

Российский физиологический журнал им. И.М. Сеченова, 2023, T. 109, № 10, стр. 1498-1514
Легочная артериальная гипертензия ослабляет вазоконстрикторные реакции, вызванные активацией альфа-1-адренорецепторов в большом круге кровообращения
А. А. Абрамов 1, *, В. Л. Лакомкин 1, Е. В. Лукошкова 1, А. В. Просвирнин 1, В. И. Капелько 1, В. С. Кузьмин 1, 2
1 Нaциональный медицинский исследовательский центр кардиологии им. акад. Е.И. Чазова
Москва, Россия
2 Московский государственный университет им. М.В. Ломоносова
Москва, Россия
* E-mail: ferk_88@list.ru
Поступила в редакцию 07.09.2023
После доработки 22.09.2023
Принята к публикации 25.09.2023
- EDN: XDTYSI
- DOI: 10.31857/S0869813923100023
Аннотация
Легочная артериальная гипертензия (ЛАГ) сопровождается подъемом давления в малом круге кровообращения, изменением его чувствительности к регуляторным факторам; для ЛАГ характерна активация симпатического отдела нервной и ренин-ангиотензин-альдостероновой систем. Изменение чувствительности сосудов системного круга кровообращения (СКК) к проконстрикторным воздействиям при ЛАГ не исследовано. В работе изучали вазоконстрикторные реакции в СКК при хронической монокроталиновой модели ЛАГ у крыс. Регистрировали среднее артериальное давление (САД) на фоне двойной автономной блокады при введении агониста α1-адренорецепторов (α1-АР) фенилэфрина (ФЭ) у бодрствующих крыс исходно и через 4 нед. после введения монокроталина (МКТ). Регистрацию САД при действии ФЭ проводили также на фоне ангиотензина-II (АТII). Оценивали максимальную величину (Аmах) изменения САД и наибольшее время полувозвращения САД (T∆САД1/2) к исходному уровню у крыс после введения ФЭ. У крыс ЛАГ через 4 нед. (n = 6) после введения МКТ не наблюдалось значимого изменения Аmах, однако T∆САД1/2 оказывалось значимо меньше, чем у контрольных животных. АТII приводит к увеличению Аmах и T∆САД1/2 у контрольных крыс. Однако у крыс с ЛАГ потенциация антиотензином Аmах и T∆САД1/2 не обнаруживается. Таким образом, у животных с ЛАГ снижается способность артерий большого круга поддерживать тонус в ответ на активацию α1-АР. ЛАГ подавляет способность АТII стимулировать симпатические ответы в СКК. Впервые in vivo на системном уровне продемонстрировано, что ремоделирование и изменение функционального состояния малого круга кровообращения приводит к перестройке регуляции сосудистого тонуса в большом круге кровообращения.
ВВЕДЕНИЕ
Легочная артериальная гипертензия (ЛАГ) является заболеванием, которое сопровождается обструктивными изменениями в сосудистом русле малого круга кровообращения, приводящими к росту правожелудочкового систолического давления, гипертрофии правого желудочка, увеличению легочного сосудистого сопротивления, нарушению циркуляции и оксигенации крови в малом круге кровообращения [1]. Большая часть как клинических, так и фундаментальных работ направлены на изучение механизмов формирования функциональных и структурных изменений в малом круге кровообращения, включая сосудистое русло легочных артерий, легочных вен, правый желудочек сердца. Однако к настоящему моменту накапливаются данные, свидетельствующие в пользу того, что множественные патологические изменениями в большом круге кровообращения сопутствуют ЛАГ. За пределами легочного круга кровообращения ЛАГ манифестирует в форме нарушенной поток-зависимой дилятации в ряде системных артерий [2], недостаточности церебральной циркуляции [3], сниженного почечного кровотока [4], миопатий, ассоциированных с уменьшением плотности микроциркуляторного русла [5]. Эти изменения, вероятно, обусловлены нарушениями нейрогуморального контроля и изменениями чувствительности элементов большого круга кровообращения к циркулирующим регуляторным факторам.
Хорошо известно, что при ЛАГ тонус симпатического отдела вегетативной нервной системы (ВНС) повышен [6]. Патологическую активацию симпатического отдела ВНС рассматривают как часть так называемого “нейрогуморального дисбаланса”, наблюдаемого при ЛАГ, который сопровождается изменениями “системной гемодинамики” [7].
Автономный нервный контроль и регуляция гемодинамики со стороны ренин-альдостерон-ангиотензиновой системы (РААС) тесно переплетены: повышенная продукция ангиотензина II (АТII) и активность РААС при ЛАГ вследствие роста легочного сосудистого сопротивления и падения ударного объема, способствует АТII-опосредованному усилению высвобождения норадреналина (НА) [8–10]. Предполагается, что симпатический отдел ВНС, как и РААС, вовлечены в патогенез ЛАГ и в проявления ЛАГ в большом круге [11]. Однако эти эффекты изучены в аспекте дисфункции и адренергической дисрегуляции правого и левого желудочков сердца за счет сдвига профиля экспрессии и вовлечения сигнальных путей β1/2/3-адренорецепторов миокарда [12].
В большинстве артерий большого круга кровообращения симпатическая стимуляция приводит к вазоконстрикции за счет активации адренорецепторов α1-типа (α1-АР) гладкомышечных клеток (ГМК) стенки сосудов [13]. Известно, что АТII потенцирует и амплифицирует вазоконстрикцию, вызванную норадреналином и опосредованную стимуляцией α1-АР [14–17]. Тем не менее остается неизвестным, изменяется ли при ЛАГ системная гемодинамика в результате перестройки чувствительности сосудистого русла большого круга кровообращения к регуляторным факторам.
Помимо РААС и ВНС, целый спектр системных регуляторных механизмов при ЛАГ может вносить вклад в “нейрогуморальный дисбаланс”. Например, при ЛАГ наблюдается рост уровня альдостерона и вазопрессина в плазме крови [18], что приводит к избыточной реабсорбции Na+ и гиперволюмии [19]. Известно, что ЛАГ сопровождается значительным повышением секреции натрийуретических пептидов (NP), изменением профиля и уровня экспрессии его рецепторов в ГМК [20], т.е. перестройкой регуляторной “оси” NP. Повышение уровня NP, вероятно, направлено на компенсацию эффектов РААС и вазопрессина.
Дисфункция эндотелия и снижение продукции вазодилятаторных факторов, в первую очередь оксида азота (NO), в системных и коронарных артериях типично для ЛАГ [5, 21]. ЛАГ-ассоциированное снижение продукции NO, вероятно, связано как с изменением транскрипционной активности [22], так и с ATII-индуцированной продукцией активных форм кислорода НАДФН-оксидазами [23, 24]. Таким образом, ЛАГ может приводить к многообразной и малопредсказуемой перестройке тесно взаимодействующих факторов, вовлеченных в регуляцию кровообращения. Механизмы и результирующие изменения реактивности сосудистого русла большого круга, обусловленные ЛАГ, практически не исследованы.
В данной работе исследуется, в какой степени вазоконстрикторные и дилятаторные реакции сосудов большого круга, вызванные симпатической стимуляцией, АТII или донорами NO, оказываются изменены при ЛАГ. Функциональные реакции сосудистого русла большого круга кровообращения исследованы in vivo с использованием хронической монокроталиновой модели легочной артериальной гипертензии на примере крыс.
МЕТОДЫ ИССЛЕДОВАНИЯ
Животные
Работа выполнена с использованием крыс (самцы стока Wistar, 2–4 мес., 290 ± 30 г, 25 животных). В ходе экспериментов были соблюдены все актуальные требования этических норм работы с лабораторными животными. Перед экспериментами животных содержали в виварии в течение 2 нед. в стандартных условиях при световом режиме 12:12 с доступом к воде и пище ad libitum. В течение 5 нед. после вживления катетеров и индукции ЛАГ животных содержали в тех же условиях, но в индивидуальных клетках.
Катетеризация
Для хронической регистрации артериального давления и инфузии вазоактивных соединений крыс контрольной и экспериментальной групп наркотизировали (золетил-100, в.б., 0.1 мл на 100 г массы тела животного). Крысам вживляли 3 сосудистых комбинированных полиуретановых катетера (Instechlabs, США): артериальный катетер (внутрисосудистая часть 4.5–5.5 см, BTPU-027(0.4 × 0.7 мм, 2 Fr), внесосудистая часть – BTPU-040 (0.6 × 1 мм, 3 Fr)) вживляли в левую бедренную артерию, 2 венозных катетера (внутрисосудистая часть 5–5.5 см, BTPU-027, 0.4 × 0.7 мм, 2 Fr) с дополнительной удлиненной силиконовой оболочкой – BTSIL-037 (для минимизации повреждения эндокарда) через левую яремную вену вводили в правое предсердия. Катетеризацию осуществляли стандартным способом. Катетеры подключали к трехканальному устройству прямого хронического сосудистого доступа (УСД, VABR3B/22, Instechlabs, США), которое имплантировали на холку в сформированный под кожей карман. Осуществляли постоперационную антисептическую обработку (стрептоцид, повидон-йод); в течение трех (пяти дней в случае гематурии) дней в период восстановления крысам вводили гентамицин (0.1 мл/100 г, в.б.).
Регистрация среднего артериального давления
Среднее артериальное давление (САД) регистрировали в модифицированной установке для работы с бодрствующими животными прямым методом. САД рассчитывали как частное площади под кривой давления за один кардиоцикл и его длительности [25]. Регистрацию САД осуществляли в одно и то же время суток в условиях приглушенного освещения и максимально возможного исключения стрессирующих факторов в “домашних” клетках, адаптированных для длительной регистрации давления и синхронной внутривенной инфузии подвижным животным (Instechlabs, CША), ответная часть – VABR3T/25, шарнир – 375/D/22, противовес – CM375BS. Для регистрации САД артериальный катетер и УСД подключали к датчику (Gould Statham 23PB, США) и мостовому усилителю (Biograph-4, Россия), сигнал от которого поступал на АЦП (National Instruments, США) и регистрировался на ПК при помощи ПО (National Instruments, LabView RunTime Engine) с частотой 10 КГц. Сигналы обрабатывали с помощью ПО (Data Acquisition, среда LabView, разработчик – Е.В. Лукошкова). Для предотвращения тромбообразования в артериальном катетере осуществляли его промывку 10%-ным раствором гепарина (5000 Ед) в физиологическом растворе (1–1.5 мкл/мин).
Индукция ЛАГ
Для индукции ЛАГ использовали стандартный подход [26], основанный на однократном введении монокроталина (МКТ, Sigma Aldrich, подкожно, 60 мг/кг, 0.3 мл). МКТ навешивали для каждого животного отдельно и растворяли в физиологическом растворе с титрованием 0.1 нормальной HCl до полного растворения осадка. Затем pH раствора МКТ доводили до 7 титрованием 0.2 нормальной NaOH. Животным контрольной группы вводили физиологический раствор (0.3 мл). В эксперимент животных брали по окончании периода восстановления через 6 сут после вживления катетеров. ЛАГ индуцировали только у тех животных, которые через 6 сут после вживления катетеров демонстрировали нормальный уровень среднего артериального давления (100 ± 10 мм рт. ст.), ЧСС покоя 360 ± 30 уд./мин, ректальную температуру 37 ± 0.5°С, а также суточный объем потребляемой жидкости (30 ± 10 мл).
Верификация ЛАГ методом эхокардиографии
Формирование экспериментальной ЛАГ верифицировали стандартным способом [27, 28] на основе ультразвуковых (УЗИ) признаков гипертрофии правого желудочка (ПЖ) и изменения функциональных показателей правого и левого желудочков (ЛЖ) сердца. УЗИ сердца проводили до инъекции МКТ, а затем на 14-й и 28-й день после введения МКТ (УЗИ аппарат Visual Sonics VEVO 1100 с линейным датчиком MS250, диапазон частот 12–24 МГц). Использовался лево- и правосторонний парастернальный доступ для построения позиций по длинной (1) и короткой (2) оси левого желудочка, по короткой оси на уровне основания сердца (3), по длинной оси легочного ствола (4). Определяли толщину свободной стенки ПЖ и диаметр легочной артерии.
При УЗИ-допплерографии измеряли систолический временной интервал AT/ET для правого желудочка как отношение длительности периода ускорения потока (АТ) к длительности фазы изгнания (ET) крови. Индекс сократимости ПЖ (TAPSE) оценивали (в М-режиме) по амплитуде систолической экскурсии трикупсидального кольца стандартным способом [29].
При эхокардиографическом исследовании (ЭХО-КГ) также определяли (В-режим) конечный диастолический и систолический объемы левого желудочка; рассчитывали фракцию выброса (ФВ) и ударный объем (УО) ЛЖ.
Инфузия вазоактивных соединений и протоколы экспериментов
Инфузию вазоактивных соединений осуществляли через внутривенные катетеры, подключенные к УСД при помощи шприцевых программируемых инфузионных помп (Harvard Apparatus Pump 11 Elite Infusion/Withdrawal, США) со шприцами Hamilton 1002 TLL (2.5 мл, байонетный затвор).
По окончании постоперационного периода восстановления, а также на 14-й и 28-й (рис. 1a) день после инъекции МКТ после 30-минутного периода адаптации и контрольной записи САД, животным вводили нитропруссид натрия (НП, Sigma Aldrich) в последовательно возрастающих дозах от 2.5 до 80 мкг/кг в объеме 0.3 мл за 3 мин с интервалами 10 или 20 мин (рис. 1b).
Рис. 1.
Моделирование ЛАГ (a) и схема (b) индукции вазомоторных ответов в сосудистом русле большого круга кровообращения. Синий цвет – формирующаяся ЛАГ (2 нед.), фиолетовый цвет – сформированная ЛАГ (4 нед.). Красная штриховка – инфузия ангиотензина II.
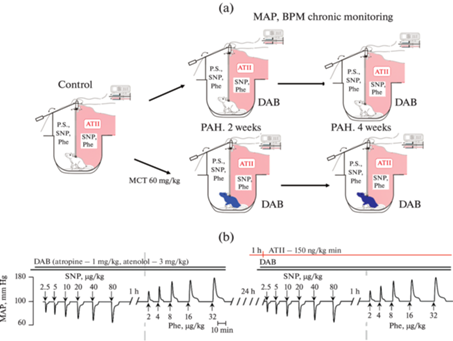
Не менее чем через 1 ч после окончания введения НП, стабилизации САД и промывки катетера осуществляли введение агониста α1-АР фенилэфрина (ФЭ, Sigma Aldrich) в последовательно возрастающих дозах от 2 до 32 мкг/кг в объеме 0.3 мл за 1 мин с интервалами 10–20 мин. Инфузию ФЭ и НП осуществляли на фоне предварительно индуцированной двойной автономной блокады (ДАБ) атенололом (3 мг/кг, в.в) и атропином (1 мг/кг, в.в), осуществляемой для подавления барорецепторного изменения ЧСС в ответ на введение вазоактивных соединений.
В части экспериментов, на следующие сутки после введения НП и ФЭ, повторяли эксперимент, но на фоне непрерывной инфузии ангиотензина II (150 нг/кг мин, разведен в 0.1%-ном растворе бычьего сывороточного альбумина), которую начинали за 60 мин до введения первой дозы НП (рис 1a, b). Для параллельной инфузии АТII использовали второй внутривенный катетер.
Оценка динамики вазоконстрикроных ответов в большом круге
Вычисляли максимальную возможную величину (амплитуду, Аmах) изменения САД и наибольшую длительность периода полувозвращения САД (T∆САД1/2) к исходному уровню у крыс в ответ на введение НП или ФЭ. Для каждого эксперимента, используя индивидуальные значения амплитуды изменения САД или индивидуальные значения времени полувозвращения САД, определяемые при введении каждой дозы вазоактивного соединения (рис. 2c), методом аппроксимации [30, 31] с помощью четырехпараметрической логистической кривой вида:
Рис. 2.
Репрезентативные примеры кривых зависимости амплитуды вазоконстрикторного ответа (a) или длительности периода “полувозвращения” САД (T∆САД1/2) (b) от дозы фенилэфрина у контрольных крыс (4 нед., группа “контроль/ДАБ”). На вставках показаны регрессионные уравнения. Красным и зеленым цветом указаны асимптотические максимальные значения амплитуды и длительности периода полувозвращения САД к исходному уровню. (с) – оригинальная запись изменения САД при действии фенилэфрина (ФЭ) и нитропруссида натрия (НП), использованная для построения репрезентативных кривых на a и b, амплитуда ответа отмечена черными вертикальными стрелками, длительность периода “полувозвращения” САД (T∆САД1/2) – горизонтальными красными стрелками.
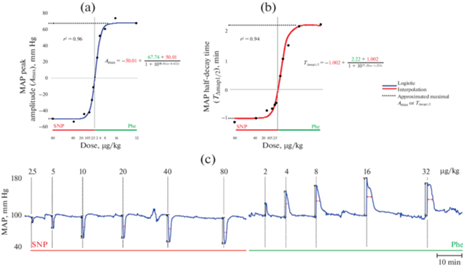
В качестве Аmах или T∆САД1/2 использовали асимптотические значения (равные коэффициентам с, d) регрессионной кривой (рис. 2a, b) для минимизации вариабельности межиндивидуальной широты диапазона реакций САД, а также вариабельности полумаксимальной эффективной концентрации вазоактивных соединений.
Статистический анализ
Статистическую обработку полученных результатов проводили в среде R [32, 33]. Подготовка данных осуществлялась средствами пакета tidyverse [34]. Статистически значимые различия между группами выявляли с помощью теста Фридмана для связанных выборок, либо теста Краскелла–Уоллеса для несвязанных (оба теста – непараметрическая альтернатива однофакторной ANOVA) с последующим применением апостериорных тестов (Вилкоксона для связанных выборок, Манна–Уитни для несвязанных выборок) для множественных сравнений в группах с повторными или независимыми, а также внесением поправки Холма. Различия считали значимыми при p < 0.05. Статистические тесты проводились с помощью пакета rstatix [35]. Данные представлены как медиана ± абсолютное отклонение медианы за исключением репрезентативных записей. Визуализация данных выполнена средствами пакетов ggplot2 [36], gghalves [37] и patchwork [38].
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Морфологические и функциональные признаки ЛАГ у крыс после введения МКТ
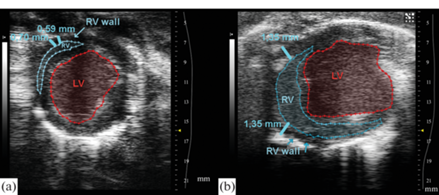
Однократная инъекция МКТ приводила к возникновению морфологических и функциональных признаков формирования ЛАГ у крыс. Толщина стенки правого желудочка (ТсПЖ) увеличивалась от 0.651 ± 0.076 до 0.9 ± 0.15 и 0.9 ± 0.27 мм через 2 и 4 нед. после введения МКТ (n = 7, p < 0.01) соответственно (рис. 3). Синхронно с изменениями ТсПЖ, увеличивался диаметр легочной артерии: от 2.46 ± 0.13 до 2.8 ± 0.19 мм через 2 нед. и до 3.4 ± 0.37 мм (n = 7, p < 0.01) через 2 и 4 нед. после введения МКТ соответственно.
Рис. 3.
Оценка толщины стенки правого желудочка (RV) при УЗИ сердца. (a) – репрезентативный пример УЗИ-изображения стенки правого желудочка крысы исходно. (b) – то же, что и на (а), для крысы через 4 нед. после инъекции монокроталина и индукции ЛАГ. Голубой линией показана толщина стенки правого желудочка, определяемая по короткой оси. Левый желудочек на уровне средних сегментов левого желудочка в момент окончания диастолы.
Индекс сократимости ПЖ (TAPSE) снижался до 1 ± 0.13 и 1.1 ± 0.15 от 1.3 ± ± 0.15 мм (n = 7, p < 0.01) соответственно к 2 и 4 нед. после инъекции МКТ. Отношение AT/ET у крыс через 2 и 4 нед. после введения МКТ снижался от 0.3 ± 0.05 до 0.23 ± 0.07 и 0.21 ± 0.04 (n = 7, p < 0.01) через 2 и 4 нед. после введения МКТ, что указывает на прогрессирующий рост сосудистого сопротивления в малом круге кровообращения.
При формировании ЛАГ у крыс не происходило значимого изменения ударного объема и фракции выброса левого желудочка: УО до введения МКТ и через 4 нед. после его инъекции составляло 280 ± 35 и 310 ± 38 мкл (n = 7, p > 0.1); ФВ до введения МКТ – 67 ± 9%; через 4 нед. – 72 ± 8% (n = 7, p > 0.1).
Максимальная величина изменения САД при действии фенилэфрина у крыс ЛАГ
В контрольной группе животных через 4 нед. после начала эксперимента САД в условиях покоя составляло 105 ± 11 мм рт. ст. (n = 6). У крыс с ЛАГ через 4 нед. после введения МКТ (n = 8) САД мало отличалось от такового у контрольных животных (109 ± 14 мм рт. ст., р > 0.1). В контрольных условиях введение ФЭ на фоне ДАБ приводило быстрому дозозависимому, но транзиторному увеличению САД. Болюсное введение ФЭ в дозах 2–32 мкг/кг вызывало рост САД, пиковое значение которого (от 100 ±1 1 до 180 ± 12 мм рт. ст. при действии 32 мкг/кг ФЭ) достигалось за 3–5 с и сопровождалось возвратом артериального давления к исходному уровню за 25–280 с (n = 6, рис. 4a). Максимальная возможная величина изменения САД (Аmах) у контрольных животных значимо не менялась в течение 4 нед. и составляла 61 ± 13 (исходно), 75 ± 13.6 мм рт. ст. (4 нед., n = 6, р > 0.05). Такой же тип реакции в ответ на болюсное введение ФЭ наблюдался у животных с ЛАГ. После введения МКТ (4 нед.) Аmах не менялся: исходно этот параметр в данной группе животных составлял 67.7 ± 1.3, а через 4 нед. 71.2 ± 12.8 мм рт. ст. (р > 0.05). Максимальная возможная величина изменения САД у крыс с ЛАГ и контрольных животных статистически значимо не различалась (р > 0.05).
Рис. 4.
Репрезентативные примеры вазоконстрикторных ответов, возникающих в русле большого круга кровообращения после активации α1-адренорецпторов фенилэфрином у контрольных крыс и крыс с ЛАГ. (a) – среднее артериальное давление (САД, в каждой панели снизу) и ЧСС (в каждой панели сверху) у контрольных крыс с двойной автономной блокадой (ДАБ) через 4 нед. после начала эксперимента (“контроль/ДАБ”) при болюсном последовательном введении возрастающих доз фенилэфрина (ФЭ, 2–32 мкг/кг). (b) – САД и ЧСС у крыс с ЛАГ (4 нед.) на фоне ДАБ (“ЛАГ/ДАБ”) при болюсном последовательном введении возрастающих доз фенилэфрина (4 нед. после введения МКТ, ФЭ, 2–32 мкг/кг). (c) – САД и ЧСС у контрольных крыс (4 нед.) на фоне ДАБ (“контроль/ДАБ/ATII”) при болюсном последовательном введении возрастающих доз фенилэфрина (ФЭ, 2–32 мкг/кг) при непрерывной внутриартериальной инфузии ангиотензина II (АТII, 150 нг/кг мин). (d) – САД и ЧСС у крыс с ЛАГ (4 нед.) на фоне ДАБ (“ЛАГ/ДАБ/ATII”) при болюсном последовательном введении возрастающих доз фенилэфрина (ФЭ, 2–32 мкг/кг) при непрерывной внутриартериальной инфузии ангиотензина II (АТII, 150 нг/кг мин).
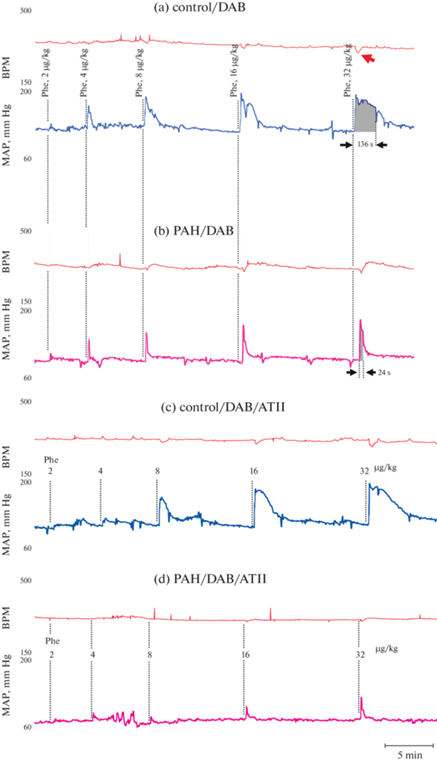
Длительность периода полувозвращения САД к исходному уровню при действии фенилэфрина у крыс с ЛАГ
У контрольных животных максимальная длительность периода полувозвращения САД к исходному уровню (T∆САД1/2) после его транзиторного роста, вызванного ФЭ на фоне ДАБ составляла 1.33 ± 0.11 мин в первый день (исходно) или 1.75 ± 0.21 мин (n = 6) через 4 нед. после начала эксперимента (рис. 4b, 5a). У крыс с ЛАГ через 4 нед. (n = 5) после введения МКТ T∆САД1/2 оказывался в три раза меньше (0.42 ± ± 0.12 мин, n = 5, p = 0.019), чем у контрольных крыс. Уменьшение T∆САД1/2 у крыс с ЛАГ по сравнению с контрольными животными наблюдалось для всего диапазона протестированных доз ФЭ (2–32 мкг/кг, рис. 4b).
Рис. 5.
(a) – наибольшая длительность периода полувозвращения САД (T∆САД1/2) к исходному уровню после вазоконстрикторной реакции, вызванной введением фенилэфрина (ФЭ) у контрольных крыс (“контроль/ДАБ”) и крыс с ЛАГ (“ЛАГ/ДАБ”, до введения и через 4 нед. после введения МКТ). (b) – то же, что на (a), но у крыс с ЛАГ до (“ЛАГ/ДАБ”) и после инфузии ангиотензина II (АТII, “ЛАГ/ДАБ/ATII”). (b) – прирост T∆САД1/2, вызываемый ATII в контроле и при ЛАГ. ns – не значимо; * – p < 0.05 (двухфакторная ANOVA с апостериорными сравнениями).
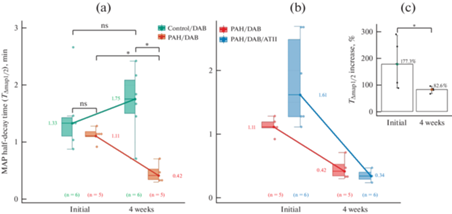
Влияние ангиотензина-II на время полувозвращения и максимальную величину изменения САД, вызванного фенилэфрином
Ангиотензин незначительно усиливал рост САД, вызванный ФЭ у контрольных животных: Аmах на фоне АТII составлял 80.5 ± 11.3 мм рт. ст. (4 нед., n = 6, р > 0.1). У крыс с ЛАГ АТII вызывал противоположный эффект – ослабление прессорного ответа, вызванного ФЭ: Аmах составлял 31.8 ± 6.5 мм рт. ст. (4 нед. после введения МКТ, n = 5, р = 0.01).
У контрольных животных АТII вызывал увеличение длительности периода полувозвращения САД к исходному уровню после введения 2–32 мкг/кг ФЭ (рис. 4b); на фоне АТII T∆САД1/2 возрастал до 1.75 ± 0.53 мин (4 нед. после начала эксперимента, n = 6, р = 0.02).
У животных группы “ЛАГ/ДАБ” до введения МКТ ангиотензин вызывал увеличение T∆САД1/2 на 77.3 ± 6.5% (n = 6, p = 0.009) по сравнению с таковым, наблюдаемым без АТII. Однако через 4 нед. после введения МКТ у животных данной группы, сформировавшей ЛАГ, АТII не оказывал усиливающего действия на прессорные эффекты ФЭ. Более того, величина T∆САД1/2, наблюдаемая на фоне инфузии ангиотензина II у крыс с ЛАГ, составляла всего 0.34 ± 0.05 мин (n = 5) или только 82 ± 7% от таковой, регистрируемой без АТII (рис. 4d, рис. 5b).
Длительность периода полувозвращения и максимальная величина изменения САД, вызванного нитропруссидом натрия у крыс с ЛАГ
В контрольных условиях введение нитропруссида натрия (НП 2.5–80 мкг/кг) на фоне ДАБ приводило к быстрому, транзиторному, дозозависимому снижению САД. Максимальная возможная величина снижения САД у контрольных животных не менялась в течение 4 нед. и составляла 51 ± 7 мм рт. ст. (n = 6). Такое же снижение САД наблюдалось у крыс через 4 нед. после введения МКТ: Аmах составляло 53 ± 8 мм рт. ст. (n = 5, p > 0.1). Кроме того, ЛАГ не влияла на длительность периода восстановления исходного уровня САД после его снижения, вызванного НП: T∆САД1/2 у контрольных крыс и крыс через 4 нед. после введения МКТ оказывалась практически одинаковым – 1.1 ± 0.08 (n = 6) и 1.05 ± 0.08 (n = 5) соответственно. Ангиотензин II не вызывал заметного изменения реакции САД (Аmах и T∆САД1/2), вызванной введением НП как у контрольных животных, так и у крыс с ЛАГ.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
В данной работе впервые проведено исследование реактивности сосудистого русла большого круга кровообращения in vivo в хронической модели ЛАГ. Впервые показано, что ЛАГ вызывает изменение вазоконстрикторных ответов в системном круге кровообращения, индуцированных активацией α1-адренорецепторов: ЛАГ уменьшает длительность прессорной реакции, ослабляет способность сосудов большого круга поддерживать тонус и САД на высоком уровне. Впервые прямо продемонстрировано, что легочная гипертензия приводит к изменению чувствительности сосудов большого круга к ангиотензину II; при ЛАГ АТII не усиливает вазоконстрикторные эффекты, обусловленные стимуляцией α1-АР, но наоборот, уменьшает величину констрикторного ответа в большом круге кровообращения. В то же время в нашей работе показано, что NO-зависимая вазодилятация и снижение САД в большом круге в хронической модели ЛАГ остаются неизмененными. Это наблюдение позволяет предположить, что ЛАГ оказывает преимущественное влияние на отдельные компоненты регуляции сосудистого тонуса в большом круге.
В наших экспериментах у крыс с ЛАГ уровень САД не отличался от такового у контрольных животных, а эффекты вазодилятатора оставались неослабленными. В работе впервые in vivo на системном уровне продемонстрировано, что ремоделирование и изменение функционального состояния малого круга кровообращения приводит к перестройке именно регуляции сосудистого тонуса в большом круге кровообращения, хотя тонус артерий, уровень САД, а также пассивные свойства сосудистой стенки, связанные с их жесткостью, могут оставаться незатронутыми.
Как указано выше, ЛАГ сопровождается нарушениями кровотока в целом ряде органов за пределами малого круга кровообращения [5]. “Нейрогуморальный дисбаланс”, нарушение адренергической/симпатической регуляции сосудистого тонуса, индуцированное ЛАГ, может служить причиной дисфункции тканей, питаемых сосудами большого круга кровообращения.
Хроническая активация симпатического отдела ВНС, усиление секреции норадреналина симпатическими постганглионарными волокнами, а также активация РААС, типичные для ЛАГ, могут приводить к изменению “реактивности” ГМК стенки артерий большого круга кровообращения, связанной с перестройкой механизмов реализации адренергических сигналов.
Комбинированная активация норадренергической “оси”, РААС и других регуляторных систем, приводит к усилению целого ряда негативных обратных связей, компенсирующих и предотвращающих проконстрикторное действие ЛАГ в большом круге. Такие компенсаторные эффекты, возможно, предотвращают чрезмерный рост базального уровня САД в большом круге, а также приводят к выявленному нами ослаблению реакции на α1-адренергическую стимуляцию. На спекулятивном уровне можно предположить, что к таким компенсаторным механизмам относится система натрийуретических пептидов. Существует значительный объем данных, подтверждающих повышение уровня секреции натрийуретических пептидов всех трех известных типов (ANP, BNP, CNP) при ЛАГ. Как указано выше, причиной усиления продукции NP предсердными кардиомиоцитами при ЛАГ является гиперволюмия, увеличение концентрации Na+ в плазме, перерастяжение стенки правого предсердия, правожелудочковая гипертрофия [39].
Известно, что ГМК экспрессируют так называемые “клиренс”-рецепторы – NPR-C, основной функцией которых считается удаление из кровотока натрийуретического пептида. Установлено, что экспрессия NPR-C находится под контролем сигнальных каскадов, сопряженных с адренорецепторами [40].
В настоящее время показано, что ГМК стенки артерий экспрессируют не только “клиренс”-, но также рецепторы натрийуретических пептидов А- и В-типа (GC-A и GC-B соответственно). Известно, что эти рецепторы являются рецепторами-гуанилатциклазами [41] и активация рецептора GC-A приводит накоплению в цитоплазме цГМФ, стимуляции протеинкиназы G первого типа (PKG-I). Среди множества мишеней PKG-I в ГМК обнаруживаются Са-АТФаза саркоплазматического ретикулюма (SERCA), фосфодиэстераза PDE3A, а также так называемый регулятор G-белок-сопряженных рецепторов (RGS-2). Фосфорилирование SERCA и PDE3A протеинкиназой G приводит к увеличению скорости удаления кальция из цитоплазмы и снижению [Ca2+]i, а также к повышению базального уровня [цАМФ]i. Указанные процессы ускоряют расслабление ГМК и, вероятно, лежат в основе наблюдаемых в наших экспериментах феноменов – уменьшения длительности периода полувозвращения САД (T∆САД1/2), т.е. к ускорению релаксация САД после вазоконстрикторного эффекта, вызванного фенилэфрином (в результате стимуляции α1-АР) у крыс с ЛАГ (рис. 6). Снижение уровня экспрессии или подавление активности NPR-C при ЛАГ может повышать активность натрийуретических пептидов и ослаблять эффекты проконстрикторных факторов.
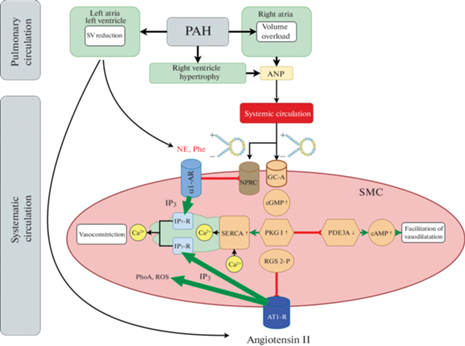
Рис. 6.
Предполагаемый механизм ослабления вазоконстрикторных ответов, вызванных активацией α1-АР и рецепторов АТII в сосудистом русле большого круга кровообращения при легочной артериальной гипертензии. УО – ударный объем; ANP – предсердный натрийуретический пептид; GC-А – рецептор ANP, обладающий гуанилатциклазной активностью; NPRC – “клиренс"-рецептор ANP; PKG I – протеинкиназа G I; RGS 2-P – фосфорилированный регулятор G-белок-сопряженных рецепторов – ингибитор рецепторов ангиотензина (AT1-P); PDE3A – фосфодиэстераза 3А; альфа-1-АР – адренорецепторы α1-типа; SERCA – кальциевая АТФаза саркоплазматического ретикулюма; RhoA – малая ГТФаза семейства Ras; ROS – активные формы кислорода.
При ЛАГ наблюдается повреждение и дисфункция эндотелия в малом круге кровообращения. Такой же эффект ЛАГ можно предположить для большого круга кровообращения. Проконстрикторное влияние ЛАГ в большом круге также может реализовываться за счет “перехвата” и инактивации NO активными формами кислорода, продукцию которых НАДФН-оксидазами стимулирует АТII. Таким образом, антиконстрикторное поток- и NO-зависимое действие эндотелия при ЛАГ может быть снижено. Как указано выше, в нашей модели ЛАГ САД в покое у контрольных крыс и крыс через 4 нед. после индукции ЛАГ оставалось на сопоставимом уровне.
Более того, в наших экспериментах наблюдается ослабление проконстрикторных реакций в большом круге. Эти результаты позволяют предположить, что, по меньшей мере, в нашей модели ЛАГ, эндотелий-зависимая регуляция в большом круге нарушена незначительно. Однако наблюдаемое отсутствие изменения САД может быть обусловлено установлением некоего баланса, возникающего в результате АФК-зависимого подавления продукции NO с одной стороны, и повышением чувствительности ГМК к NO с другой стороны.
В наших экспериментах введение донора NO приводило к одинаковому снижению САД у контрольных крыс и крыс с ЛАГ; в нашей модели ЛАГ не влияет на длительность периода восстановления САД после его снижения, вызываемого донором оксида азота. Таким образом, увеличения чувствительности сосудистого русла непосредственно к самому NO не наблюдалось. Спекулятивно, феномен сохранения нормального уровня САД может быть объяснен повышением секреции натрийуретического пептида С-типа (CNP) и рассмотрением его эффектов, опосредованных рецепторами NPR-B. Известно, что экспрессия и секреция CNP крайне высока в сосудистом эндотелии [42]; высвобождаемый конститутивно или в ответ на увеличение напряжения сдвига [43, 44] CNP стимулирует накопление cGMP, как и другие натрийуретические пептиды, в эндотелиальных клетках и ГМК. Известно, что активация сигнального каскада CNP/NPR-B/cGMP/PKG приводит к вазодилятации и/или антиконстрикторным эффектам [45]. Кроме того, известно, что CNP может стимулировать NO-синтазу и соответственно синтез NO несколькими путями [46]. Экспрессия CNP при ЛАГ может повышаться и коррелирует с дисфункцией почек [47]. Гипотетически, CNP, высвобождаемый локально в эндотелии, может являться фактором, компенсирующим недостаточность поток-зависимой дилатации при ЛАГ.
Хорошо известно, что уровень ангиотензина II при ЛАГ повышен. В текущей работе показано, что эффекты АТII, одного из наиболее значимых проконстрикторных медиаторов, изменены в большом круге кровообращения при ЛАГ. Ангиотензин II уменьшает Аmах и T∆САД1/2 у животных с ЛАГ. Таким образом, ЛАГ подавляет и, возможно, инвертирует потенциирующее действие АТII на прессорные реакции, вызванные ФЭ в большом круге кровообращения. В основе данного эффекта может лежать несколько механизмов, включающих изменение уровня экспрессии рецепторов ATII в ГМК артерий, разобщение сигнальных каскадов α1-АР и рецепторов ATII, подавление их синергизма. Ранее показано, что PKG-I-зависимое фосфорилирование белка RGS-2 приводит к его активации. Фосфорилированный RGS-2 ингибирует рецепторы ангиотензина первого типа за счет прямого взаимодействия с N-концевой областью [20, 48]. Гипотетически, ослабление либо инверсию потенциирующего дейcтвия ATII при ЛАГ можно объяснить ANP/RGS-зависимым ингибированием рецепторов ангиотензина.
В итоге, ремоделирование легочного сосудистого русла, сопровождаемое ростом давления, приводит к изменениям системной гемодинамики. Эти изменения могут быть обусловлены не только гипертрофией желудочкового миокарда, но и проявляться в форме дисрегуляции сосудистого русла большого круга.
Список литературы
Schermuly RT, Ghofrani HA, Wilkins MR, Grimminger F (2011) Mechanisms of disease: pulmonary arterial hypertension. Nat Rev Cardiol 8: 443–455. https://doi.org/10.1038/nrcardio.2011.87
Peled N, Shitrit D, Fox BD, Shlomi D, Amital A, Bendayan D, Kramer MR (2009) Peripheral arterial stiffness and endothelial dysfunction in idiopathic and scleroderma associated pulmonary arterial hypertension. J Rheumatol 36: 970–975. https://doi.org/10.3899/jrheum.081088
Malenfant S, Brassard P, Paquette M, Le Blanc O, Chouinard A, Nadeau V, Allan PD, Tzeng Y-C, Simard S, Bonnet S, Provencher S (2017) Compromised Cerebrovascular Regulation and Cerebral Oxygenation in Pulmonary Arterial Hypertension. J Am Heart Assoc 6: e006126. https://doi.org/10.1161/JAHA.117.006126
Nickel NP, O’Leary JM, Brittain EL, Fessel JP, Zamanian RT, West JD, Austin ED (2017) Kidney dysfunction in patients with pulmonary arterial hypertension. Pulm Circ 7: 38–54. https://doi.org/10.1086/690018
Nickel NP, Yuan K, Dorfmuller P, Provencher S, Lai Y-C, Bonnet S, Austin ED, Koch CD, Morris A, Perros F, Montani D, Zamanian RT, de Jesus Perez VA (2020) Beyond the Lungs: Systemic Manifestations of Pulmonary Arterial Hypertension. Am J Respir Crit Care Med 201: 148–157. https://doi.org/10.1164/rccm.201903-0656CI
Velez-Roa S, Ciarka A, Najem B, Vachiery J-L, Naeije R, van de Borne P (2004) Increased sympathetic nerve activity in pulmonary artery hypertension. Circulation 110: 1308–1312. https://doi.org/10.1161/01.CIR.0000140724.90898.D3
Maron BA, Leopold JA, Hemnes AR (2020) Metabolic syndrome, neurohumoral modulation, and pulmonary arterial hypertension. Br J Pharmacol 177: 1457–1471. https://doi.org/10.1111/bph.14968
Dendorfer A, Thornagel A, Raasch W, Grisk O, Tempel K, Dominiak P (2002) Angiotensin II induces catecholamine release by direct ganglionic excitation. Hypertension 40: 348–354. https://doi.org/10.1161/01.hyp.0000028001.65341.aa
Fabiani ME, Sourial M, Thomas WG, Johnston CI, Johnston CI, Frauman AG (2001) Angiotensin II enhances noradrenaline release from sympathetic nerves of the rat prostate via a novel angiotensin receptor: implications for the pathophysiology of benign prostatic hyperplasia. J Endocrinol 171: 97–108. https://doi.org/10.1677/joe.0.1710097
Reid IA (1992) Interactions between ANG II, sympathetic nervous system, and baroreceptor reflexes in regulation of blood pressure. Am J Physiol 262: E763–E778. https://doi.org/10.1152/ajpendo.1992.262.6.E763
Maron BA, Leopold JA (2014) The role of the renin-angiotensin-aldosterone system in the pathobiology of pulmonary arterial hypertension (2013 Grover Conference series). Pulm Circ 4: 200–210. https://doi.org/10.1086/675984
Vaillancourt M, Chia P, Sarji S, Nguyen J, Hoftman N, Ruffenach G, Eghbali M, Mahajan A, Umar S (2017) Autonomic nervous system involvement in pulmonary arterial hypertension. Respir Res 18. https://doi.org/10.1186/s12931-017-0679-6
Langer SZ, Hicks PE (1984) Alpha-adrenoreceptor subtypes in blood vessels: physiology and pharmacology. J Cardiovasc Pharmacol 6 Suppl 4: S547–S558. https://doi.org/10.1097/00005344-198406004-00001
Hu ZW, Shi XY, Okazaki M, Hoffman BB (1995) Angiotensin II induces transcription and expression of alpha 1-adrenergic receptors in vascular smooth muscle cells. Am J Physiol 268: H1006–H1014. https://doi.org/10.1152/ajpheart.1995.268.3.H1006
Malik KU, Nasjletti A (1976) Facilitation of adrenergic transmission by locally generated angiotensin II in rat mesenteric arteries. Circ Res 38: 26–30. https://doi.org/10.1161/01.res.38.1.26
Purdy RE, Weber MA (1988) Angiotensin II amplification of alpha-adrenergic vasoconstriction: role of receptor reserve. Circ Res 63: 748–757. https://doi.org/10.1161/01.res.63.4.748
Vittorio TJ, Fudim M, Wagman G, Kosmas CE (2014) Alpha-1 adrenoceptor-angiotensin II type 1 receptor cross-talk and its relevance in clinical medicine. Cardiol Rev 22: 51–55. https://doi.org/10.1097/CRD.0b013e31829ce723
Bansal S, Badesch D, Bull T, Schrier RW (2009) Role of vasopressin and aldosterone in pulmonary arterial hypertension: A pilot study. Contemp Clin Trials 30: 392–399. https://doi.org/10.1016/j.cct.2009.04.003
Samavat S, Ahmadpoor P, Samadian F (2011) Aldosterone, hypertension, and beyond. Iran J Kidney Dis 5: 71–76.
Kishimoto I, Tokudome T, Nakao K, Kangawa K (2011) Natriuretic peptide system: an overview of studies using genetically engineered animal models. FEBS J 278: 1830–1841. https://doi.org/10.1111/j.1742-4658.2011.08116.x
Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF, Rabinovitch M (2004) Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol 43: 13S–24S. https://doi.org/10.1016/j.jacc.2004.02.029
Meloche J, Lampron M-C, Nadeau V, Maltais M, Potus F, Lambert C, Tremblay E, Vitry G, Breuils-Bonnet S, Boucherat O, Charbonneau E, Provencher S, Paulin R, Bonnet S (2017) Implication of Inflammation and Epigenetic Readers in Coronary Artery Remodeling in Patients With Pulmonary Arterial Hypertension. Arterioscler Thromb Vasc Biol 37: 1513–1523. https://doi.org/10.1161/ATVBAHA.117.309156
Dikalov SI, Nazarewicz RR (2013) Angiotensin II-induced production of mitochondrial reactive oxygen species: potential mechanisms and relevance for cardiovascular disease. Antioxid Redox Signal 19: 1085–1094. https://doi.org/10.1089/ars.2012.4604
Ding J, Yu M, Jiang J, Luo Y, Zhang Q, Wang S, Yang F, Wang A, Wang L, Zhuang M, Wu S, Zhang Q, Xia Y, Lu D (2020) Angiotensin II Decreases Endothelial Nitric Oxide Synthase Phosphorylation via AT1R Nox/ROS/PP2A Pathway. Front Physiol 11: 566410. https://doi.org/10.3389/fphys.2020.566410
Boron WF, Boulpaep EL (2012) Medical Physiology, 2e Updated Edition E-Book: with STUDENT CONSULT Online Access. Elsevier Health Sciences.
Gomez-Arroyo JG, Farkas L, Alhussaini AA, Farkas D, Kraskauskas D, Voelkel NF, Bogaard HJ (2012) The monocrotaline model of pulmonary hypertension in perspective. Am J Physiol - Lung Cell Mol Physiol 302: L363–L369. https://doi.org/10.1152/ajplung.00212.2011
Spyropoulos F, Vitali SH, Touma M, Rose CD, Petty CR, Levy P, Kourembanas S, Christou H (2020) Echocardiographic markers of pulmonary hemodynamics and right ventricular hypertrophy in rat models of pulmonary hypertension. Pulm Circ 10: 2045894020910976. https://doi.org/10.1177/2045894020910976
Zhu Z, Godana D, Li A, Rodriguez B, Gu C, Tang H, Minshall RD, Huang W, Chen J (2019) Echocardiographic assessment of right ventricular function in experimental pulmonary hypertension. Pulm Circ 9: 2045894019841987. https://doi.org/10.1177/2045894019841987
Islamova MR, Lazarev PV, Safarova AF, Kobalava ZD (2018) The Value of Right Ventricular Dysfunction and Right Ventricular – Pulmonary Artery Coupling in Chronic Heart Failure: The Role of Echocardiography. Kardiologiia 58: 82–90. https://doi.org/10.18087/cardio.2018.5.10124
An H, Landis JT, Bailey AG, Marron JS, Dittmer DP (2019) dr4pl: A Stable Convergence Algorithm for the 4 Parameter Logistic Model. The R J 11: 171–190. https://doi.org/10.32614/RJ-2019-003
Ritz C, Baty F, Streibig JC, Gerhard D (2015) Dose-Response Analysis Using R. PLoS One 10: e0146021. https://doi.org/10.1371/journal.pone.0146021
R Core Team (2020) R: A Language and Environment for Statistical Computing.
RStudio Team (2020) RStudio: Integrated Development Environment for R.
Wickham H, Averick M, Bryan J, Chang W, McGowan LD, François R, Grolemund G, Hayes A, Henry L, Hester J, Kuhn M, Pedersen TL, Miller E, Bache SM, Müller K, Ooms J, Robinson D, Seidel DP, Spinu V, Takahashi K, Vaughan D, Wilke C, Woo K, Yutani H (2019) Welcome to the tidyverse. J Open Source Software 4: 1686. https://doi.org/10.21105/joss.01686
Kassambara A (2020) rstatix: Pipe-Friendly Framework for Basic Statistical Tests.
Wickham H (2016) ggplot2: Elegant Graphics for Data Analysis. Springer Int Publ.
Tiedemann F (2020) gghalves: Compose Half-Half Plots Using Your Favourite Geoms.
Pedersen TL (2020) patchwork: The Composer of Plots.
Yap LB (2004) B-type natriuretic Peptide and the right heart. Heart Fail Rev 9: 99–105. https://doi.org/10.1023/B:HREV.0000046364.68371.b0
Kishimoto I, Yoshimasa T, Suga S, Ogawa Y, Komatsu Y, Nakagawa O, Itoh H, Nakao K (1994) Natriuretic peptide clearance receptor is transcriptionally down-regulated by beta 2-adrenergic stimulation in vascular smooth muscle cells. J Biol Chem 269: 28300–28308.
Suga S, Nakao K, Hosoda K, Mukoyama M, Ogawa Y, Shirakami G, Arai H, Saito Y, Kambayashi Y, Inouye K (1992) Receptor selectivity of natriuretic peptide family, atrial natriuretic peptide, brain natriuretic peptide, and C-type natriuretic peptide. Endocrinology 130: 229–239. https://doi.org/10.1210/endo.130.1.1309330
Suga S, Nakao K, Itoh H, Komatsu Y, Ogawa Y, Hama N, Imura H (1992) Endothelial production of C-type natriuretic peptide and its marked augmentation by transforming growth factor-beta. Possible existence of “vascular natriuretic peptide system.” J Clin Invest 90: 1145–1149. https://doi.org/10.1172/JCI115933
Moyes AJ, Hobbs AJ (2019) C-type Natriuretic Peptide: A Multifaceted Paracrine Regulator in the Heart and Vasculature. Int J Mol Sci 20: 2281. https://doi.org/10.3390/ijms20092281
Okahara K, Kambayashi J, Ohnishi T, Fujiwara Y, Kawasaki T, Monden M (1995) Shear stress induces expression of CNP gene in human endothelial cells. FEBS Lett 373: 108–110. https://doi.org/10.1016/0014-5793(95)01027-c
Pernomian L, Prado AF, Silva BR, Azevedo A, Pinheiro LC, Tanus-Santos JE, Bendhack LM (2016) C-Type Natriuretic Peptide Induces Anti-contractile Effect Dependent on Nitric Oxide, Oxidative Stress, and NPR-B Activation in Sepsis. Front Physiol 7: 226. https://doi.org/10.3389/fphys.2016.00226
Andrade FA, Restini CBA, Grando MD, Ramalho LNZ, Bendhack LM (2014) Vascular relaxation induced by C-type natriuretic peptide involves the Ca2+/NO-synthase/NO pathway. PLoS One 9: e95446. https://doi.org/10.1371/journal.pone.0095446
Kaiser R, Grotemeyer K, Lepper P, Stokes C, Bals R, Wilkens H (2015) Associations of circulating natriuretic peptides with haemodynamics in precapillary pulmonary hypertension. Respir Med 109: 1213–1223. https://doi.org/10.1016/j.rmed.2015.02.014
Matsuzaki N, Nishiyama M, Song D, Moroi K, Kimura S (2011) Potent and selective inhibition of angiotensin AT1 receptor signaling by RGS2: roles of its N-terminal domain. Cell Signal 23: 1041–1049. https://doi.org/10.1016/j.cellsig.2011.01.023
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова