксии (5 мин) и реоксигенации (5 мин). Посткондиционирование оказывало инфаркт-лимитирующий эффект, 3-МА устранял кардиопротекторное действие ИПост, также действовал ингибитор NO-синтазы (NOS) L-NIO. Посткондиционирование способствовало увеличению соотношения LC3-II/LC3-I и снижению уровня P62. L-NIO устранял этот эффект ИПост. ИПост способствовало увеличению уровня эндотелиальной NOS и p-AMPK, что говорит об активации этих ферментов. Компаунд С, ингибитор AMPK, устранял эти изменения. ИПост увеличивало выживаемость H9c2 клеток в условиях Г/Р, 3-МА устранял этот эффект [20]. Следовательно, кардиопротекторный эффект ИПост связан с активацией аутофагии и повышением активности NOS и AMPK.
Представленные данные свидетельствуют, что ИПре стимулирует аутофагию за счет активации AMPK и PI3-киназы при ингибировании mTOR. Кардиопротекторный эффект ИПост связан с активацией аутофагии и повышением активности NOS и AMPK.
Половые различия в аутофагии. У крыс воспроизводили коронароокклюзию (30 мин) и реперфузию (24 ч) [54]. Размер инфаркта и количество апоптотических клеток были существенно меньше у самок, чем у самцов крыс. Ишемия/реперфузия приводила к существенному увеличению соотношения LC3-II/LC3-I у самок крыс. У самцов, напротив, происходило уменьшение соотношения LC3-II/LC3-I [54]. К сожалению, исследователи не определяли другие маркеры аутофагии, поэтому утверждать, что И/Р приводит к активации аутофагии у самок и ингибированию аутофагии у самцов крыс, нельзя. В исследовании, выполненном на изолированном перфузируемом сердце крысы, выполняли коронароокклюзию (30 мин) и реперфузию (150 мин) [55]. Соотношение РИ/ОР было меньше у самок крыс, чем у самцов этих животных. Гонадэктомия приводит к уменьшению соотношения РИ/ОР у самцов и к увеличению соотношения РИ/ОР у самок. Тестостерон и дигидротестостерон способствовали увеличению соотношения РИ/ОР у самцов, еще более выраженный инфаркт-потенцирующий эффект оказывал 17β-эстрадиол. У гонадэктомированных самок тестостерон не оказывал эффекта на размер инфаркта, 17β-эстрадиол оказывал инфаркт-лимитирующий эффект. У интактных самцов коронароокклюзия и реперфузия приводила к увеличению уровня p-AMPK, p-mTOR, беклина-1, Atg5 и увеличению соотношения LC3-II/LC3-I. У гонадэктомированных самцов после И/Р отмечался только подъем уровня p-mTOR. Представленные данные показывают, что И/Р стимулирует аутофагию, а гонадэктомия ее подавляет. Терапия тестостероном или 17β-эстрадиолом не оказывает влияния на аутофагию у самцов при И/Р сердца. У интактных самок при коронароокклюзии и реперфузии также наблюдается подъем уровня p-AMPK, p-mTOR, беклина-1, Atg5 и увеличение соотношения LC3-II/LC3-I. Гонадэктомия у самок приводила к заметному снижению уровня беклина-1, Atg5 и снижению соотношения LC3-II/LC3-I. Применение тестостерона или 17β-эстрадиола у гонадэктомированных самок крыс не вызывало заметного усиления аутофагии [55]. Авторы сделали заключение, что половые гормоны регулируют аутофагию у крыс при коронароокклюзии и реперфузии. Следует отметить, что заместительная терапия гормонами не подтверждает эту точку зрения.
Представленные данные показывают, что И/Р стимулирует аутофагию, а гонадэктомия ее подавляет. И/Р сопровождается активацией AMPK (рис. 1).
Адаптация к гипоксии и аутофагия. В исследование были включены жители Тибета, жившие на высоте более 3000 м, и 12 граждан Китая народа хань, жившие на высоте менее 500 м [56]. Этим пациентам выполнялось кардиохирургическое вмешательство по поводу дефекта межпредсердной или межжелудочковой перегородки. Операция проводилась в условиях искусственного кровообращения и кардиоплегической остановки сердца, которая по сути является ишемией миокарда. Для исследования использовали ушко правого предсердия, образцы которого забирали до и после операции. Уровень тропонина I после операции был меньше у жителей Тибета, чем у народа хань, это указывает на кардиопротекторный эффект адаптации к гипоксии. У жителей Тибета был выше уровень LC3II и LAMP2 до и после операции. Однако разницы по уровню беклина-1 выявить не удалось. Авторы полагают, что кардиопротекторный эффект адаптации к гипоксии связан с усилением аутофагии [56]. Однако им не удалось обнаружить подъема уровня беклина-1 у жителей Тибета, что не укладывается в гипотезу об усилении аутофагии при адаптации к гипоксии.
Адаптация к гипоксии оказывает кардиопротекторный эффект, который, возможно, связан с активацией аутофагии.
Метаболический синдром и аутофагия. Метаболический синдром вызывали у мышей с помощью диеты с высоким содержанием жиров (high-fat diet) [57]. Метаболический синдром способствовал увеличению соотношения РИ/ОР, стимулятор аутофагии рапамицин устранял этот негативный эффект жировой диеты. Нокаут гена, кодирующего белок беклин-1 (beclin-1+/–), устранял инфаркт-лимитирующий эффект рапамицина у мышей с метаболическим синдромом [57]. Этот факт говорит о том, что инфаркт-лимитирующий эффект ингибитора mTOR рапамицина связан с активацией аутофагии. Возможно, что негативный эффект жировой диеты на толерантность сердца к И/Р связан с ингибированием аутофагии.
Голодание и аутофагия. Установлено, что голодание перед ишемией миокарда способствовало уменьшению размера инфаркта и стимулировало аутофагию [22]. У мышей индуцировали коронароокклюзию (30 мин) и реперфузию (24 ч) [58]. Часть животных подвергали периодическому голоданию в течение 6 недель. Голодание способствовало уменьшению соотношения РИ/ОР приблизительно на 50%. Этот кардиопротекторный эффект не выявлялся у гетерозиготных мышей с нокаутом белка LAMP2, что подтверждает участие аутофагии в инфаркт-лимитирующем эффекте периодического голодания. Кратковременное (24 или 48 ч) голодание приводило к увеличению соотношения LC3-II/LC3-I и повышению уровня P62 при неизменном уровне LAMP2 и беклина-1. Периодическое голодание не влияло на уровень маркеров аутофагии [58]. К сожалению, исследователи не оценивали влияние ингибиторов аутофагии на инфаркт-лимитирующий эффект периодического голодания, поэтому нельзя утверждать, что этот эффект связан с активацией аутофагии.
Голодание стимулирует аутофагию, этот эффект сопровождается повышением толерантности сердца к действию И/Р.
Роль активных форм кислорода в аутофагии. Исследования проводили на изолированных кардиомиоцитах крыс и мышей. H2O2 усиливал аутофагию изолированных кардиомиоцитов [59]. Антиоксидант N-2-mercaptopropionyl glycine (MPG) устранял этот эффект H2O2, что говорит об участии активных форм кислорода (АФК) в индукции аутофагии. У мышей воспроизводили коронароокклюзию (45 мин) и реперфузию (2 и 24 ч). MPG вводили внутрибрюшинно за 24 и 1 ч до коронароокклюзии. Миокардиальная И/Р приводила к увеличению количества аутофагосом и аутолизосом. Предварительное введение MPG вызывало уменьшение количества аутофагосом, снижение экспрессии беклина-1 и уменьшение соотношения РИ/ОР [59]. Эти данные указывают на то, что окислительный стресс стимулирует аутофагию, которая принимает участие в И/Р повреждении сердца. Способность H2O2 стимулировать аутофагию кардиомиоцитов была подтверждена другими исследователями [60, 61]. “Ловушка” супероксидных радикалов MnTMPyP устраняла подъем уровня LC3-II после И/Р сердца [32]. В исследовании, выполненном на изолированных кардиомиоцитах, MnTMPyP устранял подъем количества аутофагосом, вызванный Г/Р [32]. Эти данные указывают на то, что супероксидный радикал может быть триггером аутофагии. Авторы связывают образование АФК при Г/Р с увеличением экспрессии беклина-1. Снижение уровня беклина-1, по их мнению, способствует процессингу аутофагосом в том числе за счет увеличения уровня белка LAMP2, что снижает гибель клеток при Г/Р [32]. В исследовании, выполненном на изолированых кардиомиоцитах, было показано, что депривация глюкозы вызывает усиление экспрессии НАДФН-оксидазы изоформы Nox-4 [62], которая, как известно, генерирует супероксидный радикал [63]. Поэтому депривация глюкозы сопровождается увеличением продукции АФК, одновременно увеличивается соотношение LC3-II/LC3-I, а уровень белка Р62, напротив, снижается. Нокаут Nox-4 приводит к уменьшению количества АФК и уменьшению соотношения LC3-II/LC3-I, а также к снижению уровня P62 и повышению количества аутолизосом [62]. Представленные данные указывают на то, что супероксидный радикал, генерируемый Nox-4, выступает в роли триггера аутофагии. Изолированное сердце морской свинки подвергали глобальной ишемии (30 мин) и реперфузии (150 мин) [64]. Сердце перед ишемией перфузировали в течение 10 мин раствором, содержащим севофлуран. В качестве “ловушки” АФК использовали MPG. Севофлуран способствовал уменьшению размера инфаркта, а MPG устранял этот эффект севофлурана. Севофлуран увеличивал количество аутофагических вакуолей, увеличивал соотношение LC3-II/LC3-I, повышал количество фосфорилированной (активной) AMPK. Антиоксидант MPG устранял все указанные эффекты [64]. Эти данные говорят о том, что АФК и AMPK играют ключевую роль в севофлуран-индуцированной аутофагии.
Таким образом, окислительный стресс стимулирует аутофагию при И/Р сердца. Супероксидный радикал, генерируемый Nox-4, выступает в роли триггера аутофагии, по-видимому, за счет активации AMPK (рис. 1).
Роль киназ в аутофагии. В экспериментах на изолированных кардиомиоцитах имитировали ишемию, удаляя из среды инкубации глюкозу, что приводило к появлению аутофагии [65]. Ингибитор аутофагии 3-метиладенин снижал выживаемость кардиомиоцитов в условиях депривации глюкозы. Ингибирование AMPK приводило к угнетению аутофагии [65]. Эти факты говорят о том, что AMPK играет пусковую роль в аутофагии, а аутофагия играет протекторную роль в условиях депривации глюкозы. Известно, что ингибирование GSK-3β (glycogen synthase kin-ase-3β) защищает сердце от действия И/Р [47]. Исследования проводили на обычных мышах (wild-type); на трансгенных мышах с доминантной мутацией гена, кодирующего GSK-3β (Tg-DnGSK-3β); гетерозиготных “нокаутированных” мышах (GSK-3β+/–) и на мышах с генетической активацией GSK-3β (βKI) [47]. Генетическое ингибирование GSK-3β приводило к уменьшению соотношения РИ/ОР. Активация GSK-3β, напротив, способствовала увеличению соотношения РИ/ОР. Ингибирование GSK-3β снижало аутофагию по рапамицин-чувствительному (mTOR-зависимому) механизму. Рапамицин усиливал аутофагию и устранял кардиопротекторный эффект генетического ингибирования GSK-3β [47]. Авторы полагают, что ингибирование GSK-3β повышает толерантность сердца к И/Р за счет ингибирования аутофагии. Следующее исследование проводили на обычных мышах (wild-type) и животных mTOR-Tg с избыточной экспрессией mTOR [66]. Ишемическое и реперфузионное повреждение сердца воспроизводили на двух моделях: (1) in vivo коронароокклюзия и реперфузия; (2) in vitro глобальная И/Р изолированного сердца. Смертность в течение 28 дней была меньше у mTOR-Tg по сравнению с обычными мышами. Эхокардиографическое исследование выявило, что функция сердца лучше сохраняется у mTOR-Tg. Исследование in vitro показало, что функциональное восстановление улучшается в сердцах mTOR-Tg [66]. У трансгенных мышей меньше соотношение РИ/ОР. Экспрессия провоспалительных цитокинов меньше у mTOR-Tg. Эти данные указывают на то, что избыточная экспрессия ингибитора аутофагии mTOR оказывает кардиопротекторный и противовоспалительный эффект. Показано, что И/Р ушка правого предсердия человека сопровождается активацией аутофагии, появлением фосфорилированной (активной) AMPK и снижением экспрессии mTOR, которая ингибирует аутофагию [12]. Следующее исследование было сделано на клетках H9c2, которые подвергали воздействию гипоксии (8 ч) реоксигенации. Ацетилхолин использовали в качестве цитопротекторного соединения [67]. Ацетилхолин-индуцированная аутофагия сопровождалась увеличением уровня фосфорилированной (активной) AMPK и снижением уровня фосфорилированной киназы mTOR. AMPK siРНК способствовала снижению LC3-II/LC3-I и сопровождалась исчезновением цитопротекторного эффекта ацетилхолина. Эти данные показывают важную роль AMPK в индукции аутофагии. Атипичная серин/треонинкиназа mTOR представлена двумя изоформами: mTORC1 и mTORC2 [68]. В регуляции аутофагии принимает участие mTORC1 [68]. Известно, что киназа JNK (c-Jun N-terminal kinase) участвует в активации апоптоза и вовлечена в повреждение сердца при И/Р [69]. Показано, что митохондриальная JNK индуцирует аутофагию, апоптоз и усугубляет ишемическое и реперфузионное повреждение сердца [69]. Вортманнин, ингибитор PI3-киназы, не влиял на уровень LC3-II в изолированных кардиомиоцитах в условиях нормоксии и аноксии/реоксигенации [39]. Установлено, что Г/Р кардиомиобластов H9c2 приводит к появлению p-AMPK и p-mTOR [36]. Коронароокклюзия без реперфузии приводила к активации аутофагии, повышению уровня p-AMPK и снижению содержания p-mTOR в миокарде [37]. Изолированные неонатальные кардиомиоциты мышей подвергали Г/Р [70]. Активатор AMPK PT1 и ингибитор mTOR 3HOI-BA-01 стимулировали аутофагию и повышали выживаемость кардиомиоцитов в условиях Г/Р. Эти данные подтверждают участие AMPK и mTOR в регуляции аутофагии. В исследовании, выполненном на изолированных кардиомиоцитах мыши, было показано, что Г/Р приводит к увеличению уровня беклина-1, LC3-II, снижению уровня P62, что рассматривается исследователями как активация аутофагии [71]. Ингибитор ERK киназы U0126 вызывал прямо противоположные изменения. Авторы полагают, что активация киназ MEK и ERK приводит к стимуляции аутофагии кардиомиоцитов в условиях Г/Р. Показано, что симвастатин оказывает кардиопротекторный эффект у пациентов с кардиохирургическим вмешательством, сопровождающимся И/Р сердца [17]. Симвастатин ингибировал аутофагию и увеличивал уровень p-AMPK. Авторы заключили, что симвастатин уменьшает повреждение миокарда за счет ингибирования аутофагии и активации AMPK [17]. Совместное культивирование васкулярных гладкомышечных клеток (ВГК) способствует выживанию клеток H9c2 в условиях гипоксии [18]. Цитопротекторный эффект осуществляется за счет секреции bFGF (basic fibroblast growth factor), который активирует PI3-киназу и киназу Akt. Эти киназы, по мнению исследователей, ингибируют аутофагию и апоптоз кардиомиоцитов [18].
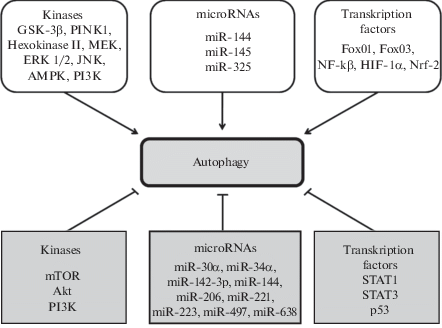
Таким образом, есть основания полагать, что AMPK, GSK-3β, JNK, MEK и ERK стимулируют аутофагию, а mTOR, Akt и PI3-киназа ингибируют аутофагию при И/Р сердца (рис. 1, 2). Вместе с тем, есть данные о том, что PI3-киназа является триггером аутофагии [48].
Рис. 2.
Роль киназ, транскрипционных факторов и микроРНК в регуляции аутофагии кардиомиоцитов в условиях ишемии/реперфузии сердца. Fig. 2. The role of kinases, transcription factors and miRNAs in the regulation of autophagy of cardiomyocytes in conditions of ischemia/reperfusion of the heart.
Роль транскрипционных факторов в аутофагии. Показано, что избыточная экспрессия транскрипционных факторов FoxO1 и FoxO3 усиливает аутофагию в кардиомиоцитах [29]. Изучали роль транскрипционного фактора STAT1 (signal transducer and activator of transcription 1) в аутофагии [72]. Было показано, что нокаут гена, кодирующего STAT1, способствует уменьшению размера инфаркта миокарда, что сопровождается увеличением соотношения LC3-II/LC3-I, увеличением экспрессии белков Atg12 и беклин-1, участвующих в аутофагии. Ингибитор аутофагии 3-метиладенин устранял кардиопротекторный эффект дефицита STAT1 [72]. Представленные данные указывают на то, что STAT1 контролирует процессы аутофагии, которые играют протекторную роль при И/Р сердца. Другое исследование проводили на обычных мышах (wild-type) и мышах с нокаутом гена TP53 p53(–/–) [73]. Через 8 ч после коронароокклюзии у мышей p53(–/–) количество аутофагических вакуолей, содержащих митохондрии, в пограничной зоне было в 5 раз больше, чем у обычных мышей. Через 28 дней после перманентной коронароокклюзии область фиброза у мышей p53(–/–) была на 34% меньше, чем у обычных мышей. У нокаутированных мышей в зоне инфаркта было меньше TUNEL-позитивных (апоптотических) клеток. Ингибитор аутофагии хлороквин устранял антапоптотический эффект у мышей p53(–/–) и устранял позитивный эффект нокаута гена TP53 в отношении постинфарктного ремоделирования сердца [73]. Эти факты говорят о том, что транскрипционный фактор p53 может регулировать процессы аутофагии и апоптоза во время ишемии миокарда. Установлено, что транскрипционный фактор NF-κB (nuclear factor κ-light-chain-enhancer of activated B cells) участвует в аутофагии [74]. У кроликов воспроизводили коронароокклюзию (1.5 ч) и реперфузию (1 ч). Ишемия/реперфузия приводила к увеличению уровня малонового диальдегида (МДА) в области риска и в зоне невосстановленного кровотока. Ингибитор NF-κB (pyrrolidine dithiocarbamate (PDTC)) устранял подъем уровня МДА. Ишемия/реперфузия приводила к увеличению в области риска уровня р65, который является субъединицей NF-κB. Уровень беклина-1 также увеличивался в зоне риска. Ингибитор NF-κB PDTC устранял подъем уровня р65 и беклина-1 [74]. Представленные данные свидетельствуют, что И/Р способствует активации NF-κB, увеличению продукции АФК и стимуляции аутофагии. Транскрипционный фактор NF-κB, по всей видимости, способствует аутофагии. Данные других исследователей подтверждают эту точку зрения [75]. В исследовании, выполненном на мышах, воспроизводили коронароокклюзию (30 мин) и реперфузию (24 ч) [75]. Исследование проводили на обычных мышах (wild-type) и трансгенных животных с доминантной негативной мутацией IκBα (2M), что приводило к нарушению экспрессии NF-κB. Кратковременная диета, богатая жирами (a high-fat diet), в течение 24 ч, 7 дней оказывала инфаркт-лимитирующий эффект, который исчезал, если диета продолжалась в течение 6 недель. Высокожировая диета приводила к усилению экспрессии беклина-1 и LC3. Мутация IκBα приводила к исчезновению инфаркт-лимитирующего эффекта жировой диеты и уменьшению уровня беклина-1 и LC3 [75]. Представленные данные свидетельствуют, что NF-κB принимает участие в кадиопротекторном эффекте жировой диеты. Кроме того, эти данные позволяют утверждать, что NF-κB регулирует аутофагию. Исследование проводили на кардиомиобластах H9c2, которые подвергали Г/Р [76]. Было показано, что Г/Р приводит к увеличению соотношения LC3-II/LC3-I, повышению уровня беклина-1, Atg5, Atg6. Избыточная экспрессия HIF-1α (hypoxia-inducible factor 1α) в этих условиях приводила к дальнейшему усилению аутофагии, а нокаут HIF-1α вызывал ингибирование аутофагии. Индуктор аутофагии рапамицин повышал выживаемость клеток H9c2 в условиях Г/Р, так же действовала избыточная экспрессия HIF-1α. Нокаут HIF-1α способствовал гибели клеток в условиях Г/Р [76]. Эти данные свидетельствуют, что HIF-1α стимулирует аутофагию кардиомиоцитов, что может способствовать увеличению их выживаемости в условиях Г/Р.
Установлено, что экспрессия транскрипционных факторов FoxO1, FoxO3, NF-κB и HIF-1α усиливает аутофагию при И/Р сердца (рис. 1, 2). Транскрипционные факторы STAT1 и p53 ингибируют аутофагию в условиях И/Р сердца.
МикроРНК и аутофагия. МикроРНК – это некодирующие короткие молекулы РНК, способные ингибировать трансляцию мРНК [77]. Показано, что коронароокклюзия (30 мин) и реперфузия (2 ч) способствуют увеличению уровня LC3-II в миокарде [78]. Кроме того, исследователи установили, что И/Р способствует снижению в миокарде уровня miR-204, которая регулирует экспрессию LC3-II. Исследование, выполненное на трансгенных мышах с избыточной экспрессией miR-325, показало, что повышение уровня miR-325 способствует увеличению соотношения LC3-II/LC3-I и размера инфаркта при коронароокклюзии и реперфузии [79]. Нокаут miR-325, напротив, ингибирует аутофагию. Дистантное прекондиционирование воспроизводили у мышей с помощью четырех циклов ишемии (5 мин) и реперфузии (5 мин) конечностей перед изоляцией сердца [80]. Изолированное сердце подвергали глобальной ишемии (30 мин) и реперфузии (60 мин). Дистантное ИПре способствовало увеличению уровня miR-144 в миокарде, а И/Р вызывала снижение уровня miR-144 почти до нуля. Прекондиционирование приводило к уменьшению размера инфаркта, а внутривенное введение ати-miR-144 устраняло этот кардиопротекторный эффект. Внутривенное введение miR-144 вызывало уменьшение размера инфаркта. miR-144 способствовала уменьшению уровня фосфорилированной mTOR и mTOR, увеличивала уровень Atg5 и катепсина L, соотношение LC3-II/LC3-I и уровень P62 при этом не менялись [80]. Представленные данные свидетельствуют о том, что miR-144 повышает толерантность сердца к действию И/Р и стимулирует аутофагию. Исследование было выполнено на мышах и изолированных кардиомиоцитах крыс [81]. У мышей воспроизводили коронароокклюзию (45 мин) и реперфузию (24 ч). Кардиомиоциты подвергали аноксии (3–24 ч) и реоксигенации (2 ч). С помощью трансфекции индуцировали избыточную экспрессию miR-497 в кардиомиоцитах или, наоборот, снижали экспрессию miR-497. С помощью аденовирусной плазмиды индуцировали избыточную экспрессию miR-497 in vivo или, напротив, подавляли экспрессию miR-497. Ингибирование экспрессии miR-497 способствовало уменьшению соотношения РИ/ОР, а избыточная экспрессия miR-497 вызывала увеличение соотношения РИ/ОР. Избыточная экспрессия miR-497 снижала выживаемость кардиомиоцитов при аноксии/реоксигенации, способствовала снижению уровня LC3-II, беклина-1, антиапоптотического белка Bcl-2, снижала количество аутофагосом и усиливала апоптоз кардиомиоцитов во время аноксии/реоксигенации. Подавление экспрессии miR-497 снижало апоптоз кардиомиоцитов во время аноксии/реоксигенации и способствовало увеличению уровня LC3-II, беклина-1, Bcl-2 и уменьшало уровень P62 и проапоптотического белка Bax [81]. Представленные данные свидетельствуют, что miR-497 снижает устойчивость кардиомиоцитов к Г/Р, усиливает апоптоз и ингибирует аутофагию кардиомиоцитов. Другое исследование было выполнено на пациентах с острым инфарктом миокарда (ОИМ) и кардиомиобластах H9c2, которые подвергали гипоксии (0.5, 2, 4, 12 и 24 ч) [41]. В экзосомах больных ОИМ было обнаружено большое количество miR-30a, эта же РНК была найдена в культуральной среде клеток H9c2 после воздействия гипоксии. Кратковременная гипоксия (4 и 12 ч) не влияла на уровень miR-30a в кардиомиобластах. В экзосомах в культуральной среде клеток H9c2 miR-30a повышалась уже через 4 ч гипоксии. Диметиламлорид, ингибитор формирования экзосом, предупреждал появление miR-30a в экзосомах. Гипоксия/реоксигенация приводила к усилению экспрессии Atg12, беклина-1 и увеличению соотношения LC3-II/LC3-I. Максимум изменений наблюдался через 30 мин и 2 ч гипоксии. Ингибирование экспрессии miR-30a вело к усилению аутофагии [41]. Представленные данные указывают на то, что miR-30a ингибирует аутофагию кардиомиоцитов в условиях Г/Р. Кардиомиобласты H9c2 и неонатальные кардиомиоциты крыс подвергали Г/Р [82]. Использовали трансфекцию miR-221 и ингибитор miR-221-i. Было показано, что miR-221 оказывает цитопротекторный эффект, снижает соотношение LC3-II/LC3-I, уменьшает количество аутофагосом, повышает уровень P62 и не влияет на уровень беклина-1. miR-221-i повышает соотношение LC3-II/LC3-I и уровень P62 [82]. Следовательно, miR-221 оказывает цитопротекторный эффект и ингибирует аутофагию. У крыс индуцировали коронароокклюзию (30 мин) и реперфузию (120 мин) [46]. Фактор роста фибробластов (FGF21) вводили внутрибрюшинно в течение 4 недель. FGF21 оказывал инфаркт-лимитирующий и антиапоптотический эффект. Исследователи обнаружили снижение в миокарде уровня miR-145, беклина-1 и снижение соотношения LC3-II/LC3-I при И/Р сердца, то есть было обнаружено угнетение аутофагии. FGF21 повышал уровень miR-145 и стимулировал аутофагию. В исследовании, выполненном на H9c2 клетках, которые подвергали Г/Р, было показано, что FGF21 увеличивает уровень беклина-1 и соотношение LC3-II/LC3-I. Трансфекция клеток ингибитором miR-145 (miR-145 inhibitor) приводила к ингибированию аутофагии, индуцированной FGF21. Ингибитор miR-145 устранял антиапоптотический эффект FGF21 in vitro [46]. Представленные данные свидетельствуют, что кардиопротекторный эффект FGF21 может быть следствием увеличения уровня miR-145, которая стимулирует аутофагию. Кардиомиоциты человека подвергали гипоксии (4 ч) и реоксигенации (4 ч) [83]. С помощью трансфекции обеспечивали избыточную экспрессию miR-638 или ингибирование экспрессии miR-638. Г/Р вызвала гибель кардиомиоцитов, апоптоз и снижение уровня miR-638. В условиях Г/Р избыточная экспрессия miR-638 способствовала увеличению выживаемости и снижала апоптоз кардиомиоцитов. Избыточная экспрессия miR-638 способствовала снижению уровня Atg5, а miR-638-ингибитор увеличивал уровень Atg5. К сожалению, исследователи не определяли другие маркеры аутофагии [83]. Представленные данные указывают на то, что miR-638 повышает толерантность кардиомиоцитов к Г/Р и, возможно, ингибирует аутофагию.
Таким образом, установлено, что miR-325 и miR-144 симулируют аутофагию при И/Р сердца. miR-325 увеличивает размер инфаркта. miR-144 повышает толерантность сердца к действию И/Р. miR-497 снижает устойчивость кардиомиоцитов к Г/Р, усиливает апоптоз и ингибирует аутофагию кардиомиоцитов. miR-30a ингибирует аутофагию кардиомиоцитов в условиях Г/Р. miR-221 оказывает цитопротекторный эффект и ингибирует аутофагию. miR-145 стимулирует аутофагию in vitro. Таким образом, miR-325, miR-145 и miR-144 симулируют аутофагию, а miR-30a, miR-221, miR-638 и miR-144 ингибируют этот процесс (рис. 1, 2).
H2S как регулятор аутофагии. В исследовании, выполненном на крысах, воспроизводили коронароокклюзию (30 мин) и реперфузию (4 ч) [19]. Использовали H2S-донор ADT (5-(4-methoxyphenyl)-3H-1,2-dithiole-3-thione) и ингибитор AMPK компаунд С. И/Р приводила к увеличению в миокарде уровня беклина-1, P62, соотношения LC3-II/LC3-I при снижении уровня LAMP-2. ADT при И/Р ограничивал размер инфаркта, способствовал увеличению уровня p-AMPK, вызывал снижение уровня беклина-1, P62, соотношения LC3-II/LC3-I и повышал уровень LAMP-2. В то же время ADT не влиял на уровень маркеров аутофагии у ложно-оперированных животных. Компаунд С устранял инфаркт-лимитирующий эффект ADT, снижал уровень p-AMPK, нивелировал изменения маркеров аутофагии, вызванные введением ADT, что свидетельствует об участии AMPK в развитии эффектов ADT [19]. Представленные данные свидетельствуют, что H2S способствует ингибированию аутофагии и повышению толерантности сердца к действию И/Р. Дальнейшие исследования проводили на изолированных неонатальных кардиомиоцитах крыс, которые подвергали гипоксии (24 ч) и реоксигенации (6 ч) [40]. В качестве донора H2S использовали NaHS. Было показано, что H2S увеличивает выживаемость кардиомиоцитов и ингибирует аутофагию. Ингибитор PI3-киназы LY294002 устранял ингибирование аутофагии и ослаблял, но не устранял цитопротекторный эффект H2S. Представленные данные свидетельствуют, что PI3-киназа участвует в цитопротекторном эффекте H2S и обеспечивает ингибирование аутофагии под действием H2S. Кроме того, способность H2S ингибировать аутофагию связывают с активацией mTOR [84]. Вместе с тем, есть данные о том, что H2S может стимулировать аутофагию, если исследование проводилось на старых животных [85].
Представленные данные свидетельствуют, что H2S повышает толерантность сердца к действию И/Р и может ингибировать аутофагию у молодых животных, но стимулирует аутофагию у старых животных (рис. 1).
Гемоксигеназа-1 и аутофагия. Известно, что гемоксигеназа-1 (ГO-1) оказывает антиапоптотический эффект за счет синтеза CO [86]. В исследовании, выполненном на клетках H9c2, которые подвергали Г/Р, было показано, что избыточная экспрессия ГO-1 способствует увеличению уровня LC3II и снижению уровня P62. Кроме того, ГO-1 предупреждала падение митохондриального мембранного потенциала, а уровень АФК в митохондриях снижался. Авторы сделали вывод, что ГO-1 стимулирует аутофагию и предупреждает повреждение митохондрий при Г/Р [86] (рис. 1).
Позитивная и негативная роль аутофагии. Изолированное перфузируемое сердце кролика подвергали гипоксической перфузии (95% N2 + 5% CO2) в течение 20, 40 и 60 мин [27]. При часовой гипоксической перфузии повреждения носили необратимый характер. Реперфузия сердца после гипоксической перфузии вела к усилению аутофагии. Авторы предположили, что аутофагия играет важную роль в репарации кардиомиоцитов во время реоксигенации [27]. В исследовании, выполненном в 2010 г, свиней подвергали коронароокклюзии (45 мин) и реперфузии (3 ч). Стимулятор аутофагии хлорамфеникол (20 мг/кг) вводили до ишемии [31]. Кроме того, хлорамфеникол вводили через 30 мин после окклюзии. Хлорамфеникол уменьшал соотношение РИ/ОР как при введении перед ишемией, так и при инъекции после ишемии. При введении перед ишемией инфаркт-лимитирующий эффект был сильнее, чем при инъекции после ишемии. Хлорамфеникол усиливал экспрессию беклина-1 и LC3-II. Эти факты указывают на то, что инфаркт-лимитирующий эффект хлорамфеникола может быть следствием стимуляции аутофагии. По мнению авторов, индукция аутофагии может быть новой стратегией, направленной на повышение толерантности сердца к действию И/Р [31, 87]. Полагают, что активация аутофагии во время ишемии важна для выживания клеток и сохранения функции сердца [23]. Считают, что активация аутофагии во время реперфузии может быть протекторной либо повреждающей в зависимости от экспериментальной модели [23]. Показано, что индуктор аутофагии рапамицин улучшал сократимость изолированного сердца во время реперфузии [88]. Бафиломицин, ингибитор аутофагии, увеличивал размер инфаркта при коронароокклюзии [22]. Показано, что рапамицин оказывает инфаркт-лимитирующий эффект у мышей [57]. Индуктор аутофагии рапамицин не влиял на смерть изолированных кардиомиоцитов во время реоксигенации, а ингибитор аутофагии хлороквин усиливал гибель кардиомиоцитов во время реоксигенации [32]. Вместе с тем, есть данные о том, что хлороквин не влияет на соотношение РИ/ОР при коронароокклюзии и реперфузии [21, 33]. Показано, что рапамицин у мышей с коронароокклюзией (35 мин) и реперфузией (4 ч) стимулирует аутофагию, уменьшает соотношение РИ/ОР на 45% и снижает интенсивность апоптоза в зоне риска [89]. Эти факты указывают на то, что аутофагия имеет протекторное значение при И/Р сердца. Однако к результатам исследований с использованием рапамицина нужно относиться осторожно, поскольку есть данные о том, что его кардиопротекторный эффект может быть связан с активацией PI3-киназы и митохондриальных АТФ-чувствительных К+-каналов, а не с блокадой mTOR [90]. Клетки H9c2 подвергали воздействию гипоксии (8 ч) реоксигенации (4, 8, 12 и 16 ч). По мере увеличения продолжительности реоксигенации снижалось количество аутофагосом и уменьшалось количество жизнеспособных клеток [67]. Ацетилхолин во время реоксигенации увеличивал количество аутофагосом и соотношение LC3-II/LC3-I, но уменьшал количество белка P62, что авторы расценивают как усиление аутофагического потока. Атропин устранял эти эффекты ацетилхолина, что говорит об участии М-холинорецепторов. Индукция аутофагии коррелировала с увеличением выживаемости клеток и уменьшением апоптоза. Ингибирование аутофагии с помощью хлороквина или Atg7 siРНК сопровождалось ослаблением цитопротекторного эффекта ацетилхолина [67]. Эти данные свидетельствуют о цитопротекторном эффекте аутофагии. Другое исследование было выполнено на неонатальных кардиомиоцитах мыши, которые подвергались Г/Р [91]. Показано, что активатор AMPK D942 и неселективный ингибитор mTOR куркумин стимулировали аутофагию и повышали выживаемость кардиомиоцитов в условиях Г/Р. Эти данные указывают на протекторную роль аутофагии. У крыс воспроизводили коронароокклюзию (30 мин) и реперфузию (120 мин) [92]. Антагонист рецепторов ангиотензина II валсартан вводили ежедневно в течение 7 дней до коронароокклюзии. Валсартан способствовал уменьшению соотношения РИ/ОР и увеличивал соотношение LC3-II/LC3-I. Ингибитор аутофагии 3-МА устранял инфаркт-лимитирующий эффект валсартана и устранял подъем соотношения LC3-II/LC3-I [92]. Эти данные указывают на то, что кардиопротекторный эффект валсартана может быть следствием активации аутофагии. Исследования проводили на обычных мышах (wild-type) и нокаутированных животных (Perkin–/–) [93]. Симвастатин вводили внутрибрюшинно за 4 ч до коронароокклюзии (20 мин) и реперфузии (22 ч). Симвастатин оказывал инфаркт-лимитирующий эффект у обычных мышей, но не влиял на размер инфаркта у нокаутированных животных. Представленные данные показывают, что паркин и митофагия играют позитивную роль в инфаркт-лимитирующем эффекте симвастатина. Следующее исследование проводили на мышах и кроликах с коронароокклюзией и реперфузией [94]. Кроме того, проводили исследования на изолированных неонатальных кардиомиоцитах крыс, которые подвергались Г/Р. Использовали ингибитор гистондиацилазы SAHA (suberoylanilide hydroxamic acid), который ограничивал размер инфаркта. SAHA снижал смерть клеток на 40%. SAHA индуцировал увеличение LC3-II, повышал количество аутофагосом и аутолизосом в пограничной зоне инфаркта. Atg5 siРНК и Atg7 siРНК устраняли цитопротекторный SAHA [94]. Эти данные указывают на то, что усиление аутофагии под действием SAHA может иметь отношение к его цитопротекторному эффекту. Изолированное предсердие крысы подвергали ишемии (75 мин) и реперфузии (75 мин) [95]. Ингибирование аутофагии с помощью 3-МА способствовало увеличению частоты возникновения тахиаритмий во время реперфузии и снижало инотропный эффект стимуляции β-адренорецепторов [95]. Перманентная коронароокклюзия вызывала у мышей в первый день повышение уровня беклина-1 и соотношения LC3-II/LC3-I, позднее эти показатели возвращались к значениям, характерным для ложнооперированных животных [96]. Ежедневно животным вводили рапамицин или 3-МА. Активатор аутофагии рапамицин способствовал уменьшению размера инфаркта через 7 и 21 дней после окклюзии. Кроме того, рапамицин способствовал увеличению фракции выброса левого желудочка. 3-Метиладенин, напротив, увеличивал размер инфаркта через 7 и 21 дней после окклюзии. Авторы заключили, что рапамицин ослабляет, а 3-МА усугубляет постинфарктное ремоделирование сердца. Аутофагия является механизмом, который защищает сердце от неблагоприятного ремоделирования [96]. Будет ли фармакологический агент, стимулирующий аутофагию, повышать толерантность кардиомиоцитов к Г/Р или, напротив, усугублять гибель клеток, во многом зависит от использованной модели Г/Р. Так, показано, что индуктор аутофагии рапамицин увеличивал выживаемость неонатальных кардиомиоцитов крысы в условиях аноксии (6 ч) и реоксигенации (4 ч), но снижал выживаемость кардиомиоцитов, если продолжительность аноксии составляла 24 ч с последующей реоксигенацией [39]. Показано, что рапамицин повышает выживаемость клеток H9c2 в условиях Г/Р [76]. Кардиопротекторный эффект многих соединений связывают с усилением аутофагии [97–103].
Вместе с тем, есть данные о том, что аутофагия усугубляет ишемическое и реперфузионное повреждение сердца. Изолированные неонатальные кардиомиоциты и кардиомиоциты взрослых крыс подвергали Г/Р, что вызывало активацию аутофагии [25]. Авторы использовали siРНК, которая ингибирует экспрессию беклина-1. Оказалось, что эта РНК ингибировала аутофагию кардиомиоцитов и увеличивала выживаемость клеток в условиях Г/Р. Пептид урокортин, который повышает толерантность кардиомиоцитов к действию Г/Р, также ингибировал аутофагию и экспрессию беклина-1 [25]. Этот эффект урокортина был связан с активацией PI3-киназы/Akt-киназы и не зависел от киназы ERK1/2. Исследования проводили на изолированном перфузируемом сердце мыши, которое подвергали глобальной ишемии (25 мин) и реперфузии (80 мин) [104]. Было обнаружено, что уровень беклина-1 и соотношение LC3-II/LC3-I выше в сердцах с фибрилляцией по сравнению с сердцами без фибрилляции. Этот результат позволил авторам сделать вывод о возможной взаимосвязи фибрилляции желудочков и аутофагии [104]. Гипоксия/реоксигенация клеток H9c2 приводила к увеличению экспрессии беклина-1 и увеличению соотношения LC3-II/LC3-I [105]. Липоиковая кислота ингибировала аутофагию и увеличивала выживаемость клеток. Эти данные говорят о негативной роли аутофагии в регуляции толерантности кардиомиоцитов к действию Г/Р [105]. Показано, что белок нетрин-1 в дозе 5 и 10 мкг/кг уменьшал соотношение РИ/ОР, но не влиял на размер инфаркта в дозе 50 мкг/кг [106]. Кроме того, нетрин-1 снижал соотношение LC3-II/LC3-I, что авторы расценивают, как ингибирование аутофагии. Эти данные показывают, что препарат может ограничивать размер инфаркта и одновременно угнетать аутофагию. Показано, что улинастатин (ulinastatin) повышает толерантность сердца крыс к действию И/Р in vivo [107]. Это эффект сопровождался снижением соотношения LC3-II/LC3-I и фосфорилированием mTOR. Следовательно, инфаркт-лимитирующий эффект улинастатина сопровождается снижением интенсивности аутофагии. Есть данные о том, что кальцитриол, активная форма витамина D, повышает устойчивость сердца к И/Р, ингибируя апоптоз и аутофагию кардиомиоцитов [108]. Список статей, в которых различные соединения оказывают кардиопротекторный эффект и ингибируют аутофагию, можно продолжить [16, 18, 19, 34, 36, 38, 83, 109–111]. Эти соединения часто ингибируют не только аутофагию, но и апоптоз [16, 18, 34, 38, 83, 110].
Следовательно, активация аутофагии во время И/Р может быть протекторной или повреждающей в зависимости от экспериментальной модели.
ЗАКЛЮЧЕНИЕ
Большинство данных свидетельствует, что И/Р сердца приводит к усилению аутофагического потока. Представленные данные показывают, что ИПре стимулирует аутофагию за счет активации AMPK и PI3-киназы при ингибировании mTOR. Кардиопротекторный эффект ИПост связан с активацией аутофагии и повышением активности NOS и AMPK. И/Р стимулирует аутофагию, а гонадэктомия ее подавляет. Адаптация к гипоксии оказывает кардиопротекторный эффект, который, возможно, связан с активацией аутофагии. Возможно, что негативный эффект жировой диеты на толерантность сердца к И/Р связан с ингибированием аутофагии. Голодание стимулирует аутофагию, этот эффект сопровождается повышением толерантности сердца к действию И/Р. Окислительный стресс стимулирует аутофагию при И/Р сердца. Супероксидный радикал, генерируемый Nox-4, выступает в роли триггера аутофагии, по-видимому, за счет активации AMPK. Есть основания полагать, что AMPK, GSK-3β, JNK, MEK и ERK стимулируют аутофагию, а mTOR, Akt и PI3-киназа ингибируют этот процесс при И/Р сердца. Вместе с тем, есть данные о том, что PI3-киназа является триггером аутофагии. Установлено, что экспрессия транскрипционных факторов FoxO1, FoxO3, NF-κB и HIF-1α усиливает аутофагию при И/Р сердца. Транскрипционные факторы STAT1 и p53 ингибируют аутофагию в условиях И/Р сердца. miR-325, miR-145 и miR-144 стимулируют аутофагию, а miR-30a, miR-221, miR-638 и miR-144 ее ингибируют при И/Р сердца. Представленные данные свидетельствуют, что H2S повышает толерантность сердца к действию И/Р и может ингибировать аутофагию у молодых животных, но стимулирует аутофагию у старых животных. HO-1 стимулирует аутофагию и предупреждает повреждение митохондрий при Г/Р кардиомиоцитов. В зависимости от модели, которую выбрали исследователи, аутофагия может носить защитный или повреждающий характер. Конечный результат, то есть ограничение размера инфаркта, зависит не только от того, как влияет то или иное соединение на аутофагию, но и от того, как оно влияет на апоптоз, некроптоз и некроз кардиомиоцитов при И/Р сердца.
Список литературы
De Duve C., Pressman B.C., Gianetto R., Wattiaux R., Appelmans F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J. 60(4): 604–617. 1955.
Bainton D.F. The discovery of lysosomes. J. Cell Biol. 91(3 Pt 2): 66s–76s. 1981.
De Duve C. Lysosomes revisited. Eur. J. Biochem. 137(3): 391–397. 1983.
Ashford T.P., Porter K.R. Cytoplasmic components in hepatic cell lysosomes. J. Cell Biol. 12: 198–202. 1962.
Klionsky D.J. Autophagy revisited: A conversation with Christian de Duve. Autophagy. 4(6): 740–743. 2008.
Radewa J. Observations on autophagocytosis phenomena in the blood. Z Rheumaforsch. 22: 36–46. 1963.
Deter R.L., Baudhuin P., De Duve C. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J. Cell Biol. 35(2): C11–C16. 1967.
Deter R.L., De Duve C. Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J. Cell Biol. 33(2): 437–449. 1967.
Sciarretta S., Maejima Y., Zablocki D., Sadoshima J. The role of autophagy in the heart. Annu. Rev. Physiol. 80: 1–26. 2018.
Shintani T., Klionsky D.J. Autophagy in health and disease: a double-edged sword. Science. 306(5698): 990–995. 2004.
Ravikumar B., Sarkar S., Davies J.E., Futter M., Garcia-Arencibia M., Green-Thompson Z.W., Jimenez-Sanchez M., Korolchuk V.I., Lichtenberg M., Luo S., Massey D.C., Menzies F.M., Moreau K., Narayanan U., in physiology and pathophysiology. Physiol. Rev. 90(4): 1383–1435. 2010.
Renna M., Siddiqi F.H., Underwood B.R., Winslow A.R., Rubinsztein D.C. Regulation of mammalian autophagy Singh K.K., Yanagawa B., Quan A., Wang R., Garg A., Khan R., Pan Y., Wheatcroft M.D., Lovren F., Teoh H., Verma S. Autophagy gene fingerprint in human ischemia and reperfusion. J. Thorac. Cardiovasc. Surg. 147(3): 1065–1072. 2014.
Huang C., Andres A.M., Ratliff E.P., Hernandez G., Lee P., Gottlieb R.A. Preconditioning involves selective mitophagy mediated by parkin and p62/SQSTM1. PLoS One. 6(6): e20975. 2011.
Vélez D.E., Hermann R., Barreda Frank M., Mestre Cordero V.E., Savino E.A., Varela A., Marina Prendes M.G. Effects of wortmannin on cardioprotection exerted by ischemic preconditioning in rat hearts subjected to ischemia-reperfusion. J. Physiol. Biochem. 72(1): 83–91. 2016.
García-Rúa V., Feijóo-Bandín S., Rodríguez-Penas D., Mosquera-Leal A., Abu-Assi E., Beiras A., María Seoane L., Lear P., Parrington J., Portolés M., Roselló-Lletí E., Rivera M., Gualillo O., Parra V., Hill J.A., Rothermel B., González-Juanatey J.R., Lago F. Endolysosomal two-pore channels regulate autophagy in cardiomyocytes. J. Physiol. 594(11): 3061–3077. 2016.
Wang B., Zhong S., Zheng F., Zhang Y., Gao F., Chen Y., Lu B., Xu H., Shi G. N-n-butyl haloperidol iodide protects cardiomyocytes against hypoxia/reoxygenation injury by inhibiting autophagy. Oncotarget. 6(28): 24709–24721. 2015.
Hua P., Liu J., Tao J., Zou R., Lin X., Zhang D., Yang S. Efficacy and mechanism of preoperative simvastatin therapy on myocardial protection after extracorporeal circulation. Biomed. Res. Int. 2017: 6082430. 2017.
Ye G., Fu Q., Jiang L., Li Z. Vascular smooth muscle cells activate PI3K/Akt pathway to attenuate myocardial ischemia/reperfusion-induced apoptosis and autophagy by secreting bFGF. Biomed. Pharmacother. 107: 1779–1785. 2018.
Xie H., Xu Q., Jia J., Ao G., Sun Y., Hu L., Alkayed N.J., Wang C., Cheng J. Hydrogen sulfide protects against myocardial ischemia and reperfusion injury by activating AMP-activated protein kinase to restore autophagic flux. Biochem. Biophys. Res. Commun. 458(3): 632–638. 2015.
Shao J., Miao C., Geng Z., Gu M., Wu Y., Li Q. Effect of eNOS on ischemic postconditioning-induced autophagy against ischemia/reperfusion injury in mice. Biomed. Res. Int. 2019: 5201014. 2019.
Zhao T., Huang X., Han L., Wang X., Cheng H., Zhao Y., Chen Q., Chen J., Cheng H., Xiao R., Zheng M. Central role of mitofusin 2 in autophagosome-lysosome fusion in cardiomyocytes. J. Biol. Chem. 287(28): 23615–23625. 2012.
Kanamori H., Takemura G., Goto K., Maruyama R., Ono K., Nagao K., Tsujimoto A., Ogino A., Takeyama T., Kawaguchi T., Watanabe T., Kawasaki M., Fujiwara T., Fujiwara H., Seishima M., Minatoguchi S. Autophagy limits acute myocardial infarction induced by permanent coronary artery occlusion. Am. J. Physiol. Heart Circ. Physiol. 300(6): H2261–H2271. 2011.
Qian J., Ren X., Wang X., Zhang P., Jones W.K., Molkentin J.D., Fan G.C., Kranias E.G. Blockade of Hsp20 phosphorylation exacerbates cardiac ischemia/reperfusion injury by suppressed autophagy and increased cell death. Circ. Res. 105(12): 1223–1231. 2009.
De Meyer G.R., Martinet W. Autophagy in the cardiovascular system. Biochim. Biophys. Acta. 1793(9): 1485–1495. 2009.
Valentim L., Laurence K.M., Townsend P.A., Carroll C.J., Soond S., Scarabelli T.M., Knight R.A., Latchman D.S., Stephanou A. Urocortin inhibits Beclin1-mediated autophagic cell death in cardiac myocytes exposed to ischaemia/reperfusion injury. J. Mol. Cell Cardiol. 40(6): 846–852. 2006.
Yan L., Vatner D.E., Kim S.J., Ge H., Masurekar M., Massover W.H., Yang G., Matsui Y., Sadoshima J., Vatner S.F. Autophagy in chronically ischemic myocardium. Proc. Natl. Acad. Sci. USA. 102(39): 13807–13812. 2005.
Decker R.S., Wildenthal K. Lysosomal alterations in hypoxic and reoxygenated hearts. I. Ultrastructural and cytochemical changes. Am. J. Pathol. 98(2): 425–444. 1980.
Sybers H.D., Ingwall J., DeLuca M. Autophagy in cardiac myocytes. Recent Adv. Stud. Cardiac. Struct. Metab. 12: 453–463. 1976.
Sengupta A., Molkentin J.D., Yutzey K.E. FoxO transcription factors promote autophagy in cardiomyocytes. J. Biol. Chem. 284(41): 28319–28331. 2009.
Yan L., Sadoshima J., Vatner D.E., Vatner S.F. Autophagy in ischemic preconditioning and hibernating myocardium. Autophagy. 5(5): 709–712. 2009.
Sala-Mercado J.A., Wider J., Undyala V.V., Jahania S., Yoo W., Mentzer R.M. Jr., Gottlieb R.A., Przyklenk K. Profound cardioprotection with chloramphenicol succinate in the swine model of myocardial ischemia-reperfusion injury. Circulation. 122(11 Suppl): S179–S184. 2010.
Ma X., Liu H., Foyil S.R., Godar R.J., Weinheimer C.J., Hill J.A., Diwan A. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation. 125(25): 3170–3181. 2012.
Zhang Y.L., Yao Y.T., Fang N.X., Zhou C.H., Gong J.S., Li L.H. Restoration of autophagic flux in myocardial tissues is required for cardioprotection of sevoflurane postconditioning in rats. Acta Pharmacol. Sin. 35(6): 758–769. 2014.
Wang Z.G., Wang Y., Huang Y., Lu Q., Zheng L., Hu D., Feng W.K., Liu Y.L., Ji K.T., Zhang H.Y., Fu X.B., Li X.K., Chu M.P., Xiao J. bFGF regulates autophagy and ubiquitinated protein accumulation induced by myocardial ischemia/reperfusion via the activation of the PI3K/Akt/mTOR pathway. Sci. Rep. 5: 9287. 2015.
Guo L., Xu J.M., Mo X.Y. Ischemic postconditioning regulates cardiomyocyte autophagic activity following ischemia/reperfusion injury. Mol. Med. Rep. 12(1): 1169–1176. 2015.
Huang Z., Han Z., Ye B., Dai Z., Shan P., Lu Z., Dai K., Wang C., Huang W. Berberine alleviates cardiac ischemia/reperfusion injury by inhibiting excessive autophagy in cardiomyocytes. Eur. J. Pharmacol. 762: 1–10. 2015.
Ma M.Q., Thapalia B.A., Lin X.H. A 6 hour therapeutic window, optimal for interventions targeting AMPK synergism and apoptosis antagonism, for cardioprotection against myocardial ischemic injury: an experimental study on rats. Am. J. Cardiovasc. Dis. 5(1): 63–71. 2015.
Yu P., Zhang J., Yu S., Luo Z., Hua F., Yuan L., Zhou Z., Liu Q., Du X., Chen S., Zhang L., Xu G. Protective effect of sevoflurane postconditioning against cardiac ischemia/reperfusion injury via ameliorating mitochondrial impairment, oxidative stress and rescuing autophagic clearance. PLoS One. 10(8): e0134666. 2015.
Xu Q., Li X., Lu Y., Shen L., Zhang J., Cao S., Huang X., Bin J., Liao Y. Pharmacological modulation of autophagy to protect cardiomyocytes according to the time windows of ischaemia/reperfusion. Br. J. Pharmacol. 172(12): 3072–3085. 2015.
Jiang H., Xiao J., Kang B., Zhu X., Xin N., Wang Z. PI3K/SGK1/GSK3β signaling pathway is involved in inhibition of autophagy in neonatal rat cardiomyocytes exposed to hypoxia/reoxygenation by hydrogen sulfide. Exp. Cell Res. 345(2): 134–140. 2016.
Yang Y., Li Y., Chen X., Cheng X., Liao Y., Yu X. Exosomal transfer of miR-30a between cardiomyocytes regulates autophagy after hypoxia. J. Mol. Med. (Berl). 94(6): 711–724. 2016.
Wang J.J., Bie Z.D., Sun C.F. Long noncoding RNA AK088388 regulates autophagy through miR-30a to affect cardiomyocyte injury. J. Cell Biochem. 120(6): 10155–10163. 2019.
Gurusamy N., Lekli I., Mukherjee S., Ray D., Ahsan M.K., Gherghiceanu M., Popescu L.M., Das D.K. Cardioprotection by resveratrol: a novel mechanism via autophagy involving the mTORC2 pathway. Cardiovasc. Res. 86(1): 103–112. 2010.
French C.J., Taatjes D.J., Sobel B.E. Autophagy in myocardium of murine hearts subjected to ischemia followed by reperfusion. Histochem. Cell Biol. 134(5): 519–526. 2010.
Cao X., Wang X., Ling Y., Song X., Yang P., Liu Y., Liu L., Wang L., Guo J., Chen A. Comparison of the degree of autophagy in neonatal rat cardiomyocytes and H9c2 cells exposed to hypoxia/reoxygenation. Clin. Lab. 60(5): 809–814. 2014.
Hu S., Cao S., Tong Z., Liu J. FGF21 protects myocardial ischemia-reperfusion injury through reduction of miR-145-mediated autophagy. Am. J. Transl. Res. 10(11): 3677–3688. 2018.
Heusch G. Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ. Res. 116(4): 674–699. 2015.
Gurusamy N., Lekli I., Gorbunov N.V., Gherghiceanu M., Popescu L.M., Das D.K. Cardioprotection by adaptation to ischaemia augments autophagy in association with BAG-1 protein. J. Cell Mol. Med. 13(2): 373–387. 2009.
Gedik N., Thielmann M., Kottenberg E., Peters J., Jakob H., Heusch G., Kleinbongard P. No evidence for activated autophagy in left ventricular myocardium at early reperfusion with protection by remote ischemic preconditioning in patients undergoing coronary artery bypass grafting. PLoS One. 9(5): e96567. 2014.
Rohailla S., Clarizia N., Sourour M., Sourour W., Gelber N., Wei C., Li J., Redington A.N. Acute, delayed and chronic remote ischemic conditioning is associated with downregulation of mTOR and enhanced autophagy signaling. PLoS One. 9(10): e111291. 2014.
Dosenko V.E., Nagibin V.S., Tumanovskaya L.V., Zagoriy V.Y., Moibenko A.A., Vaage J. Proteasome inhibitors eliminate protective effect of postconditioning in cultured neonatal cardiomyocytes. Fiziol. Zh. 52(3): 15–24. 2006.
Wagner C., Tillack D., Simonis G., Strasser R.H., Weinbrenner C. Ischemic post-conditioning reduces infarct size of the in vivo rat heart: role of PI3-K, mTOR, GSK-3beta, and apoptosis. Mol. Cell Biochem. 339(12): 135–147. 2010.
Wei C., Li H., Han L., Zhang L., Yang X. Activation of autophagy in ischemic postconditioning contributes to cardioprotective effects against ischemia/reperfusion injury in rat hearts. J. Cardiovasc. Pharmacol. 61(5): 416–422. 2013.
Chen C., Hu L.X., Dong T., Wang G.Q., Wang L.H., Zhou X.P., Jiang Y., Murao K., Lu S.Q., Chen J.W., Zhang G.X. Apoptosis and autophagy contribute to gender difference in cardiac ischemia-reperfusion induced injury in rats. Life Sci. 93(7): 265–270. 2013.
Le T.Y., Ashton A.W., Mardini M., Stanton P.G., Funder J.W., Handelsman D.J., Mihailidou A.S. Role of androgens in sex differences in cardiac damage during myocardial infarction. Endocrinology. 155(2): 568–575. 2014.
Hu Y., Sun Q., Li Z., Chen J., Shen C., Song Y., Zhong Q. High basal level of autophagy in high-altitude residents attenuates myocardial ischemia-reperfusion injury. J. Thorac. Cardiovasc. Surg. 148(4): 1674–1680. 2014.
Sciarretta S., Zhai P., Shao D., Maejima Y., Robbins J., Volpe M., Condorelli G., Sadoshima J. Rheb is a critical regulator of autophagy during myocardial ischemia: pathophysiological implications in obesity and metabolic syndrome. Circulation. 125(9): 1134–1146. 2012.
Godar R.J., Ma X., Liu H., Murphy J.T., Weinheimer C.J., Kovacs A., Crosby S.D., Saftig P., Diwan A. Repetitive stimulation of autophagy-lysosome machinery by intermittent fasting preconditions the myocardium to ischemia-reperfusion injury. Autophagy. 11(9): 1537–1560. 2015.
Hariharan N., Zhai P., Sadoshima J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid. Redox. Signal. 14(11): 2179–2190. 2011.
Sengupta A., Molkentin J.D., Paik J.H., DePinho R.A., Yutzey K.E. FoxO transcription factors promote cardiomyocyte survival upon induction of oxidative stress. J. Biol. Chem. 286(9): 7468–7478. 2011.
Liu L., Jin X., Hu C.F., Li R., Zhou Z., Shen C.X. Exosomes derived from mesenchymal stem cells rescue myocardial ischaemia/reperfusion injury by inducing cardiomyocyte autophagy via AMPK and Akt pathways. Cell Physiol. Biochem. 43(1): 52–68. 2017.
Sciarretta S., Zhai P., Shao D., Zablocki D., Nagarajan N., Terada L.S., Volpe M., Sadoshima J. Activation of NADPH oxidase 4 in the endoplasmic reticulum promotes cardiomyocyte autophagy and survival during energy stress through the protein kinase RNA-activated-like endoplasmic reticulum kinase/eukaryotic initiation factor 2α/activating transcription factor 4 pathway. Circ. Res. 113(11): 1253–1264. 2013.
Krylatov A.V., Maslov L.N., Voronkov N.S., Boshchenko A.A., Popov S.V., Gomez L., Wang H., Jaggi A.S., Downey J.M. Reactive oxygen species as intracellular signaling molecules in the cardiovascular system. Curr. Cardiol. Rev. 14(4): 290–300. 2018.
Shiomi M., Miyamae M., Takemura G., Kaneda K., Inamura Y., Onishi A., Koshinuma S., Momota Y., Minami T., Figueredo V.M. Sevoflurane induces cardioprotection through reactive oxygen species-mediated upregulation of autophagy in isolated guinea pig hearts. J. Anesth. 28(4): 593–600. 2014.
Matsui Y., Takagi H., Qu X., Abdellatif M., Sakoda H., Asano T., Levine B., Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res. 100(6): 914–922. 2007.
Aoyagi T., Kusakari Y., Xiao C.Y., Inouye B.T., Takahashi M., Scherrer-Crosbie M., Rosenzweig A., Hara K., Matsui T. Cardiac mTOR protects the heart against ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 303(1): H75–H85. 2012.
Zhao M., Sun L., Yu X.J., Miao Y., Liu J.J., Wang H., Ren J., Zang W.J. Acetylcholine mediates AMPK-dependent autophagic cytoprotection in H9c2 cells during hypoxia/reoxygenation injury. Cell Physiol. Biochem. 32(3): 601–613. 2013.
Sciarretta S., Volpe M., Sadoshima J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ. Res. 114(3): 549–564. 2014.
Xu J., Qin X., Cai X., Yang L., Xing Y., Li J., Zhang L., Tang Y., Liu J., Zhang X., Gao F. Mitochondrial JNK activation triggers autophagy and apoptosis and aggravates myocardial injury following ischemia/reperfusion. Biochim. Biophys. Acta. 1852(2): 262–270. 2015.
Huang L., Dai K., Chen M., Zhou W., Wang X., Chen J., Zhou W. The AMPK agonist PT1 and mTOR inhibitor 3HOI-BA-01 protect cardiomyocytes after ischemia through induction of autophagy. J. Cardiovasc. Pharmacol. Ther. 21(1): 70–81. 2016.
Wang A., Zhang H., Liang Z., Xu K., Qiu W., Tian Y., Guo H., Jia J., Xing E., Chen R., Xiang Z., Liu J. U0126 attenuates ischemia/reperfusion-induced apoptosis and autophagy in myocardium through MEK/ERK/EGR-1 pathway. Eur. J. Pharmacol. 788: 280–285. 2016.
McCormick J., Suleman N., Scarabelli T.M., Knight R.A., Latchman D.S., Stephanou A. STAT1 deficiency in the heart protects against myocardial infarction by enhancing autophagy. J. Cell Mol. Med. 16(2): 386–393. 2012.
Hoshino A., Matoba S., Iwai-Kanai E., Nakamura H., Kimata M., Nakaoka M., Katamura M., Okawa Y., Ariyoshi M., Mita Y., Ikeda K., Ueyama T., Okigaki M., Matsubara H. p53-TIGAR axis attenuates mitophagy to exacerbate cardiac damage after ischemia. J. Mol. Cell Cardiol. 52(1): 175–184. 2012.
Zeng M., Wei X., Wu Z., Li W., Li B., Zhen Y., Chen J., Wang P., Fei Y. NF-κB-mediated induction of autophagy in cardiac ischemia/reperfusion injury. Biochem. Biophys. Res. Commun. 436(2): 180–185. 2013.
Haar L., Ren X., Liu Y., Koch S.E., Goines J., Tranter M., Engevik M.A., Nieman M., Rubinstein J., Jones W.K. Acute consumption of a high-fat diet prior to ischemia-reperfusion results in cardioprotection through NF-κB-dependent regulation of autophagic pathways. Am. J. Physiol. Heart Circ. Physiol. 307(12): H1705–P1713. 2014.
Gui L., Liu B., Lv G. Hypoxia induces autophagy in cardiomyocytes via a hypoxia-inducible factor 1-dependent mechanism. Exp. Ther. Med. 11(6): 2233–2239. 2016.
Fromm B., Billipp T., Peck L.E., Johansen M., Tarver J.E., King B.L., Newcomb J.M., Sempere L.F., Flatmark K., Hovig E., Peterson K.J. A uniform system for the annotation of vertebrate microRNA genes and the evolution of the human microRNAome. Annu. Rev. Genet. 49: 213–242. 2015.
Xiao J., Zhu X., He B., Zhang Y., Kang B., Wang Z., Ni X. MiR-204 regulates cardiomyocyte autophagy induced by ischemia-reperfusion through LC3-II. J. Biomed. Sci. 18: 35. 2011.
Bo L., Su-Ling D., Fang L., Lu-Yu Z., Tao A., Stefan D., Kun W., Pei-Feng L. Autophagic program is regulated by miR-325. Cell Death Differ. 21(6): 967–977. 2014.
Li J., Rohailla S., Gelber N., Rutka J., Sabah N., Gladstone R.A., Wei C., Hu P., Kharbanda R.K., Redington A.N. MicroRNA-144 is a circulating effector of remote ischemic preconditioning. Basic Res. Cardiol. 109(5): 423. 2014.
Li X., Zeng Z., Li Q., Xu Q., Xie J., Hao H., Luo G., Liao W., Bin J., Huang X., Liao Y. Inhibition of microRNA-497 ameliorates anoxia/reoxygenation injury in cardiomyocytes by suppressing cell apoptosis and enhancing autophagy. Oncotarget. 6(22): 18829–18844. 2015.
Chen Q., Zhou Y., Richards A.M., Wang P. Up-regulation of miRNA-221 inhibits hypoxia/reoxygenation-induced autophagy through the DDIT4/mTORC1 and Tp53inp1/p62 pathways. Biochem. Biophys. Res. Commun. 474(1): 168–174. 2016.
Zhao P., Zhang B.L., Liu K., Qin B., Li Z.H. Overexpression of miR-638 attenuated the effects of hypoxia/reoxygenation treatment on cell viability, cell apoptosis and autophagy by targeting ATG5 in the human cardiomyocytes. Eur. Rev. Med. Pharmacol. Sci. 22(23): 8462–8471. 2018.
Xiao J., Zhu X., Kang B., Xu J., Wu L., Hong J., Zhang Y., Ni X., Wang Z. Hydrogen sulfide attenuates myocardial hypoxia-reoxygenation injury by inhibiting autophagy via mTOR activation. Cell Physiol. Biochem. 37(6): 2444–2453. 2015.
Chen J., Gao J., Sun W., Li L., Wang Y., Bai S., Li X., Wang R., Wu L., Li H., Xu C. Involvement of exogenous H2S in recovery of cardioprotection from ischemic post-conditioning via increase of autophagy in the aged hearts. Int. J. Cardiol. 220: 681–692. 2016.
Chen D., Jin Z., Zhang J., Jiang L., Chen K., He X., Song Y., Ke J., Wang Y. HO-1 protects against hypoxia/reoxygenation-induced mitochondrial dysfunction in H9c2 cardiomyocytes. PLoS One. 11(5): e0153587. 2016.
Przyklenk K., Undyala V.V., Wider J., Sala-Mercado J.A., Gottlieb R.A., Mentzer R.M Jr. Acute induction of autophagy as a novel strategy for cardioprotection: getting to the heart of the matter. Autophagy. 7(4): 432–433. 2011.
Loos B., Genade S., Ellis B., Lochner A., Engelbrecht A.M. At the core of survival: autophagy delays the onset of both apoptotic and necrotic cell death in a model of ischemic cell injury. Exp. Cell Res. 317(10): 1437–1453. 2011.
Chen H.H., Mekkaoui C., Cho H., Ngoy S., Marinelli B., Waterman P., Nahrendorf M., Liao R., Josephson L., Sosnovik D.E. Fluorescence tomography of rapamycin-induced autophagy and cardioprotection in vivo. Circ. Cardiovasc. Imaging. 6(3): 441–447. 2013.
Yang S.S., Liu Y.B., Yu J.B., Fan Y., Tang S.Y., Duan W.T., Wang Z., Gan R.T., Yu B. Rapamycin protects heart from ischemia/reperfusion injury independent of autophagy by activating PI3 kinase-Akt pathway and mitochondria KATP channel. Pharmazie. 65(10): 760–765. 2010.
Yang K., Xu C., Li X., Jiang H. Combination of D942 with curcumin protects cardiomyocytes from ischemic damage through promoting autophagy. J. Cardiovasc. Pharmacol. Ther. 18(6): 570–581. 2013.
Wu X., He L., Cai Y., Zhang G., He Y., Zhang Z., He X., He Y., Zhang G., Luo J. Induction of autophagy contributes to the myocardial protection of valsartan against ischemia‑reperfusion injury. Mol. Med. Rep. 8(6): 1824–1830. 2013.
Andres A.M., Hernandez G., Lee P., Huang C., Ratliff E.P., Sin J., Thornton C.A., Damasco M.V., Gottlieb R.A. Mitophagy is required for acute cardioprotection by simvastatin. Antioxid. Redox Signal. 21(14): 1960–1973. 2014.
Xie M., Kong Y., Tan W., May H., Battiprolu P.K., Pedrozo Z., Wang Z.V., Morales C., Luo X., Cho G., Jiang N., Jessen M.E., Warner J.J., Lavandero S., Gillette T.G., Turer A.T., Hill J.A. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation. 129(10): 1139–1151. 2014.
Hermann R., Vélez D.E., Rusiecki T.M., Fernández Pazos Mde L., Mestre Cordero V.E., Marina Prendes M.G., Perazzo Rossini J.C., Savino E.A., Varela A. Effects of 3-methyladenine on isolated left atria subjected to simulated ischaemia-reperfusion. Clin. Exp. Pharmacol. Physiol. 42(1): 41–51. 2015.
Wu X., He L., Chen F., He X., Cai Y., Zhang G., Yi Q., He M., Luo J. Impaired autophagy contributes to adverse cardiac remodeling in acute myocardial infarction. PLoS One. 9(11): e112891. 2014.
Zhang J., Nadtochiy S.M., Urciuoli W.R., Brookes P.S. The cardioprotective compound cloxyquin uncouples mitochondria and induces autophagy. Am. J. Physiol. Heart Circ. Physiol. 310(1): H29–H38. 2016.
Liu L., Wu Y., Huang X. Orientin protects myocardial cells against hypoxia-reoxygenation injury through induction of autophagy. Eur. J. Pharmacol. 776: 90–98. 2016.
Ma Y., Gai Y., Yan J., Li J., Zhang Y. Puerarin attenuates anoxia/reoxygenation injury through enhancing Bcl-2 associated athanogene 3 expression, a modulator of apoptosis and autophagy. Med. Sci. Monit. 22: 977–983. 2016.
Wang Y., Yang Z., Zheng G., Yu L., Yin Y., Mu N., Ma H. Metformin promotes autophagy in ischemia/reperfusion myocardium via cytoplasmic AMPKα1 and nuclear AMPKα2 pathways. Life Sci. 225: 64–71. 2019.
Zhao R., Xie E., Yang X., Gong B. Alliin alleviates myocardial ischemia-reperfusion injury by promoting autophagy. Biochem. Biophys. Res. Commun. 512(2): 236–243. 2019.
Ren Z., Xiao W., Zeng Y., Liu M.H., Li G.H., Tang Z.H., Qu S.L., Hao Y.M., Yuan H.Q., Jiang Z.S. Fibroblast growth factor-21 alleviates hypoxia/reoxygenation injury in H9c2 cardiomyocytes by promoting autophagic flux. Int. J. Mol. Med. 43(3): 1321–1330. 2019.
Qiao S.G., Sun Y., Sun B., Wang A., Qiu J., Hong L., An J.Z., Wang C., Zhang H.L. Sevoflurane postconditioning protects against myocardial ischemia/reperfusion injury by restoring autophagic flux via an NO-dependent mechanism. Acta Pharmacol. Sin. 40(1): 35–45. 2019.
Meyer G., Czompa A., Reboul C., Csepanyi E., Czegledi A., Bak I., Balla G., Balla J., Tosaki A., Lekli I. The cellular autophagy markers Beclin-1 and LC3B-II are increased during reperfusion in fibrillated mouse hearts. Curr. Pharm. Des. 19(39): 6912–6918. 2013.
Cao X., Chen A., Yang P., Song X., Liu Y., Li Z., Wang X., Wang L., Li Y. Alpha-lipoic acid protects cardiomyocytes against hypoxia/reoxygenation injury by inhibiting autophagy. Biochem Biophys. Res. Commun. 441(4): 935–940. 2013.
Bouhidel J.O., Wang P., Siu K.L., Li H., Youn J.Y., Cai H. Netrin-1 improves post-injury cardiac function in vivo via DCC/NO-dependent preservation of mitochondrial integrity, while attenuating autophagy. Biochim. Biophys. Acta. 1852(2): 277–289. 2015.
Xiao J., Zhu X., Ji G., Yang Q., Kang B., Zhao J., Yao F., Wu L., Ni X., Wang Z. Ulinastatin protects cardiomyocytes against ischemia‑reperfusion injury by regulating autophagy through mTOR activation. Mol. Med. Rep. 10(4): 1949–1953. 2014.
Yao T., Ying X., Zhao Y., Yuan A., He Q., Tong H., Ding S., Liu J., Peng X., Gao E., Pu J., He B. Vitamin D receptor activation protects against myocardial reperfusion injury through inhibition of apoptosis and modulation of autophagy. Antioxid. Redox. Signal. 22(8): 633–650. 2015.
Xie H., Liu Q., Qiao S., Jiang X., Wang C. Delayed cardioprotection by sevoflurane preconditioning: a novel mechanism via inhibiting Beclin 1-mediated autophagic cell death in cardiac myocytes exposed to hypoxia/reoxygenation injury. Int. J. Clin. Exp. Pathol. 8(1): 217–226. 2015b.
Cao J., Xie H., Sun Y., Zhu J., Ying M., Qiao S., Shao Q., Wu H., Wang C. Sevoflurane post-conditioning reduces rat myocardial ischemia reperfusion injury through an increase in NOS and a decrease in phopshorylated NHE1 levels. Int. J. Mol. Med. 36(6): 1529–1337. 2015.
Zhou L.Y., Zhai M., Huang Y., Xu S., An T., Wang Y.H., Zhang R.C., Liu C.Y., Dong Y.H., Wang M., Qian L.L., Ponnusamy M., Zhang Y.H., Zhang J., Wang K. The circular RNA ACR attenuates myocardial ischemia/reperfusion injury by suppressing autophagy via modulation of the Pink1/ FAM65B pathway. Cell Death Differ. 26(7): 1299–1315. 2019.
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова