

Теоретические основы химической технологии, 2020, T. 54, № 1, стр. 3-9
Распределение изотопов водорода между фазами при парожидкостном равновесии водных солевых растворов
Н. Н. Кулов a, *, А. В. Полковниченко a, Е. В. Лупачев a, **, А. А. Вошкин a, Э. П. Магомедбеков b
a Институт общей и неорганической химии им. Н.С. Курнакова РАН
Москва, Россия
b Российский химико-технологический университет им. Д.И. Менделеева
Москва, Россия
* E-mail: kulovnn@mail.ru
** E-mail: egorlu91@gmail.com
Поступила в редакцию 08.10.2019
После доработки 09.10.2019
Принята к публикации 10.10.2019
Аннотация
Экспериментально изучен однократный разделительный эффект между дейтерием и протием в условиях фазового равновесия системы водяной пар–водный солевой раствор в диапазоне температур от 59 до 94°С. Для исследования парожидкостного равновесия использовали динамический метод, реализованный в кубе Бушмакина. Установлено, что присутствие в воде карбамида существенно увеличивает коэффициент разделения αL–V в рассматриваемом диапазоне температур. Показано, что характерное увеличение коэффициента относительной летучести изотопов водорода воды при понижении температуры в присутствии солей сохраняется. Изотопный солевой эффект, как отрицательный, так и положительный, возрастает с увеличением концентрации солей.
ВВЕДЕНИЕ
Применение солей для улучшения условий разделения жидких смесей известно давно [1]. Однако лишь в связи с развитием теории и практики использования экстрактивной ректификации потенциальные возможности солевой ректификации стали все больше привлекать внимание химиков-технологов [2].
Среди представляющих практический интерес близкокипящих смесей солевая ректификация может оказаться полезной, в частности, как способ разделения изотопов водорода и кислорода при ректификации природной воды. В настоящее время этот процесс, экологически чистый и безопасный с практически неисчерпаемым запасом целевых продуктов, становится основным при производстве изотопов водорода и кислорода, но требует снижения энергетических затрат [3].
В принципе, добавление незначительного количества водорастворимой соли в качестве разделяющего агента может существенно улучшить энергетический баланс ректификационной колонны. Введение нелетучей соли в жидкий раствор, состоящий из двух и более летучих компонентов, приводит к изменению состава равновесного этой жидкости пара без присутствия соли и ее ионов в парах [4, 5]. Компонент разделяемой жидкости, которым обогащается паровая фаза, называют “высоленым” введенной в жидкость солью, тогда как другой компонент питания, оставшийся в жидкости, называют “всоленым” [6].
При разделении изотопов водорода молекулы воды, координирующиеся в растворе вблизи иона или молекулы разделяющего солевого агента, энергетически отличаются от молекул воды, которые не включены в гидратную оболочку растворенной частицы [7–9]. Структура гидратных сфер растворителя в водных растворах была целью исследования в течение многих лет [6, 10, 11], однако в настоящее время поиск рациональных разделяющих агентов все еще в значительной мере приходится осуществлять экспериментально.
Целью настоящей работы является экспериментальное изучение однократного разделительного эффекта между дейтерием и протием при равновесии в системе водяной пар–водно-солевой раствор для отыскания рационального разделяющего агента в процессе солевой ректификации воды.
ЧИСТАЯ ВОДА
Рассматривая периодическую литературу, посвященную разделению изотопов методом солевой ректификации, следует отметить малое количество работ в этой области. Чаще всего разделение изотопов различных элементов в водно-солевых системах жидкость–пар рассматривается с позиций геохимии. Для эффективной реализации солевой ректификации воды под вакуумом верхний температурный диапазон процесса ограничивается значениями, близкими к 100°С.
В работе [12] приводятся обширные результаты прецизионных измерений однократного равновесного коэффициента разделения изотопов водорода между жидкой природной водой и водяным паром в диапазоне температур от 25 до 350°С. Результаты измерения находятся в хорошем согласии с литературными данными других исследователей [13–19] и позволяют получить стандартную зависимость для характеристики чистой природной воды. Полученные в работе [12] измерения с привлечением большого числа литературных данных описываются предлагаемой авторами этой работы регрессией
(1)
$\begin{array}{*{20}{c}} \begin{gathered} {{10}^{3}}{\text{ln}}_{{l - v}}^{{pw}}\alpha = 1158.8\left( {\frac{{{{T}^{3}}}}{{{{{10}}^{9}}}}} \right) - 1620.1\left( {\frac{{{{T}^{2}}}}{{{{{10}}^{6}}}}} \right) + \hfill \\ + \,\,794.84\left( {\frac{T}{{{{{10}}^{3}}}}} \right) - 161.04 + 2.9992\left( {\frac{{{{{10}}^{9}}}}{{{{T}^{3}}}}} \right)~ \hfill \\ \end{gathered} \end{array}$в области температур от 0°С до критической температуры воды (374.1°С) с высокой точностью. Авторы работы отмечают, что коэффициент разделения изотопов водорода между жидкостью и паром $\alpha _{{l--v}}^{{pv}} = {{\alpha }_{{L--V}}}$ для чистой воды быстро падает с ростом температуры, достигая единицы в области 220–230°С, после чего претерпевает инверсию. Выше отмеченной точки температур водяные пары обогащаются дейтерием в большей степени, чем соприкасающаяся с ними жидкая вода.
Киршенбаум [20] отмечает, что в большинстве случаев H2O, D2O и HDO образуют идеальные растворы, причем упругость паров HDO можно рассчитывать как среднегеометрическую между H2O, D2O, т.е. РHDO = (${{Р}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}{{Р}_{{{{{\text{D}}}_{{\text{2}}}}{\text{O}}}}}$)1/2. Заметим, что результаты эксперимента с водой, содержащей около 1 ат. % дейтерия, указывают на то, что наблюдаемая величина ${{\alpha }_{(}}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O/НDO)}}}}$ = 1.029 ± 0.0018 была выше расчетной 1.026, из чего следует, что поведение смесей H2O и HDO несколько отклоняется от идеальности. В рассмотренном эксперименте также были приведены оценки ошибок, вносимых изменением концентрации 18О и содержанием D2O. Было показано, что обе ошибки не выходят за пределы погрешности эксперимента. На это обстоятельство также указано в работе [21], где приведено отклонение коэффициента относительной летучести αL–V от идеальности для воды природного изотопного состава в диапазоне температур от 0 до 100°С.
ФАЗОВОЕ РАВНОВЕСИЕ ВОДНО-СОЛЕВОЙ РАСТВОР–ВОДЯНОЙ ПАР
Следует отметить многообразие изученных в литературе водно-солевых систем, как по типу фазового равновесия, так и по количеству объектов исследования в достаточно широком концентрационном и температурном диапазоне. Однако, к сожалению, обнаруженные эффекты, в большинстве своем, не представляют практического интереса с точки зрения процесса ректификации, поскольку приводят к уменьшению αL–V.
Форма представления опубликованных данных в основном сводится к построению зависимостей так называемых изотопных (солевых) эффектов от температуры или концентрации соли. Основная форма представления солевого эффекта, где αi – коэффициент разделения при наличии соли, αj – для чистой воды, имеет следующий вид:
Следует отметить, что минеральные соли в большинстве своем уменьшают коэффициент разделения αL–V [22]. Наибольший интерес для целей настоящего исследования представляют работы [21, 23], в которых было рассмотрено влияние карбамида на значение ${{\alpha }_{{L - V}}}$ при 15 и 25°С. Полученные в этих работах результаты приведены на рис. 1. и в табл. 1.
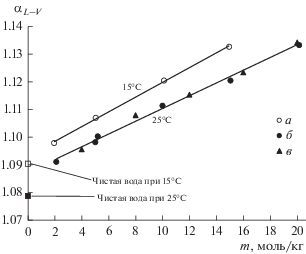
Таблица 1.
Литературные данные
| m, моль/(кг H2O) | αL–V | T, K | lnαL–V | Литературный источник |
|---|---|---|---|---|
| 0 | 1.0880 | 288 | 0.08434 | [23] |
| 1.95 | 1.0979 | 0.09340 | ||
| 5.04 | 1.1070 | 0.10165 | ||
| 10.11 | 1.1205 | 0.11378 | ||
| 14.95 | 1.1327 | 0.12460 | ||
| 0 | 1.0778 | 298 | 0.07492 | [23] |
| 2.11 | 1.0911 | 0.08719 | ||
| 5.01 | 1.0982 | 0.09367 | ||
| 5.19 | 1.1003 | 0.09558 | ||
| 10 | 1.1114 | 0.10562 | ||
| 15.5 | 1.1205 | 0.11378 | ||
| 20.15 | 1.1333 | 0.12513 | ||
| 0 | 1.0787 | 298 | 0.07580 | [12] |
| 4 | 1.0956 | 0.09130 | [21] | |
| 8 | 1.1079 | 0.10250 | ||
| 12 | 1.1154 | 0.10920 | ||
| 16 | 1.1234 | 0.11640 | ||
| 20 | 1.1343 | 0.12600 |
Из рис. 1 следует, что значение коэффициента разделения ${{\alpha }_{{L - V}}}$ в присутствии карбамида в растворе выше, чем для чистой воды и увеличивается с ростом концентрации соли. Таким образом, карбамид может выступать в качестве потенциального солевого агента для разделения изотопов D/H в процессе солевой ректификации природной воды. К сожалению, экспериментальные данные о воздействии карбамида на фракционирование изотопов водорода, в технологически приемлемом для процесса ректификации диапазоне температур, отсутствуют.
В настоящей работе объектом исследования выбраны растворы карбамида в температурном диапазоне 60–100°С, а также растворы N-метилпирролидона и ряда органических и неорганических солей, влияние которых на коэффициент разделения изотопов водорода ранее не изучалось.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Приборы и реактивы. Для приготовления исходных солевых растворов использовали дистиллированную воду с содержанием дейтерия 140–145 млн–1, а также KBr, NaBr, N-метилпирролидон и формиат натрия категории “Ч.”, СuSO4 категории “ч.д.а.”, СsCl, KI, ацетат натрия, бензоат натрия, Na2SO4, K2SO4, ZnSO4 и карбамид категории “х.ч.”. Навески готовили с помощью компаратора массы AND МС-1000 с точностью измерения 0.0005 г. Состав исходной воды замеряли каждый раз перед приготовлением раствора.
Изотопный анализ проб проводили с использованием спектрального жидкостного изотопного анализатора T-LWA-45-EP, предназначенного для анализа воды на уровне природного содержания изотопов с погрешностью измерения ±0.0001 ат. % (1 млн–1).
Экспериментальная установка и методика проведения эксперимента. В качестве установки для исследования фазового равновесия жидкость–пар использовали модифицированный куб Бушмакина, представленный на рис. 2. Используемая в настоящей работе установка, в отличие от классической, позволяет отбирать пробы дистиллята не только из внутреннего контура, но и из внешнего. Во избежание контакта солевого раствора с металлическим корпусом, в куб прибора помещен тефлоновый стакан 2, а также установлен обратный клапан 3, не позволяющий солевому раствору стекать из куба.
Рис. 2.
Модифицированный куб Бушмакина: 1 – обечайка, 2 – тефлоновая вкладка (тефлоновый стакан), 3 – обратный клапан, 4 – электрообогреватель, 5 – соединение КЛЭМП, 6 – обечайка, 7 – внутренний купол, 8 – трубка для отвода паров из внешнего контура, 9 – трубка для отвода паров из внутреннего контура, 10 – холодильник для конденсации паров внешнего контура, 11 – холодильник для конденсации паров внутреннего контура, 12 – кран для отбора проб, 13, 14 – теплоизоляция.
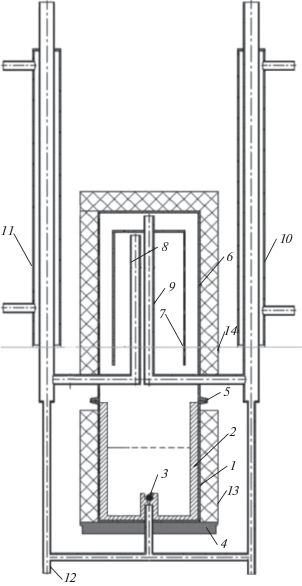
Объем загрузки составлял 350–400 мл. Если того требовали условия эксперимента, установка подключалась к вакуумной системе через холодильники 10 и 11. Давление в системе определяли с помощью электронного манометра с точностью 0.1 кПа, равновесная температура определялась с помощью термопар с точностью 0.5°С.
Предварительные эксперименты по определению однократного коэффициента разделения ${{\alpha }_{{L - V}}}$ для чистой воды показали, что фазовое равновесие между жидкостью и паром наступает примерно через 20 ч после начала эксперимента. Исходя из этого, каждый опыт проводили в течение суток (24 ч). Время эксперимента отсчитывали с момента стекания первой капли дистиллята из холодильника 11.
Используемый нами метод анализа позволяет определять изотопный состав только чистой воды. Как следствие, определять изотопный состав кубовой жидкости непосредственно невозможно, поскольку она представляет собой водный раствор соли. По этой причине изотопный состав воды в кубе определяли по материальному балансу системы. Анализу подвергали воду, из которой готовился исходный солевой раствор и паровой конденсат обоих контуров. Поскольку используемые нами солевые агенты являются нелетучими, они не присутствуют в паровом конденсате.
Выбор динамического метода исследования фазового равновесия жидкость–пар в аппарате Бушмакина обусловлен приближением полученных в настоящей работе данных к условиям ректификационного процесса.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Поскольку атомная концентрация дейтерия в рассматриваемых нами системах близка к природной, следует отметить, что практически весь дейтерий находится в составе молекул HDO, а концентрация D2O в системе, согласно приведенным в [20] данным, пренебрежительно мала. Таким образом, в условиях настоящей работы можно считать, что вода представляет собой двухкомпонентную систему, состоящую из Н2О и HDO.
В настоящей работе коэффициент разделения изотопов αL–V между жидкой и паровой фазами рассчитывали по формуле
(3)
${{\alpha }_{{L - V}}} = \frac{{{{{{x}_{D}}} \mathord{\left/ {\vphantom {{{{x}_{D}}} {(1 - {{x}_{D}})}}} \right. \kern-0em} {(1 - {{x}_{D}})}}}}{{{{{{y}_{D}}} \mathord{\left/ {\vphantom {{{{y}_{D}}} {(1 - {{y}_{D}})}}} \right. \kern-0em} {(1 - {{y}_{D}})}}}},$Таблица 2.
Данные настоящей работы по фазовому равновесию водных растворов минеральных солей
| Соль | m, моль/(кг Н2O) | αL–V | t, °С |
|---|---|---|---|
| CsCl | 1 | 1.04507 | 63 |
| 1.03572 | 81 | ||
| 1.03147 | 89 | ||
| 3 | 1.03671 | 63 | |
| 1.03129 | 79 | ||
| 1.02380 | 92 | ||
| 5 | 1.02748 | 69 | |
| 1.02181 | 82 | ||
| 1.01256 | 94 | ||
| KBr | 3 | 1.03596 | 63 |
| 1.02736 | 83 | ||
| 1.02349 | 91 | ||
| NaBr | 3 | 1.03909 | 61 |
| 1.02974 | 80 | ||
| 1.02518 | 90 | ||
| KI | 1 | 1.04051 | 61 |
| 1.03238 | 78 | ||
| 1.02710 | 91 | ||
| CuSO4 | 1.2 | 1.02134 | 60 |
| Na2SO4 | 0.4 | 1.03762 | 60 |
| K2SO4 | 0.5 | 1.03165 | 60 |
| ZnSO4 | 1.4 | 1.01644 | 61 |
Таблица 3.
Экспериментальные данные настоящей работы
| Соль | m, моль/(кг Н2O) | αL–V | t, °С |
|---|---|---|---|
| N-метилпирролидон | 1 | 1.04478 | 60 |
| 1.04490 | 61 | ||
| 1.05189 | 61 | ||
| 1.03508 | 70 | ||
| 1.04776 | 71 | ||
| 1.04952 | 71 | ||
| 1.04304 | 80 | ||
| 1.05107 | 80 | ||
| 1.03836 | 81 | ||
| 1.03592 | 90 | ||
| Ацетат натрия | 2.6 | 1.04126 | 60 |
| Бензоат натрия | 3 | 1.03383 | 61 |
| Формиат натрия | 3 | 1.04950 | 60 |
| Карбамид | 3 | 1.05290 | 60 |
| 1.04502 | 77 | ||
| 1.04055 | 89 | ||
| 7 | 1.07230 | 59 | |
| 1.06787 | 71 | ||
| 1.0629 | 79 | ||
| 1.06356 | 80 | ||
| 1.06005 | 87 | ||
| 9 | 1.07350 | 67 | |
| 1.07096 | 79 | ||
| 1.06498 | 92 | ||
| 1.07526 | 61 | ||
| 1.07742 | 59 | ||
| 1.06500 | 87 |
Поиск эффективного солевого агента для разделения изотопов водорода показал, что в рабочем диапазоне температур для водных растворов большинства минеральных солей значение коэффициента относительной летучести ${{\alpha }_{{L - V}}}$ снижается, а пар, в свою очередь, в большей степени обогащен дейтерием по сравнению с соответствующим значением для чистой воды. Некоторые сульфаты приводят к незначительному повышению величины ${{\alpha }_{{L - V}}}$ при температурах ниже 50°С, однако слабый положительный эффект делает нецелесообразным их применение в качестве разделяющего агента в рассматриваемом процессе.
Полученные результаты по органическим солям также указывают на снижение ${{\alpha }_{{L - V}}}$ в присутствии бензоата и ацетата натрия. Эксперименты с формиатом натрия и N-метилпирролидоном дают значение ${{\alpha }_{{L - V}}}$, близкое к значению для чистой воды.
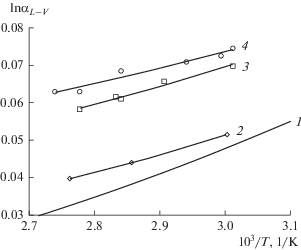
Заметный положительный эффект наблюдается у растворов карбамида. Экспериментально полученные данные ${{\alpha }_{{L - V}}}$ в зависимости от концентрации карбамида в сравнении с чистой водой представлены на рис. 3.
Рис. 3.
Зависимость атомного коэффициента разделения αL–V между D и Н для растворов карбамида концентрацией 3 (2), 7 (3) и 9 (4) моль/(кг Н2О) в сравнении с чистой водой (1).
Уравнения, описывающие зависимость величины изотопного солевого эффекта карбамида от температуры, имеют вид
Коэффициенты уравнения приведены в табл. 4.
Таблица 4.
Коэффициенты уравнения (4)
| m, моль/(кг Н2О) | A | B |
|---|---|---|
| 3 | –22026.94 | 92.033 |
| 7 | –15069.67 | 67.101 |
| 9 | –14929.92 | 48.446 |
Из рис. 3 нетрудно убедиться, что наличие карбамида существенно повышает коэффициент относительной летучести ${{\alpha }_{{L - V}}}$ по сравнению с чистой водой. С ростом концентрации соли в растворе при постоянной температуре значение ${{\alpha }_{{L - V}}}$ увеличивается, а с понижением температуры раствора при постоянной концентрации соли сохраняется характерная для чистой воды тенденция к увеличению коэффициента разделения. Отметим, что величина коэффициента разделения изотопов водорода для 9 моляльного раствора карбамида при 60°C соизмерима с коэффициентом фракционирования чистой воды при 26°C.
ЗАКЛЮЧЕНИЕ
Исследовано разделение природной воды для условий вакуумной солевой ректификации. Настоящая работа является одной из первых попыток в данной области, поэтому поиск подходящих солевых агентов для разделения изотопов водорода производился опытным путем.
Исследован ряд минеральных солей и органических соединений. Найден потенциальный разделяющий агент – карбамид, который по полученному однократному коэффициенту разделения следует считать эффективным. Влияние карбамида на ${{\alpha }_{{L - V}}}$ исследовано в области концентраций от 3 до 9 моль/(кг воды) в приемлемом с точки зрения процесса ректификации диапазоне температур. Приведена зависимость изотопного солевого эффекта от температуры при различных концентрациях.
Работа выполнена при финансовой поддержке Российского научного фонда (проект № 18-13-00475).
ОБОЗНАЧЕНИЯ
| A, B | коэффициенты уравнения (4) |
| m | моляльность, моль/(кг воды) |
| P | давление, кПа |
| T | температура, K |
| t | температура, °С |
| xi | состав жидкой фазы, ат. д. |
| yi | состав паровой фазы, ат. д. |
| α | коэффициент разделения |
| Γ | изотопный солевой эффект |
ИНДЕКСЫ
Список литературы
Ципарис И.Н., Добросердов Л.Л., Коган В.Б. Солевая ректификация. М.: Химия, 1969.
Furter W.F. Extractive distillation by salt effect // Chem. Eng. Commun. 1992. V. 116. P. 35.
Miller A.I. Heavy water: a manufacturers' guide for the hydrogen century // Can. Nucl. Soc. Bull. 2001. V. 22. № 1. P. 1.
Lei Z., Wang H., Zhou R., Duan Z. Influence of salt added to solvent on extractive distillation // Chem. Eng. J. 2002. V. 87. P. 149.
Soares R.B., Pessoa F.L.P., Mendes M.F. Dehydration of ethanol with different salts in a packed distillation column // Process Saf. Environ. Prot. 2015. V. 93. P. 147. https://doi.org/10.1016/j.psep.2014.02.012
Самойлов О.В. Структура водных растворов электролитов и гидратация ионов. М.: АН СССР, 1957.
Pupezin J., Jakli G., Jancso G., Van Hook W.A. The vapor pressure isotope effect in aqueoous systems // J. Phys. Chem. 1979. V. 76. № 6. P. 743.
Jakli G., Van Hook W.A. Isotope Effects in Aqueous Systems. 12. Thermodynamics of Urea-h4/H2O and Urea-d4/D2O Solutions // J. Phys. Chem. 1981. V. 85. № 23. P. 3480. https://doi.org/10.1021/j150623a025
O'Neil J.R., Truesdell A.H. Oxygen isotope fractionation studies of solute-water interactions // Stable Isotope Geochemistry: A Tribute to Samuel Epstein (The Geochemical Society. Special Publication No. 3) / Eds. Taylor H.P.Jr., O’Neil J.R., Kaplan I.R. Washington, DC: Geochemical Society, 1991. P. 17.
Barrer R.M., Denny A.F. Water in hydrates. Part I. Fractionation of hydrogen isotopes by crystallisation of salt hydrates // J. Chem. Soc. 1964. P. 4677. https://doi.org/10.1039/JR9640004677
Driesner T., Ha T.-K., Seward T.M. Oxygen and hydrogen isotope fractionation by hydration complexes of Li+, Na+, K+, Mg2+, F–, Cl–, and Br–: A theoretical study // Geochim. Cosmochim. Acta. 2000. V. 64. № 17. P. 3007. https://doi.org/10.1016/S0016-7037(00)00407-5
Horita J., Wesolowski D.J. Liquid–vapor fractionation of oxygen and hydrogen isotopes of water from the freezing to the critical temperature // Geochim. Cosmochim. Acta. 1994. V. 58. P. 3425.
Bottinga Y., Craig H. High temperature liquid-vapor fractionation factors for H2O–HDO–H218O// Trans., Am. Geophys. Union. 1968. V. 49. P. 356.
Von Ehhalt D., Knott K. Kinetische Isotopentrennung bei der Verdampfung von Wasser // Tellus. 1965. V. 17. P. 389. https://doi.org/10.1111/j.2153-3490.1965.tb01431.x
Kakiuchi M., Matsco S. Direct measurements of D/H and 18O/16O fractionation factors between vapor and liquid water in the temperature range from 10 to 40°C // Geochem. J. 1979. V. 13. P. 307. https://doi.org/10.2343/geochemj.13.307
Kazahaya K. Chemical and isotopic studies on hydrothermal solutions. Ph.D. Dissertation. Tokyo: Tokyo Institute of Technology, 1986.
Majoube M. Fractionnement en oxygène 18 et en deutérium entre l’eau et sa vapeur // J. Chim. Phys. 1971. V. 68. P. 1423. https://doi.org/10.1051/jcp/1971681423
Merlivat L., Botter R., Nief G. Fractionnement isotopique au cours de la distillation de l’eau // J. Chim. Phys. 1963. V. 60. P. 56. https://doi.org/10.1051/jcp/1963600056
Rennow M. Gleichgewichts- und kinetische Trennfaktoren bei der Verdunstung von Wasser. Staatsexamensarbeit: Univ. Heidelberg, 1970.
Kirshenbaum I. Physical Properties and Analysis of Heavy Water. New York: McGraw-Hill, 1951.
Kakiuchi M. Distribution of isotopic water molecules, H2O, HDO, and D2O, in vapor and liquid phases in pure water and aqueous solution systems // Geochim. Cosmochim. Acta. 2000. V. 64. № 9. P. 1485.
Hoiuta J., Wesolowski D.J., Cole D.R. The activity-composition relationship of oxygen and hydrogen isotopes in aqueous salt solutions: I. Vapor-liquid water equilibration of single salt solutions from 50 to 100°C // Geochim. Cosmochim. Acta. 1993. V. 57. P. 2797.
Kakiuchi M., Matsuo S. Fractionation of hydrogen and oxygen isotopes between hydrated and free water molecules in aqueous urea solution // J. Phys. Chem. 1985. V. 89. P. 4627.
Дополнительные материалы отсутствуют.
Инструменты
Теоретические основы химической технологии