

Успехи современной биологии, 2019, T. 139, № 1, стр. 3-24
Оценки генетической интрогрессии, ретикуляции генных деревьев, дивергенции таксонов и состоятельности ДНК-штрихкодирования по молекулярным маркерам генов
Ю. Ф. Картавцев 1, 2, *, А. Д. Редин 1, 2
1 Национальный научный центр морской биологии ДВО РАН
Владивосток, Россия
2 Дальневосточный федеральный университет
Владивосток, Россия
* E-mail: yuri.kartavtsev48@hotmail.com
Поступила в редакцию 05.07.2018
После доработки 25.07.2018
Принята к публикации 25.07.2018
Аннотация
Рассмотрены литературные источники и собственные публикации по молекулярной эволюции, а также приложение новых молекулярно-генетических данных к систематике и проблематике генетической дивергенции различных группировок организмов, преимущественно животных. Проведен сравнительный анализ и даны оценки состоятельности использования молекулярных маркеров для идентификации видов, включая предложенный одним из авторов подход для описания биологического разнообразия в рамках глобальной программы ДНК-штрихкодирования. Затронуты вопросы идентификации гибридов и распространенности генетической интрогрессии. Выяснено: 1. Для идентификации гибридов и оценки интрогрессии или потока генов наиболее подходит комбинация маркеров ядерной ДНК (яДНК) и митохондриальной ДНК (мтДНК). 2. Имеющиеся факты об изменчивости яДНК и мтДНК убеждают в существовании генетической интрогрессии для многих таксонов животных и растений. Однако даже для широких гибридных зон, например комплекса мидий Mytilus ex. group edulis, интрогрессия может быть весьма ограниченной для значительной части ареала или асимметричной, таким образом сохраняя нетронутыми, по меньшей мере, некоторые таксоны. 3. Принимая, что бисексуальные морские и наземные виды подвержены генетической интрогрессии, как это обнаружено для довольно большого числа таксонов, надо согласиться также с тем, что ортодоксальная биологическая концепция вида (БКВ), постулирующая отсутствие потока генов между видами, не является адекватной, в том смысле, что многие зоологические или таксономические виды в настоящий момент времени еще не достигли стадии биологического вида. Тем не менее, также очевидно, что раньше или позже, но они становятся биологическими видами. Данное заключение поддерживается с нескольких позиций: увеличенными генетическими расстояниями в иерархии таксономических категорий и наименьшей дивергенцией на внутривидовом уровне по отдельным генам мтДНК, по полным митохондриальным геномам (митогеномам), а также по генам яДНК. 4. Исследование генетической дивергенции среди рыб с использованием обширной базы данных BOLD (www.boldsystems.org) обнаружило, что генные деревья для таксонов до уровня семейства являются преимущественно монофилетичными, а межвидовые ретикуляции, соответственно, редкими для большинства генных деревьев. 5. Имеющиеся данные позволяют заключить, что молекулярная эволюция и, в частности, генетическая дивергенция в иерархии таксонов в целом хорошо согласуются с БКВ и неодарвинизмом, составляя теоретическую базу успешности ДНК-штрихкодирования в мире животных, а также применимости и для других организмов.
ВВЕДЕНИЕ
ДНК-штрихкодирование получило широкое применение в биологии с 2003 г. (Hebert et al., 2003a,b). Однако истоки приложения изменчивости биологических макромолекул, например, к систематике и к эволюционной биологии имеют давние корни (Антонов, Белозерский, 1961; Антонов и др., 1971; Zuckerkandl, Pauling, 1965; Hubby, Throckmorton, 1965; Nanney, 1982). В целом, биологические молекулярные маркеры нашли многочисленные сферы применения в зависимости от потребностей современного общества. Биологический молекулярный маркер (ММ) – это любая макромолекула живого организма, способная быть идентификатором определенной функции, результатов биохимического, популяционного или эволюционного процесса. Изучение и использование ММ уже стало новой ветвью биомедицинской науки, о чем свидетельствует наличие специальных журналов (Biomarkers, Journal of Current Biomarker Findings, Biomarker Insights, DNA barcodes и др.). ММ применяются в биологии и медицине во многих областях. Можно отметить три наиболее значимые.
1. ДНК-штрихкодирование (DNA barcoding). ММ нашли применение в глобальной программе переописания биологического разнообразия на современной молекулярной и биоинформационной основе (iBOL, international barcode of life project; www.ibol.org). В качестве стандартного маркера, или ДНК-штрихкода, для большинства беспозвоночных и позвоночных животных используют последовательность нуклеотидов (далее для краткости – последовательность) гена Co-1, кодирующего субъединицу 1 цитохромоксидазы c митохондриальной ДНК (мтДНК). Для простоты работы, в качестве штрихкода используется первая половина гена длиной порядка 650 пар нуклеотидов (п.н.). Для растений больше подходят другие ММ или штрихкоды (Шнеер, Родионов, 2018; Жохова и др., 2019). Основа успешной идентификации изученных видов эукариот – низкая внутривидовая изменчивость (слабые различия последовательностей между особями одного вида), но на порядок большая межвидовая дивергенция образцов (между особями разных видов) – в среднем примерно 0.5–1.0% и 10%, по данным для животных (Картавцев, Ли, 2006; Картавцев, 2013; Kartavtsev, 2009, 2011a,b, 2013a; Stoeckle, Thaler, 2018, fig. 2).
2. ММ для идентификации стад, линий и пород животных. Для этого уровня Co-1 и другие ММ мтДНК не вполне подходят, поскольку относительно мало изменчивы внутри вида (хотя имеются исключения); обычно более консервативные у животных ММ ядерной ДНК (яДНК) еще менее применимы на этом уровне. Наибольшую результативность для выявления различий между популяциями, породами, линиями животных и для их паспортизации у высших организмов проявляют локусы микросателлитных ДНК и однонуклеотидные замены.
Одно из прикладных применений ММ – это идентификация гибридов и инвазивных видов. В связи с глобализацией и интенсификацией международной торговли продуктами питания огромное значение приобретает идентификация образцов при экспортно-импортных операциях. Фальсификация товарных марок, например филе рыбы, икры и других продуктов может быть точно установлена по ММ, что помогает государственным и частным предприятиям избежать значительных экономических и репутационных потерь (Nedunoori et al., 2017).
3. Наибольшее значение ММ приобрели в сфере медицины при диагностике заболеваний (в частности, рака молочной железы, предстательной железы, толстой кишки и др.), в криминалистике с целью исключения из подозреваемых тех или иных индивидуумов. В сферу применения ММ входит и мониторинг генетической безопасности для оценки рисков использования рекомбинантных ДНК и генетически модифицированных продуктов/объектов в пищевой и медицинской промышленности (Жохова и др., 2019).
Вышеперечисленные направления работ, кроме очевидных – применения в медицине и описания биоразнообразия, имеют исключительное значение для парадигм общей биологии, эволюционной генетики, а также для научной составляющей программы iBOL. В базе данных iBOL на 21.06.2018 аккумулированы результаты исследований 8 452 436 образцов живых организмов, при этом число образцов со штрихкодами – 6 108 027, а число идентифицированных по штрихкодам видов – 279 679 (http://v4.boldsystems.org/index.php/TaxBrowser_Home). Все эти данные сопровождаются унифицированной документацией по стандартам iBOL и доступны любому пользователю через интернет. Вклад РФ и RUS-BOL (http://www.imb.dvo.ru/misc/barcoding/index.htm) в исследования по ДНК-штрихкодированию отражен в централизованной базе данных BOLD (barcode of life database) (www.boldsystems.org) и составляет 32 177 опубликованных в базе записей, образующих 7097 кластеров штрихкодов, которые представлены 241 организацией (лабораторией). Сделанные в BOLD записи относятся к 16 764 видовым именам, представляют в целом 4438 видов. РФ по активности находится в середине списка участников программы, на уровне Бразилии и Франции.
Встречающаяся в подавляющем большинстве таксонов успешная идентификация видов на основе ДНК-штрихкодов составляет сейчас обширную естественнонаучную базу и требует объяснения и теоретического обоснования (Kartavtsev, 2013a, 2018; Stoeckle, Thaler, 2018). В одном из подходов для представления биологической основы этого феномена предлагается ориентироваться преимущественно на метрику “попарные расстояния”, эквивалентную р-расстоянию или доле различающихся нуклеотидов в паре случайно выбранных последовательностей (Картавцев, 2013; Nei, Kumar, 2000), а также оценивать молекулярные особенности Co-1 и мтДНК в целом (Stoeckle, Thaler, 2018). В другом подходе, реализуемом в данной работе, предполагается, что подобная особенность существует, в основном, благодаря преобладанию в природе географического способа видообразования, допускающего накопление организмами стохастических мутаций и уникальных замен нуклеотидов в цепях ДНК и других макромолекул при формировании дочерних популяций (таксонов) в условиях изоляции. При реализации этой модели особи разных видов экспериментально идентифицируемы посредством ДНК-штрихкодов, и, кроме того, при соответствующем анализе можно обнаружить корреляцию р-расстояний и рангов таксонов (Картавцев, 2013; Kartavtsev et al., 2011a,b).
Чтобы разобраться в этих вопросах требуется специальное рассмотрение эмпирических данных, разносторонний их анализ и сопряжение с генетическими основами видообразования, а также с соответствующими положениями Биологической Концепции Вида (БКВ) и, более широко, с неодарвинизмом, или Синтетической Теорией Эволюции (СТЭ). Актуальность такой работы также определяется необходимостью рассмотреть критику парадигм БКВ/СТЭ, базирующуюся на представлениях об обширной интрогрессии (Arnold, 1997, 2008; Arnold, Emms, 1998; Arnold, Fogarty, 2009) и ретикулярной эволюции (Боркин, Литвинчук, 2013; Arnold, Fogarty, 2009). Эти вопросы уже частично поднимались (Картавцев, 2005, 2009, 2013; Kartavtsev, 2011a,b; 2013 a,b).
В данной работе представлено совместное рассмотрение интрогрессии и ретикуляции, а также проведен анализ генетических расстояний для ММ 16S рРНК и для полных митохондриальных геномов (митогеномов) по выборке последовательностей из GenBank (www.ncbi.gov). Эти маркеры ранее не были использованы в сравнительно-эволюционном аспекте. Изменчивость генетических расстояний в иерархии таксонов по гену 16S рРНК вообще не представлена в литературе, а по митогеномам – ранее рассмотрена только для трех конкретных таксонов (Kartavtsev et al., 2016; Turanov et al., 2016; Redin, Kartavtsev, 2017). Одна из ключевых задач данного краткого обзора состоит в поиске ответа на вопрос, позволяют ли имеющиеся молекулярно-генетические данные сделать обобщения о широком присутствии генетической интрогрессии и ретикуляции в исследуемых генных деревьях, или же, наоборот, они согласуются с БКВ/СТЭ. Безусловно, сама СТЭ не догма и требует дальнейшего развития. В действительности, для биологии СТЭ – это общая эволюционная концепция и поэтому может именоваться теорией. Но с формально-научных позиций вряд ли она соответствует критериям теории. Теория должна содержать аналитическое описание в математических терминах или/и должна представлять строгую модель, скажем, компьютерную, и иметь прогностические компоненты. Понимание этого недостатка в соответствующей литературе имеется, как и периодически появляются фрагменты теоретических наработок различной степени подробности (Картавцев, 2009; Bush, 1975; Nei, 1987; Templeton, 1981, 1998; Kondrashov, 1998; Zhuravlev, Avetisov, 2006; Kartavtsev, 2009, 2011a,b, 2013a, 2015).
Данные о возможном влиянии интрогрессии генов на эволюцию видов, эволюционную судьбу таксонов, включая ретикуляции филогенетических деревьев, а также согласованность современных молекулярно-генетических данных в целом с главной современной эволюционной парадигмой, неодарвинизмом, представлены во многих публикациях (Barton, Hewitt, 1985; Campton, 1987; Arnold, 1997, 2008; Avise, 2000; Gerber et al., 2001; Arnold, Fogarty, 2009; Kartavtsev, 2013b; Stoeckle, Thaler, 2018). Учитывая проблематику обзора, важно уточнить некоторую филогенетическую терминологию в самом начале статьи. Термин “генное дерево” был введен довольно давно (Картавцев, 2009; Tateno et al., 1982; Nei, 1987). Генное дерево – это филогенетическое дерево, построенное по данным для одного гена. Этот термин противопоставляется понятию видового дерева (Картавцев, 2009, с. 189; Nei, 1987), которое включает филогенетический сигнал по нескольким генам, а также может инкорпорировать и другие признаки. Филогенетическое дерево, включая генное дерево, может иметь различную топологию, в том числе иметь единые корни для ветвей/узлов/кластеров (монофилия) или иметь иные ветвления (пара- или полифилия). Безусловно, имеются различные дискуссионные вопросы относительно БКВ/СТЭ. В данном обзоре рассматриваются в основном четыре вопроса: 1. Какие методы обнаружения гибридов и генетической интрогрессии, или потока генов, являются наиболее подходящими? 2. О чем свидетельствуют факты, полученные на основе маркеров мтДНК и яДНК? 3. Имеются ли в литературе данные о соответствии молекулярной изменчивости в филетических линиях или таксонах с БКВ/СТЭ? 4. Насколько часто встречаются ретикуляции и политомии генных деревьев, а также какой главный информационный сигнал выявляет их топология? Преимущественно в статье проанализированы данные для таксонов животных, но многие идеи относятся и к другим группам организмов.
КОНЦЕПЦИЯ, ТЕРМИНЫ И МЕТОДЫ ДЛЯ АДЕКВАТНОЙ ОЦЕНКИ ГЕНЕТИЧЕСКОЙ ИНТРОГРЕССИИ
Гибриды и гибридизация: уточнение этой и сопряженной терминологии
Для понимания сути генетической интрогрессии важны два понятия: гибрид и гибридизация. Гибрид – это генетическая помесь или потомство от скрещивания между генетически различными организмами. Гибридом можно также считать особь со смешанной родословной, метиса. В предельном случае гетерозигота по одному или нескольким локусам – это гибридная особь. Таким образом, скрещивание P1: A1A1 B2B2 × A2A2 B3B3 дает гибрид F1 = FH: A1A2 B2B3. Гибридизация – это процесс, посредством которого появляются гибриды. Между тем, следует отметить разницу между простым внутрипопуляционным скрещиванием и скрещиванием особей различных линий, популяций и видов. Однако в обычном понимании гибриды представляются потомками более отдаленных скрещиваний. Причем отдаленность скрещивания – условное понятие, зависящее от конкретного вида организмов и от сложившейся для него нормальной системы скрещиваний. Кроме F1, могут возникать другие типы гибридов: F1 × F1 = F2, F1 × P1 = Fb и т.д.
Гибридизация может быть искусственной или естественной природы. В данной статье изложение касается преимущественно естественной гибридизации. Как отмечено выше, важно понимание гибридизации и гибридов как генетических сущностей, определяемых в терминах генотипов и скрещиваний в природной среде, в норме между перекрестно размножающимися организмами. Агамные и клональные формы здесь не рассматриваются.
Гибриды не обязательно должны быть промежуточными между родительскими формами, но в зависимости от сложности скрещивания могут быть более близкими к одному из родителей, и в природе они, обычно, менее приспособлены по сравнению с родителями. Соответственно, гибридный индекс, например IH (Campton, 1987), может быть далек от 0.5 (Картавцев, 2009; Kartavtsev, 2015, ch. 10). При искусственном воспроизведении и линейной селекции возможен даже противоположный эффект с возникновением гетерозиса у гибридов при отдаленных скрещиваниях между инбредными линиями или определенными типами пород и сортов. Однако в природных популяциях избыточная изменчивость чаще не имеет положительного эффекта, создавая дополнительный генетический груз (Картавцев, 2009).
Необходимо уточнить еще несколько терминов. Во-первых, примем поток генов как процесс, маркируемый селективно нейтральными (или почти нейтральными) аллелями. Репродуктивная изоляция между биологическими видами означает отсутствие какого бы то ни было потока генов. Так, если гибридов нет или же F1, и особенно F2 или Fb, не плодовиты или не жизнеспособны (низко плодовиты, слабо жизнеспособны), то и потока генов нет, или он пренебрежимо мал. Наоборот, присутствие существенной доли особей F2 или Fb, а не F1, следует рассматривать как индикатор потока генов. Эта формулировка подходит под строгую или ортодоксальную БКВ (Тимофеев-Ресовкий и др., 1977; Dobzhansky, 1955; Mayr, 1982). Во-вторых, примем, что гибридная зона – это географическое пространство, где встречаются гибриды естественного происхождения между предполагаемыми родительскими формами. В такой зоне часто возникает клина. В‑третьих, будем считать, что клина – это постепенное (градиентное) или резкое изменение частот аллелей в прямом или обратном направлении: вид 1 (популяция 1) → гибриды → вид 2 (популяция 2), поддерживаемое балансом между расселением и отбором против гибридов (модифицировано из Barton, Hewitt, 1985).
Из представленных выше определений следует, что точное установление наличия гибридов возможно с помощью ядерных маркеров генов, позволяющих идентифицировать аллели обоих родителей в генотипе. Маркеры мтДНК, которые в норме наследуются у большинства животных как компоненты клеточной плазмы овоцитов, то есть по материнской линии, могут использоваться, но при определенных условиях. Так, присутствие в образцах фрагмента мтДНК вида X или полного митогенома при исследовании вида Y объяснимо произошедшим в прошлом событием гибридизации. Однако надо исключить при этом такие события, как горизонтальный перенос, рекомбинацию и др. Свидетельства присутствия гибридов, полученные только по маркерам мтДНК, следует рассматривать как предварительные, поскольку, как определено выше, генотип гибрида может быть точно идентифицирован лишь по ядерному геному. Иногда такие свидетельства могут отражать рекомбинацию участка гена мтДНК с яДНК. Например, такие события отмечены у карповой рыбы комплекса Gila robusta (Gerber et al., 2001). Также описан перенос мтДНК от ленка Brachymystax lenok в геном сибирского тайменя Hucho taimen (Balakirev et al., 2013). Оба этих примера являются только предварительными индикаторами возможной гибридизации, поскольку не содержат данных по маркерам яДНК.
Описания, показывающие, что таксономически различные рыбы могут скрещиваться и производить плодовитое потомство, хорошо документированы (Hubbs, 1955). Исследователи (Алтухов и др., 1997; Schwartz, 1972, 1981; Barley et al., 2001) суммировали данные из более четырех тысяч работ с примерами искусственной и естественной гибридизации у рыб, но в большинстве случаев генотипического документирования при этом не выполнено. Некоторые данные на эту тему представлены также в статье (Kartavtsev, 2013b).
Считается, что естественная гибридизация более обычна у рыб, чем в других группах позвоночных. К морским беспозвоночным подобное заключение, возможно, относится еще в большей степени. На первое место среди причин более частой гибридизации в данных группах надо поставить не столь развитую, как, например, у млеко-питающих, систему хромосомного определения пола. У большинства позвоночных животных пол определяется детерминирующим геном (Devlin, Nagahama, 2002), локализованным на Y-хромосоме. У рыб пол определяется несколькими причинами и лишь иногда половыми хромосомами, которые могут отсутствовать вовсе (Кирпичников, 1979). К этому можно добавить, что недавние молекулярные исследования выявили слабую связь фенотипа самцов и самок с генными маркерами пола у лососевых рыб (Podlesnykh et al., 2017). В основе повышенного уровня гибридизации в этих таксонах могут лежать несколько особенностей биологии рыб и беспозвоночных: внешнее оплодотворение, слабые поведенческие изолирующие механизмы, неравная численность двух потенциальных родительских видов, конкуренция за ограниченные нерестовые биотопы и, наконец, восприимчивость к вторичному контакту недавно дивергировавших форм (Avise, Sanders, 1984; Campton, 1987; Avise, 2001). В зависимости от локальных условий эти особенности могут сильно варьировать. Естественные и вызванные вмешательством человека изменения во внешней среде часто приводятся в качестве факторов, способствующих гибридизации у рыб (Алтухов и др., 1997). Например, гибридизация относительно обычна у пресноводных рыб из умеренных широт, где геологические и климатические условия начиная с плейстоцена резко изменялись, преобразуя пресноводную среду, тогда как морская среда оставалась относительно стабильной (Campton, 1987). Порожденные цивилизацией изменения экосистем в Северной Америке также коррелируют с повышенной гибридизацией, встречающейся между исходно аллопатрическими и естественно симпатрическими парами видов (Hubbs et al., 1953; Nelson, 1966, 1973; Stevenson, Buchanon, 1973). Для лососевых рыб такие примеры были суммированы отдельно (Алтухов и др., 1997; Simon, Nobble, 1968; Altukhov, Salmenkova, 1991). Имеющиеся оценки указывают, что гибридизация влияет на судьбу около 25% видов растений и до 10% видов животных (Arnold, 1997). Предполагается, что случаи гибридизации и последующей генетической интрогрессии, относятся, как правило, к молодым недавно дивергировавшим видам. Как отмечено ранее, основное внимание будет уделено видам животных, которые предположительно более удовлетворяют положениям БКВ в оригинальном понимании биологического вида (Mayr, 1982) или его версиям (Тимофеев-Ресовский и др., 1977; Картавцев, 2009, с. 86; Kartavtsev, 2015, p. 95). Близкие публикации рассматривают природоохранные аспекты (Genovart, 2008), недавние исторические изменения (Seehausen, 2004), а также частоту встречаемости естественных гибридов в различных таксонах: например у птиц (Grant, Grant, 1992) в сравнении с другими позвоночными (Prager, Wilson, 1975; Fitzpatrick, 2004).
Методы, используемые для идентификации гибридов
Имеются, по меньшей мере, четыре метода идентификации гибридов: 1) морфологический, 2) кариологический, 3) генетико-биохимический, 4) молекулярно-генетический (Картавцев, 2009; Campton, 1987; Kartavtsev, 2015). В принципе, методы 3 и 4 можно объединить в один, поскольку оба относятся к ММ. Первые два метода описаны ранее (Картавцев, 2009; Kartavtsev, 2015), и в обзоре для краткости изложения они будут опущены, хотя оба могут быть информативными в плане возможного присутствия гибридов в популяциях. Однако первый – можно рассматривать только как дающий непрямые индикаторы присутствия гибридизации, а второй – трудно применять на практике ввиду небольшого размера хромосом во многих группах организмов и трудоемкости методик. Тем не менее, хромосомные методы неоднократно с успехом применялись в природоохранных работах (Васильев, 1985; Фролов, 2000; Setzer, 1970; Greenfield, Greenfield, 1972; Greenfield et al., 1973; Busack et al., 1980; Phillips, Zajicek, 1982; Delaney, Bloom, 1984; Phillips et al., 1985).
ММ наиболее полно отвечают требованиям идентификации гибридов и для ответа на вопрос о существовании генетической интрогрессии. Установление фактов гибридизации и интрогрессии по таким ММ, как аллозимы и яДНК, является относительно точным, если два родительских предка имели различные мономорфные (фиксированные) аллели для одного или более локусов. Например, для двух локусов X и Y с двумя аллелями в каждом, X1 и X2 плюс Y2 и Y3, гибридная особь, FH: X1X2Y2Y3, однозначно отличима от родительских форм поскольку обе родительские формы имеют различные гомозиготные генотипы, а гибрид представлен гетерозиготным генотипом по всем диагностическим локусам. Однако если гибридизация продолжается дальше поколения F1, то потомство будет иметь широкий спектр рекомбинантных генотипов, включая генотипы, идентичные обеим родительским формам. Соответственно, особь с составным генотипом, которая идентична по генотипу одной из родительских форм, может быть гибридом F1, полученным при самовоспроизводстве или при возвратном скрещивании с одним из родителей. Присутствие рекомбинантных генотипов, иными словами, может быть признано свидетельством плодовитости гибридов и наличия второго поколения гибридов (F2 или Fb). В таких ситуациях становится совершенно необходимым выяснение характеристик генотипической изменчивости в популяционно-генетических терминах, с оценкой параметров миграции, Nm и m, дифференциации, Fst и т.п. В существующей литературе имеется не так много работ, использующих такие точные популяционно-генетические подходы, а обзоры отражают широкий спектр других примеров (Боркин, Литвинчук, 2013; Arnold, Fogarty, 2009). Краткое рассмотрение некоторых выводов, следующих из этих работ, будет дано ниже.
Эмпирические данные подтверждают, что аллозимы и маркеры яДНК наиболее эффективны, когда родители имеют различные фиксированные аллели (Картавцев, 2009; Campton, 1987; Kartavtsev, 2015; Kartavtsev et al., 2018) и/или когда проанализированы мультигеномные данные (Fong, Chen, 2010; Nevado et al., 2011) с оценками таких параметров, как Nm, m, Fst и др. Комбинированные подходы, например, с использованием мтДНК и яДНК могут быть даже более успешными, поскольку выявляют, например, направление скрещивания родителей по полу ввиду преобладания материнского наследования мтДНК у животных (Avise, 2001). Комплексный подход с использованием ММ и морфометрии позволяет оценить наличие генотипических эффектов, в частности гетерозиготности, на фенотип (Kartavtsev et al., 2005, 2018), что может иметь выход в марикультуру. Другие примеры успешного использования комбинации ММ были представлены для черепах рода Mauremys (Fong, Chen, 2010), для цихлид рода Ophthalmotilapia из оз. Танганьика (Nevado et al., 2011), для мидий комплекса Mytilus ex. group edulis, с использованием GLU-5 и других ММ (Картавцев, 2009; Heath et al., 1995; Inoue et al., 1995; Skurikhina et al., 2001; Kartavtsev, 2015; Kartavtsev et al., 2014, 2018), а также для некоторых других таксонов (Avise, 2001).
Подводя итоги вышеизложенного, следует отметить следующее.
1) Идентификация гибридов и выявление присутствия генетической интрогрессии являются сложными задачами. Прежде всего, эти задачи требуют выполнения точного генетического анализа с идентификацией гибридов на основе многих локусов и сопоставления потомков различных типов (F1, F2, Fb и т.п.). Впоследствии предполагается получение оценок частот аллелей, потока генов и обобщение всех вычлененных компонентов.
2) В этом контексте БКВ является базовой концепцией для выборочного тестирования групп организмов в генетических терминах; инбредные линии и агамные виды (линии организмов) не могут рассматриваться как представительные для понимания сути процессов.
3) Имеющиеся в распоряжении генетиков экспериментальные средства для анализа в целом являются прямыми и достаточными для выявления гибридов и для оценки уровня интрогрессии.
Осуществление генетической интрогрессии через видовые барьеры
Обнаружить гибридов часто является затруднительным или даже невозможным, если гибридизация успешна и длительна во времени, вследствие чего в потомстве имеются F1, F2, Fb и т.п. В смешанной популяции, если потомки возвратных скрещиваний или гибриды последующих за F1 поколений обычны, то возможно присутствие рекомбинантных генотипов с достаточно высокой частотой, и становится затруднительно отличить гибридов F1 от редких родительских гетерозигот, даже при использовании нескольких диагностических ядерных маркеров (Campton, 1987). Имеются специализированные программные средства, например Structure (http://pritch.bsd.uchicago.edu/structure.html) и т.п., которые помогают решить часть проблем с идентификацией гибридов при обилии данных. Однако надо заметить, что сам предмет исследования и численное моделирование являются настолько сложными, что найти однозначное популяционно-генетическое решение при массовой гибридизации бывает затруднительно. Особенно сложны случаи оценки уровня генетической интрогрессии и ее последствий во времени. Добавляет сложностей в решении задач отсутствие знания, достигли ли анализируемые гибридизующие формы видового статуса или нет. Речь идет о той ситуации, когда гибридизация обнаружена, но исследователь не обладает убедительными данными о таксономическом ранге объектов, которые при этом могут иметь разные видовые имена. В связи с этим точное разграничение видов является очень важным вызовом, стоящим перед современной эволюционной генетикой и эволюционной биологией в целом (Brower, 1999; Sites, Marshall, 2004; Kartavtsev, 2009, 2011a,b, 2015). Чтобы подступиться к этому весьма сложному предмету, удалось реализовать подход, который позволяет протестировать конгруэнтность двух информационных групп данных в BOLD. Первая группа содержит материалы зоологических коллекций (с образцами, отнесенными к определенным таксонам: видам, родам и семействам), а вторая – представляет собой образцы, идентифицированные по ММ, точнее, на основе ДНК-штрихкодов (Ratnasingham, Hebert, 2007), с присвоением каждому образцу уникального идентификатора (шифра) или специального индекса BIN (barcode index number).
Одно из первых исследований частоты встречаемости гибридов в природных популяциях выполнено на организмах водной биоты, на рыбах и беспозвоночных животных. В 1980-е гг. использовали мтДНК в комбинации с аллозимными локусами для исследования частоты гибридизации между 9 видами солнечных рыб (род Lepomis), населяющих две географические локальности на юго-востоке США (Avise, Saunders, 1984). Исследователи пришли к следующим заключениям: 1. Гибридизация происходит с относительно низкой частотой, но в нее вовлечены пять из девяти исследованных видов. 2. Не обнаружено ни мтДНК, ни аллозимных доказательств существования интрогрессии генов между видами рода Lepomis; все гибриды оказались исключительно потомством F1. 3. Каждый обнаруженный гибрид представляет скрещивание между наиболее распространенным и редким видом. 4. В шести из семи возможных гибридных находок материнский родитель был представителем редкого вида, как было установлено по генотипу мтДНК. Эта особенность была объяснена интенсивной конкуренцией среди самцов за партнера по спариванию и общим промискуитетом самок. С того времени число работ, в которых исследовались случаи межвидовой гибридизации в природе, многократно возросло. В большинстве из них показано присутствие в природных популяциях только гибридов F1, реже встречаются гибриды нескольких следующих поколений и потомки возвратных скрещиваний, именно посредством которых и происходит интрогрессия чужеродных генов в геномы родительских видов (Avise, 2001).
Примеры анализа мтДНК можно легко увеличить. Некоторые из них будут рассмотрены ниже. В частности, морские моллюски мидии Mollusca, Mytilidae часто использовались для исследований гибридных зон. Рассмотрим некоторые собственные данные для Японского моря, относящиеся к тихоокеанской мидии Mytilus trossulus и к инвазивному атлантическому виду M. galloprovincialis. В одной из ранних работ использовали комбинированный аллозимно-морфометрический подход, который выявил приблизительно 5% гибридов в водах России, Южной Кореи и Японии, с варьированием их доли по годам от 1.6 ± 0.9% до 8.9 ± 1.7% (Kartavtsev et al., 2005). На основе данных о преобладании потока генов в направлении M. trossulus → M. edulis → M. galloprovincialis, авторами выдвинуто предположение, что видовой ранг M. trossulus в комплексе Mytilus ex. group edulis, безусловно, достигнут. При этом таксоны M. edulis и M. galloprovincialis следует рассматривать в ранге подвиды/полувиды, если следовать определению ортодоксальной БКВ (Kartavtsev et al., 2005). Такое заключение хорошо согласуется с палеонтологическими датировками, согласно которым M. trossulus является наиболее древним элементом комплекса Mytilus ex. group edulis (Кафанов, 1987; Kartavtsev et al., 2005), а также с молекулярно-филогенетическими оценками дивергенции в семействе Mytilidae (Картавцев и др., 2018). В недавних работах вновь представлено комплексное популяционно-генетическое и морфометрическое исследование мидий этой группы из северо-западной части Японского моря (Kartavtsev et al., 2014, 2018). Генотипирование особей выполнено по восьми полиморфным ферментным локусам и двум маркерам яДНК (GLU-5 и ITS-1,2). Ферментные и ядерные маркеры обнаружили согласованную генетическую изменчивость частот у родительских видов и их гибридов. Генотипы местного вида M. trossulus преобладают в исследованных выборках, а гибриды встречаются с относительно невысокой частотой (рис. 1). Суммарная частота встречаемости инвазивного вида M. galloprovincialis была относительно низкой, но она достигала значения 42 ± 2% в одной из выборок в заливе Посьета, вблизи пос. Зарубино, являющегося также портом, в том числе для судов международной пассажирской линии Россия–Корея. В этом районе обнаружено также наибольшее число гибридов. Выполненный совместный анализ генетической и морфологической изменчивости выявил в целом низкую дифференциацию между поселениями мидий. Выраженный в величинах потока мигрантов на поколение (Nm) обмен между популяциями в обследованном регионе составил Nm = 5 (Kartavtsev et al., 2018). Доля мигрантов в предположении, что межвидовой поток генов обеспечивается, в основном, потомством F2, F3 и Fb, а не F1, оценена как Fb + F2, которая составила 0.9 ± 0.7%. Ранее поток генов для более обширного района сравнения в Японском море был оценен величиной Nm = 0.5–2.0 (Kartavtsev et al., 2005). Таким образом, полученные данные убеждают в сохранении инвазии M. galloprovincialis в северо-западной части Японского моря. Судя по встречаемости гибридов всех типов, можно полагать, что уровень генетической интрогрессии между двумя таксонами в регионе находится на низком уровне, варьируя за 14-летний период наблюдений в заливе Восток (залив Петра Великого, Японское море) в следующем диапазоне: 0% в 2012 и 2013 гг. (Kartavtsev et al., 2018) и 8.95 ± 1.68% в 1999 г. (Skurikhina et al., 2001). Данные поддерживают концепцию бимодальной гибридной зоны с ограниченным уровнем гибридизации между двумя номинальными видами в зоне контакта. В других участках ареала ситуация может отличаться. Однако, например, уровень рекомбинации, судя по аллозимному локусу MPI*, был выше среди edulis-подобных морфотипов, чем среди trossulus-подобных фенотипов (Penney, Hart, 1999). Эти результаты хорошо согласуются с данными исследования мидий Калифорнии, как упоминалось ранее, где авторами также отмечен низкий уровень интрогрессии с небольшим преобладанием ее направления M. trossulus → M. galloprovincialis (Saarman, Pogson, 2015). Можно полагать, что вообще асимметрия интрогрессии является общим правилом, следующим из разновозрастности видов (различия возникновения во времени) (Kartavtsev, 2013b; Kartavtsev et al., 2018).
Рис. 1.
Пример изменчивости частоты трех типов гибридов (Fh, Fb, F1) комплекса мидий Mytilus ex. group edulis в Японском море при отсутствии потока генов между видами и/или при его малом эффекте. По оси Х – номера выборок из поселений мидий в заливе Петра Великого Японского моря и в близких акваториях, по оси Y – частота гибридных особей (по Kartavtsev et al., 2018).

Имеются многочисленные данные, свидетельствующие, что маркеры мтДНК могут проникать из генофонда одного вида в другой, где существуют достаточно длительное время. Проникшие чужие гены могут оставаться в генофонде вида; при этом сохраняется биологическая интегрированность вида, которая была документирована с использованием других ММ и фенотипических признаков. Последние факты хорошо подтверждены данными для мышей, лягушек, рыб, мидий и других организмов (Yonekawa et al., 1981, 2000; Ferris et al., 1983; Avise, 2001; Gerber et al., 2001). Исследования генотипов мтДНК в комбинации с маркерами яДНК или аллозимными локусами дали экспериментальные доказательства способности мтДНК проникать из генома одного вида в другой, минуя видовые барьеры, при условии, что гибриды между видами и их потомство являются жизнеспособными и плодовитыми. Приведенные примеры могут указывать на отсутствие нарушений ядерно-цитоплазматического баланса (отрегулированного взаимодействия двух геномов). Это особенность интрогрессии генов мтДНК. Интрогрессия генов мтДНК проходит независимо от рекомбинационных и сегрегационных событий в ядерном геноме. Таким образом, компоненты мтДНК могут избегать естественного отбора, поддерживающего ядерно-цитоплазматическую совместимость, отмеченную давно (Takahata, Slatkin, 1984; Nei, 1987). Тем не менее, также давно известны факты другого рода, которые указывают на существование ряда селективных механизмов, определяющих ядерно-цитоплазматическое взаимодействие (Powell, 1983). Вопреки этой последней возможности, примеры реальной интрогрессивной гибридизации показывают, что подобный отбор, если и существует, не является настолько сильным, чтобы остановить генетическую интрогрессию посредством мтДНК, даже если само событие гибридизации достаточно редко. Соответственно, случаи нахождения чужеродной мтДНК у природных гибридов, которые были идентифицированы, скажем, по ММ яДНК, могут рассматриваться как выверенные доказательства событий гибридизации (предположительно, между близко родственными видами/таксонами). Хотя также очевидно, что и события гибридизации, и интрогрессия генов мтДНК могут не столь значимо влиять на эволюционную судьбу вовлеченных видов. Межвидовая передача мтДНК отмечена для широкого спектра организмов беспозвоночных (Drosophila) и позвоночных (Mus, Apodenus и Rana) (Yonekawa et al., 1981, 2000; Ferris et al., 1983; Powell, 1983; Clark, 1984; Takahata, Slatkin, 1984; Spolsky, Uzzell, 1984; Nei, 1987; Avise, 2001; Suzuki et al., 2004, 2008). Литература на эту тему уже была подвергнута сравнительному анализу (Картавцев, 2013; Campton, 1987; Avise, Wollenberg, 1997; Yonekawa et al., 2000; Suzuki et al., 2008; Kartavtsev, 2013b, 2015; Kartavtsev et al., 2018). С учетом анализа ядерно-цитоплазматического равновесия (Clark, 1984; Asmussen et al., 1987) для оценки интенсивности интрогрессии был предложен (Avise, 2001) оригинальный метод. Данная область достаточно быстро развивается, так что новые средства анализа появляются постоянно (Wiens et al., 2010; Joly, 2012); в настоящем обзоре представлен только краткий экскурс на тему интрогрессии мтДНК в связи с возможным потенциалом для оценки статуса вида в рамках БКВ.
Асимметричная интрогрессия, описанная для двух видов лягушек рода Hyla, является частой в природе (Avise, 2001). Выявленная однонаправленность подтверждается и многими другими работами (Kartavtsev et al., 2005, 2014, 2018; McGuire et al., 2007; Alves et al., 2008; Plotner et al., 2008; Keck, Near, 2009; Nevado et al., 2009; Saarman, Pogson, 2015). Это обеспечивается несколькими биотическими и абиотическими факторами. Подобная асимметрия для определенных пар видов, уже отмеченная выше и в ранее опубликованных работах (Kartavtsev et al., 2005, 2014, 2018; Kartavtsev, 2013b, 2018), оставляет нетронутым генофонд, по меньшей мере, одного из видов пары. Часто самопроникновение через видовые барьеры маркеров мтДНК, пластидных генов и, очевидно, некоторых мобильных элементов из чужих геномов может не приводить к дезинтеграции генома особей вида и не вызывать какого-либо прямого негативного эффекта. Однако возможно последующее опосредованное влияние на геном хозяина, например, через плейотропное действие внедрившихся генов или генетических элементов. Как предполагается БКВ, новые гены мтДНК и другие наследуемые элементы, захваченные в ходе интрогрессии, в некоторых случаях могут принимать участие в формировании репродуктивных изолирующих барьеров. Возникновение репродуктивных изолирующих барьеров зависит от становления дальнейших ядерно-цитоплазматических отношений и от других биологических и климатических событий, как это, очевидно, реализуется сейчас в комплексе видов мидий Mytilus ex. group edulis. Примером текущего развития репродуктивно-изолирующих барьеров в присутствии гибридизации, урбанизации, глобального мореходства и изменения климата могут служить различные пары видов упомянутого выше комплекса мидий (Skibinski et al., 1983; Gardner, Skibinski, 1988; Rawson et al., 1996, 1999; Skurikhina et al., 2001; Kartavtsev et al., 2005, 2014, 2018; Väinölä, Strelkov, 2011; Saarman, Pogson, 2015; Katolikova et al., 2016).
Мониторинг гибридизации и генетической интрогрессии можно считать одной из важнейших задач эволюционной генетики, общей генетики и общей биологии. Это становится особенно очевидным при исследовании многочисленных примеров генетической интрогрессии, а также ретикуляционных изменений в филогениях морских организмов (Avise, 2001; Arnold, Fogarty, 2009; Balakirev et al., 2012, 2013). Анализ некоторых данных обнаружения гибридов у животных (Боркин, Литвинчук, 2013; Arnold, Fogarty, 2009) показал, что многие факты в реальности не представляют межвидовую интрогрессию, но относятся к случаям внутривидовой интрогрессии, к интрогрессии между таксонами неясного таксономического ранга или между такими таксонами как подвиды/полувиды. Очевидно, что такие случаи не могут рассматриваться как противоречащие БКВ (Kartavtsev, 2013b, 2018). Данные некоторых исследований (Balakirev et al., 2013) не ясны в отдельных нюансах и, скорее, представляют случай очень редкой двойной рекомбинации, произошедшей при искусственном скрещивании отдаленных родителей или, вероятно, при редкой естественной гибридизации, произошедшей в далеком прошлом. Так, в одном случае гены мтДНК ленка B. lenok предположительно проникли от самки другого вида, что обнаруживается по присутствию последовательностей двух генов генома тайменя Hucho taimen (Balakirev et al., 2013). Однако, как следует из другого источника, это направление скрещивания дает несовместимость гибридов (Wang et al., 2011), тогда как обратное направление скрещивания дает даже гетерозис (Xu et al., 2011). В описанном случае (Balakirev et al., 2013) существует возможность, что обнаруженная рекомбинация, интерпретируемая как следствие широкой естественной гибридизации, на самом деле является последствием искусственных скрещиваний или лабораторных манипуляций с геномами. Эти данные, в любом случае, не имеют никаких доказательств распространенности гибридизации среди описываемых видов лососевых рыб из разных семейств, как и вообще не доказывают факт гибридизации, поскольку не опираются на диагностические маркеры яДНК. В другой обзорной работе (Arnold, Fogarty, 2009) приводятся примеры интрогрессии, часть из которых указывает на присутствие гибридов, но не доказывает факт генетической интрогрессии как таковой. Как отмечено ранее, присутствие гибридов F1 не доказывает существования интрогрессии генов (Campton, 1987). Для некоторых видов рыб гибридизация обычна (Campton, 1987), но только спорадически это ведет к собственно генетической интрогрессии (Avise, Saunders, 1984). Также были представлены убедительные данные, что события гибридизации и последующая интрогрессия были сопряжены с масштабными изменениями климата, как это следует из примеров для гольцов рода Salvelinus (Олейник, 2013; Glemet et al., 1998; Oleinik, Skurikhina, 2010; Roberts et al., 2010). Подобные события также прямо или опосредованно могут быть обусловлены активностью человека (Altukhov, Salmenkova, 1991; Avise, 2001). В упомянутом обзоре (Arnold, Fogarty, 2009) приведены примеры обнаружения гибридов по морфологическим признакам, но это вряд ли следует принять как доказательство не только генетической интрогрессии, но даже присутствия гибридов (Campton, 1987; Grant, Grant, 1992).
Тщательные исследования видов рыб и мидий, основанные на маркерах мтДНК и яДНК, обнаружили, что в большинстве выявленные гибриды являются F1-потомками, что можно рассматривать как слабость или полное отсутствие реальной генетической интрогрессии (Avise, Sunders, 1984; Kartavtsev et al., 2005, 2014, 2018; Oleinik, Skurikhina, 2010; Saarman, Pogson, 2015). Однако имеются работы, надежно доказывающие массовость продвинутых гибридов у рыб F2, F3 и т.д. (Roberts et al., 2010; Nevado et al., 2011). В одном случае маркеры мтДНК и ядерные микросателлитные локусы показали присутствие потомков возвратных скрещиваний особей F1 с одним из родительских видов с высокой частотой, равной 44.9% (Roberts et al., 2010). В другом случае с использованием различных комбинаций маркеров яДНК и мтДНК отмечен поток генов между четырьмя видами цихлид рода Ophthalmotilapia из оз. Танганьика (Nevado et al., 2011). Авторы в цитированной работе сообщают о значительной пропорции особей повсеместного в озере вида O. nasuta, обладающего частично маркерами мтДНК и яДНК, типичными для трех других номинальных видов рода. Все такие особи были обнаружены в популяциях, обитающих симпатрично с остальными видами рода. Авторы предполагают, что эта особенность возникла вследствие последовательных и независимых эпизодов генетического обмена в разных частях озера с однонаправленной интрогрессией в геном O. nasuta. Из представленных материалов следует, что даже в случаях тщательного анализа, не всегда имеются точные оценки пропорций F2 и подобных потомков, как это требуется в терминах оценки генетической интрогрессии. В отношении второй из цитированных выше работ также имеется вопрос о соответствии изученных таксонов видовому статусу в согласии с БКВ, хотя авторы и рассматривают род Ophthalmotilapia как старую валидную ветвь таксономических видов (Nevado et al., 2011). Но по поводу цихлид имеются и противоположные мнения (Mayer et al., 1998; Turner et al., 2001). Можно добавить к этой критике, что в некоторых работах разграничение потомства по категориям F1, F2, F3 и т.п. отсутствует. К примеру, в упомянутом обзоре (Arnold, Fogarty, 2009) в сводной таблице 1 представлены генетические данные об интрогрессии для 27 аутбредных видов из 32 сравниваемых видов и других категорий. То есть в подавляющем большинстве данные должны свидетельствовать, по мнению авторов, о существовании генетической интрогрессии. Однако если проанализировать эту информацию в соответствии с требованиями, представленными ранее, тогда приемлемой для доказательства генетической интрогрессии остается лишь малая доля из всего массива данных, а остальные относятся к обменам на внутривидовом уровне, между подвидами или таксонами неясного статуса.
Случаи интрогрессии, представленные, например, для морской флоры, водорослей и другой растительности (Balakirev et al., 2012), в целом согласуются с данными, известными для наземных растений (Шнеер, Родионов, 2018); то есть они подтверждают слабость репродуктивных барьеров у многих растительных видов. В любом случае, следует быть осторожными, заявляя об обнаружении гибридов по маркерам мтДНК: выявление гибридов по данным изменчивости мтДНК может привести к ложным заключениям. Например, мидии имеют двойное однородительское наследование мтДНК. В ходе эволюционной истории нуклеотидные последовательности 16S рДНК материнской и отцовской линий дивергировали на 8.3% в пределах одного и того же вида (Rawson, Hilbish, 1995). Соответственно, если в каком-то исследовании провести анализ дивергенции видов без учета различий по полу, можно прийти к ошибочным заключениям.
Дополнительные подробности теоретических и эмпирических исследований встречаемости гибридов, гибридизации и генетической интрогрессии можно найти (Боркин, Литвинчук, 2013; Barton, Hewitt, 1985; Smith, 1992; Arnold, 1997, 2008; Hewitt, 2011; Saarman, Pogson, 2015). Из изложенного ясно, что многие аспекты гибридизации достаточно сложны и не вполне ясны и, соответственно, вряд ли стоит ожидать быстрых и четких ответов на все вопросы. Среди позвоночных животных птицы чаще упоминаются в примерах встречаемости гибридизации 10–19% (Панов, 1989; Grant, Grant, 1992; Aliabadian, Nijman, 2007), тогда как амфибии и рыбы в этом плане цитируются реже, хотя чаще, чем рептилии и млекопитающие (Боркин, Литвинчук, 2013). Хотя на эту тему нет четкой статистики, так что это впечатление может быть связано с плохой репрезентативностью выборки таксонов в исследованиях по данному вопросу. В представленном выше материале приведены некоторые примеры количественной оценки уровня генетической интрогрессии. После анализа данных в сводной таблице (Arnold, Fogarty, 2009) следует, что реальное число случаев генетической интрогрессии завышено. Как упоминалось ранее, частота интрогрессивной гибридизации у животных оценена величиной 10% (Arnold, 1997), так что доля генетической интрогрессии в реальности должна быть ниже. В любом случае, в поддержку поставленной задачи настоящей работы можно заключить, что, вопреки соответствию или не соответствию БКВ, существующие в природе виды как биологические сущности в большинстве способны поддерживать свою интегрированность и самостоятельность; по меньшей мере, это прослеживается в тестируемой ретроспективе и предполагаемой перспективе. Следующие ниже данные должны помочь понять связь таксономии, и шире – БКВ/СТЭ, с молекулярной эволюцией.
ПРЕОБЛАДАЮЩАЯ ТОПОЛОГИЯ ГЕННЫХ ДЕРЕВЬЕВ, УРОВНИ ГЕНЕТИЧЕСКОЙ ДИВЕРГЕНЦИИ МЕЖДУ ТАКСОНАМИ И СТЕПЕНЬ СОГЛАСОВАННОСТИ МОЛЕКУЛЯРНЫХ ДАННЫХ С БКВ/СТЭ И С ПРАКТИКОЙ ДНК-ШТРИХКОДИРОВАНИЯ
Уточнение понятия вида
Решая поставленные в статье задачи, нельзя не уточнить сущность вида, о котором идет речь. По одному из первых определений в рамках БКВ, принято считать: вид – это репродуктивное сообщество популяций (репродуктивно изолированных от других совокупностей), занимающее специфическую нишу в природе (Mayr, 1982, p. 273). Такое концептуальное понятие вида в рамках БКВ принято в работе как основа излагаемых фактов и идей, хотя оно ограничено главным образом только бисексуальными организмами и имеет несколько уточнений (Майр, 1968; Тимофеев-Ресовкий и др., 1977; Картавцев, 2005; Templeton, 1998). В целом можно перечислить, по меньшей мере, семь различных определений понятия вид (Mayr, 1982; Simpson, 1961; Paterson, 1978, 1985; Wiley, 1978; Crawcraft, 1983; van Vallen, 1976; Templeton, 1998). Известная идея (Dobzhansky, 1955) позволила обосновать (Bush, 1975) ключевую роль процесса прекращения (разрыва) потока генов, который разъединяет исходный вид на две или более репродуктивно изолированные единицы, что ведет в дальнейшем к видообразованию. Можно даже заявить, что если будут представлены неоспоримые доказательства, что возникновение вида и его эволюция возможны без прекращения потока генов между предковыми формами, а вид может возникать при существовании широкого генетического обмена, тогда, конечно, придется отвергнуть БКВ/СТЭ-концепции. Аналогично, если существует и преобладает в природе модель видообразования, по которой новый вид может быть сформирован без долговременного разобщения генофондов родительской и дочерней форм, без возникновения репродуктивных изолирующих барьеров, тогда парадигмы БКВ/СТЭ должны быть заменены. Принимая концепции БКВ/СТЭ, также следует признать прямую связь между генетическим расстоянием и временем, как это ясно сформулировано для белковых локусов (Nei, 1987), а позже расширено для любых ММ (Drummond, Bouckaert, 2015).
Эмпирические сведения о топологии генных деревьев, уровни генетической дивергенции между таксонами
Имеются различные типы генных деревьев, но большинство из них выглядят как монофилетичные или иногда парафилетичные (полифилетичные) дендрограммы. Прямые оценки конгруэнтности различных генных деревьев или ожидаемой топологии, скажем, видового дерева не часты, а когда выполняются, показывают результаты, варьирующие в диапазоне от хороших до отличных (Birky, 2013; Hedges et al., 2015; Turanov et al., 2016; Kartavtsev et al., 2017). Неудовлетворительную “сходимость” топологии деревьев разных генов тоже можно наблюдать, но чаще это обусловлено информационными и техническо-лабораторными возможностями реконструкции. Например, недостаточной информационной емкостью используемых последовательностей (малой длиной) при большом числе анализируемых операционных таксономических единиц (ОТЕ), неудачным подбором ММ для выявления филогенетического сигнала при реконструкции дерева и другими погрешностями работы. В связи со случайной сортировкой филетических линий по отдельным генам более подходящим подходом может быть использование митогеномов. Уточним попутно, что операционной (операбельной) таксономической единицей исторически наименовали некоторые группы, предназначенные для анализа в нумерической систематике (Sneath, Sokal, 1963), представляющие в реальности отдельные организмы популяции, определенные таксономические группы, такие как виды и роды, которые обладают набором сходных признаков (Бейли, 1970, с. 156). Обычно смысловое понимание ОТЕ задается для конкретного исследования.
Успешность конструирования филогенетических деревьев на основе полного митогенома продемонстрирована для 100 таксонов рыб (Miya et al., 2003), а также для многих других групп и вариантов анализа, включая 13 белковых генов митогенома камбал Pleuronectiformes (Kartavtsev et al., 2016) или карповых рыб Cypriniformes, Cyprinidae (Imoto et al., 2013; Kartavtsev et al., 2017) и для многих других таксонов рыб, чьи представительные выборки яДНК-маркеров также были включены в анализ (Berendzen, Dimmick, 2002; Pardo et al., 2003, 2005; Saitoh et al., 2006; Mayden et al., 2009; Betancur-R, Orti, 2014). Сложный подход с цифровым моделированием и использованием временных деревьев для большого числа подобранных генов-кандидатов яДНК эукариот из 2274 исследований, представляющих 50632 видов (образцов последовательностей) глобального древа жизни, обнаружил, что генетическая дивергенция увеличивается с увеличением ранга таксона (Hedges et al., 2015). Главные выводы из представленных материалов и преобладающего топологического сигнала генных деревьев таковы: 1) в большинстве деревьев имеются очевидные(ая) ветви(вь) внешней группы; 2) внутри таксонов ранга отряда основные ветви (узлы, кластеры) представлены семействами/подсемействами; 3) ниже в иерархии имеются отчетливые ветви, представляющие различные рода семейств; 4) имеются наборы наиболее близких ветвей, состоящие из единичных особей, которые классифицируются как образцы одного и того же вида. Некоторая доля деревьев содержит неясные внутриродовые и внутрисемейные кластеры, представляющие в большинстве случаи парафилии или полифилии внутри таксона, которые требуют объяснения (обычно, последние данные ведут к ревизии таксона в систематике).
Непростой вопрос, как быть со всем массивом этой информации? На текущий момент, к примеру, нет общего подхода для оценки числа ошибочных отнесений к одному кластеру или ошибок идентификации соседей по ОТЕ в генных деревьях, выбранных из исследований и представленных в источниках литературы. Соответственно, нет очевидных путей оценки степени ретикуляции или политомии внутри деревьев. Попытки найти общее решение для количественной оценки биоразнообразия предпринимались на основе нескольких методов в рамках инструментария ДНК-штрихкодирования (Bringloe et al., 2016; Kartavtsev, 2018; Stoeckle, Thaler, 2018), хотя каждая из цитированных работ сфокусирована на различных целях. Авторам известно четыре общих подхода для решения этой задачи: 1) на основе использования Пуассоновского древоподобного процесса PTP (Poisson tree processes) (Zhang et al., 2013); 2) посредством метода, схожего с PTP, или коалесцентного подхода по генерализованной смешанной теории Юла GMYC (generalized mixed Yule coalescent) (Fujisawa, Barraclough, 2013); 3) на основе сравнения бифуркаций в видовых деревьях, реконструируемых по последовательностям (Joly, 2012); 4) посредством GMYC-подобной идеологии, K/θ-подхода (Birky, 2013).
Например, имеются осложнения в интерпретации данных генных деревьев Co-1 и Cyt-b камбал, которые содержат внутриродовые парафилетические кластеры для родов Hippoglossoides и Pseudopleuronectes (Kartavtsev et al., 2016, figs 1–2). Хотя для этих двух родов данный факт просто отражает неправильную морфологическую идентификацию некоторых образцов; другими словами, приведенный пример иллюстрирует синонимию имен для одних и тех же видов, что уже обсуждалось (Kartavtsev et al., 2007, 2016). Это один из вопросов, которые ДНК-штрихкоды помогают успешно разрешать. Возможны и другие проблемы ДНК-штрихкодирования, отмеченные при исследовании дождевых червей, среди которых по ДНК-штрихкодам выявлено криптическое биоразнообразие, но без четкого таксономического сигнала. Различными авторами также ставится вопрос в более широком контексте с диагностикой живых форм в слабо проработанных систематиками таксонах (Stoeckle, Thaler, 2018). Чаще, однако, проблема идентификаций решаема при более внимательном подборе ММ созданием референсной библиотеки штрихкодов для конкретного таксона или рассматриваемой задачи и уточнением понимания вида в таксоне. Обнаруженные по ММ ошибочные классификации в пределах ОТЕ обострили проблему, которая была известна в систематике для специалистов, а именно, существование многочисленной синонимии имен (Bringloe et al., 2016; Bayne et al., 2017; Kartavtsev, 2018), и которая обычно провоцирует многочисленные таксономические ревизии. Другая очевидная проблема возникает из-за невозможности иногда различить таксономическую ошибку идентификации образца и реальную ложную кластеризацию, выполненную по ММ в генном или видовом дереве и вызванную генетической ретикуляцией. Особенно эту задачу трудно решить для таксонов, далеких от непосредственной компетенции авторов. Имеются и другие осложнения, например, несогласованность темпов эволюции и, как следствие, несовпадение деревьев, полученных по маркерам мтДНК и яДНК (Wiens et al., 2010), различия в темпах замен для разных генов, сортировка филетических линий, связанная с генетически эффективным размером популяций-основателей, и некоторые другие (Nei, 1987; Avise, 2000; Drummond, Bouckaert, 2015).
В данной работе предложен подход к обозначенной проблеме на основе ММ и BOLD (Kartavtsev, 2018). Предлагается найти ответ на вопрос: имеется ли согласованность между 1) молекулярными классификациями по данным ДНК-штрихкодирования, выраженным в значениях метрики BIN, и 2) образцами, классифицированными экспертами в систематике и также размещенными в BOLD (Kartavtsev, 2018). Как отмечено ранее, BIN – это присвоение каждому образцу уникального идентификатора (или специального индекса), позволяющего отнести вновь тестируемый образец к тому или иному таксону из BOLD. Значения BIN сейчас определены главным образом на основе последовательностей Co-1. То есть спецификация или кодирование ОТЕ через BIN в BOLD выполнена, в основном, по Co-1. Но база пополняется быстро и другими маркерами. Значения BIN являются независимыми от предыдущих таксономических идентификаций образцов; то есть имеются два ряда независимых переменных, пригодных для сравнения. Иными словами, при использовании BIN появляется возможность оценить соответствие между кластерами ДНК-штрихкодов и описаниями тех же образцов в базе данных, обозначенных в рамках традиционной систематики. Значения BIN могут выявлять согласие двух независимых классификаций или несогласованность классификаций в сопоставляемых массивах образцов в BOLD, что и составляет основу для статистической оценки степени сходства этих двух рядов данных. Поскольку получаемый сигнал относится к иерархическим древовидным реконструкциям, то можно говорить об их конгруэнтности.
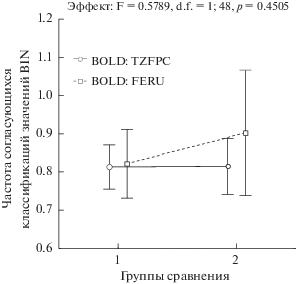
В цитированной работе (Kartavtsev, 2018) проанализированы данные из BOLD, представленные тремя проектами: TZFPC (Steinke et al., 2009), FERU (Turanov et al., 2014) и SCFAA (McCusker et al., 2013), в которых таксономическая экспертиза обеспечена авторами представленных проектов. Именно эти данные по рыбам были выбраны для простоты работы как наиболее близкие к профессиональным интересам авторов исследования. Результаты анализа, проведенного по значениям BIN, выявили, что 81.4 ± 2.3% образцов видов, 84.0 ± 3.9% образцов родов (рис. 2) и 88.0 ± 5.8% образцов семейств (Kartavtsev, 2018) этих проектов BOLD согласовывались с предварительными зоологическими классификациями представленных в них таксонов рыб. Выяснилось, что даже для уровня семейства единственный ММ Co-1 с неполной последовательностью нуклеотидов хорошо подходит для идентификации образцов. Оказалось также, что нет значительных различий между тремя таксономическими уровнями (Kartavtsev, 2018, tab. 1S), а данные из двух разных проектов статистически значимо не отличаются (рис. 2). Дополнительные детали этого анализа можно найти в цитированной работе и прилагаемых электронных материалах (Kartavtsev, 2018, tab. 1S). В представленном анализе log-преобразованные численности всех значений BIN (образцы, определенные морфологически) и согласующиеся классификации среди значений BIN (образцы, идентифицированные по ММ) показали пропорционально-положительную изменчивость, которая хорошо аппроксимировалась уравнением линейной регрессии; результаты анализа справедливы для комбинированных данных из нашего собственного исследования и данных коллег FERU/TZFPC, а также только из литературных данных по проекту SCFAA. Коэффициент детерминации этой линейной зависимости составляет около 90% (rp = = 0.989, p < 0.001, для наименьшего эффекта; Kartavtsev, 2018). Из информации, представленной на рис. 2, и этих данных следует, что все последовательности/особи одного и того же вида определяются в подавляющем большинстве случаев как кластеры одного вида, тогда как различные виды классифицируются в отдельные роды, в согласии с традиционной таксономической практикой. Такой же вывод справедлив в отношении кластеризации образцов различных родов. Эти два типа ветвей на внутри- и межвидовом уровнях ясно обособлены на основе любой из шкал генетических расстояний (Картавцев, Ли, 2005; Kartavtsev, 2009) и используются как одно из главных средств ДНК-штрихкодирования в iBOL и близких проектах. Исходя из изложенного материала, теперь понятно, почему существует явное генеральное свойство формирования кластеров ММ, включая ДНК-штрихкоды по Co-1, которые представляют биологический вид. Имеются исключения из общего правила, когда вид представлен единой репродуктивной единицей. Исключения включают, прежде всего, случаи с филогеографической подразделенностью видов, за счет достаточно изолированных подвидов (Avise, 2000). Существуют также ситуации, когда виды имеют общие или перекрывающиеся кластеры ДНК-штрихкодов вследствие сложной истории их формирования и интрогрессии мтДНК (Stoeckle, Thaler, 2018, figs 2,3), что усложняет задачу идентификации. Возможны связанные с биологией исключения из общей закономерности, приводящие к отсутствию дивергенции по ММ, например, при видообразовании не по географической модели D1 (по классификации Templeton, 1981) с аккумулированием мутаций-замен во время длительного существования в изоляции, а за счет регуляторных генных изменений, хромосомных трансформаций и т.п. – это способы видообразования D3, T2–T4 (по Templeton, 1981).
Рис. 2.
Двухфакторный дисперсионный анализ распределения средних значений BIN в двух исследованиях рыб, суммированных по BOLD. Средние значения для BIN показывают согласованные классификации (по оси Y, %) по молекулярному маркеру Co-1 мтДНК видов/образцов и его согласованность с независимым диагнозом по традиционным признакам, представленным в BOLD. Группы сравнения (ось X) представлены двумя категориями: отдельные виды (1) и самостоятельные роды (2). Значения согласующихся классификаций не различаются в двух сопоставленных группах (1 и 2). Они также не отличаются между оценками в двух исследованиях (Steinke et al., 2009, TZFPC; Turanov et al., 2014, FERU), которые представили 1219 и 285, а также 199 и 64 проанализированных образцов и видов соответственно. Суммарная согласованность классификаций на основе оценок BIN составила более 82% (точки с доверительными интервалами на графике); следовательно, величина ошибочных классификаций составляет менее 18% (по Kartavtsev, 2018).
Прежде чем выполнить последующий анализ, в котором будут рассмотрены данные о генетических расстояниях, вычисленных на основе последовательностей ММ, обратимся еще раз к филогении на уровне семейств. Это весьма важный пункт. Наличие монофилии на этом уровне, особенно для обширных семейств камбалообразных рыб Pleuronectidae, Soleidae и Bothidae, базовая информация для которых сегодня является морфолого-анатомической (FishBase; www.fishbase.org), имеет принципиальную значимость. К счастью, сравнительная информация для этих таксонов также получена и на основе молекулярной филогенетики (Berendzen, Dimmick, 2002; Pardo et al., 2005; Sharina, Kartavtsev, 2010; Betancur-R, Orti, 2014). Монофилия как минимум трех филетических линий (семейств) в пределах Pleuronectoidei исходно базировалась на данных 12S и 16S рДНК (Berendzen, Dimmick, 2002) и 16S рДНК для нескольких семейств (Pardo et al., 2005), а позже дополнена данными для Co-1, Cyt-b и митогенома преимущественно для Pleuronectidae (Kartavtsev et al., 2016). Полное понимание филогении для последнего семейства развивается и недавно было пополнено ММ по нескольким генам мтДНК и яДНК (Betancur-R, Orti, 2014; Vinnikov et al., 2018). Причем монофилия и отряда по некоторым данным выглядит достаточно убедительно (Betancur-R, Orti, 2014; Kartavtsev et al., 2016). Некоторые осложнения, как например присутствие парафилии в подсемействе Pleuronectinae и в роде Limanda, которые были недавно установлены на основе личиночной морфологии и молекулярно-филогенетических данных, очевидно, неизбежны (Roje, 2010; Kartavtsev et al., 2016). Есть сообщение о межвидовой гибридизации некоторых таксонов камбал Европы (Kijewska et al., 2009). Однако пока не ясно, есть ли парафилия даже для этих таксонов, или, может быть, политомию следует отнести к неадекватности морфологических признаков для идентификации, а не к гибридизации и формированию полностью гибридных популяций (и потенциальных таксонов) в зоне последнего исследования на Балтике, а также в соседних бассейнах Европы. Хотя подобные ситуации здесь и не исключены, как отмечено, например, для мидий в этом регионе (Smietanka et al., 2014; Väinölä, Strelkov, 2011).
Другой таксон, карпообразные рыбы Cypriniformes, был подвергнут генетико-биохимическим и молекулярно-филогенетическим исследованиям даже более интенсивно, чем камбалы (Hanzawa et al., 1984; Kartavtsev et al., 2002; Miya et al., 2006; Sakai et al., 2006; Semina et al., 2006; Saitoh et al., 2006, 2010; Sasaki et al., 2007; Mayden et al., 2009; Batischeva et al., 2011; Imoto et al., 2013; Kartavtsev et al., 2017). Для этих рыб, вопреки многочисленным находкам гибридов, существованию полиплоидных форм и предположениям о видообразовании посредством гибридизации (Saitoh et al., 2010; Yang et al., 2015), имеющиеся данные для деревьев отдельных генов и митогеномов указывают на отсутствие преобладания ретикулярной эволюции. Деревья имеют, как правило, бифуркационный тип и монофилию подавляющего большинства ветвей таксонов. Конечно, определенные проблемы в молекулярной систематике этого весьма разнообразного таксона существуют. Они будут возникать и далее при конструировании больших деревьев и при их сравнении с небольшими деревьями. Например, было обнаружено, что большие генные деревья ельцов Leuciscinae имеют гораздо меньшую конгруэнтность по оценке на основе средств программного пакета Dendroscope, по сравнению с небольшими деревьями (Kartavtsev et al., 2017). Однако, очевидно, что это лишь требует увеличения информационной емкости сигнала для лучшего разрешения топологии.
Кроме упомянутых, имеются сведения, противоречащие изложенным выше данным, которые поддерживают наличие ретикуляций в топологии деревьев. Подобный сигнал обнаружен в деревьях, построенных для таксонов из гибридных зон, как это недавно документировано для комплекса Mytilus ex. group edulis (Smietanka et al., 2014; Zbawicka et al., 2014, 2018). Комплексы богатой разнообразием тропической и неотропической фауны также представляют примеры ретикуляций (Nevado et al., 2011; Pereira et al., 2013). В уже цитированном обзоре (Arnold, Fogarty, 2009, tab. 1) имеются 17 находок филогенетических рассогласований. Однако эти факты не опровергают главный сигнал, свидетельствующий о преобладании бифуркаций и монофилии в генных деревьях, а также способность ДНК и других ММ идентифицировать таксоны рыб с точностью выше 80% (Pereira et al., 2013; Turanov et al., 2014; Kartavtsev, 2018). Доказательства, базирующиеся на ДНК-штрихкодировании, основанные на обширных материалах BOLD, поддерживают эти заключения для иерархических категорий до родового уровня (на что преимущественно и ориентирована программа iBOL). Однако приведенные выше факты неожиданно доказывают распространение этого сигнала и до уровня семейства. Хорошее соответствие топологического сигнала генных деревьев по данным ДНК-штрихкодов (то есть для неглубоких филогений) и существующих таксономических иерархий недавно продемонстрировано для нескольких филумов, включая Chordata, Arthropoda, Mollusca, Echinodermata, представляющих 75% всех описанных видов этих таксонов (Stoeckle, Thaler, 2018).
Как предполагалось ранее, обратимся к данным о генетических расстояниях и к их соответствию рангам эволюционной дивергенции. Один набор такой информации был суммирован для двух генов мтДНК Co-1 и Cyt-b (Kartavtsev 2009, 2011a, b, 2013a, 2015, Ch. 7). Эти данные будут приобщены к выводам, даваемым ниже. В настоящей сводке приведены новые данные, которые показывают изменчивость мтДНК для гена 16S рРНК и полного митогенома животных в иерархии таксонов или групп сравнения (рис. 3а,б). Последовательности 16S рРНК длиной после выравнивания 191–444 п.н. различных видов животных извлечены из GenBank, 01.05.2018 и подвергнуты выравниванию в программном пакете MEGA-6 (Tamura et al., 2013). Затем в этом же программном пакете авторами были рассчитаны попарные р-расстояния для двух выборок размерами 6673 и 693 особи, соответственно для первой и второй половины последовательностей всего исходного набора (данная процедура понадобилась ввиду невозможности выполнить выравнивание одновременно из-за слишком больших различий последовательностей в исходном файле GenBank). После этого в программном пакете Statistica-6 (STATISTICA, 2001) с помощью однофакторного дисперсионного анализа проведено сравнение изменчивости внутри и между сравниваемыми группами (рис. 3а). Анализ 7459 последовательностей полного митогенома животных со средней длиной 15173 п.н. (данные GenBank, 13.02.2006–27.04.2018, длина: 15667, 15347, 14761, 16117, 13974 п.н.) выполнен примерно в таком же порядке. Однако расчеты р‑расстояний ввиду больших массивов данных первоначально выполнены по отдельности для каждой из групп сравнения, а затем сведены в общую таблицу для всех групп, на основе которой затем проведен однофакторный дисперсионный анализ в Statistica-6 (рис. 3б). Учитывая отклонения от нормального распределения вариационных рядов р-расстояний (хотя для представленных файлов они одномодальны и пригодны для этого анализа), кроме параметрического ANOVA, проиллюстрированного на рис. 3, также выполнен непараметрический дисперсионный анализ по Краскалу–Уоллису. Это тестирование подтвердило значимость различия сравниваемых групп, как для последовательностей гена 16S рРНК, так и для последовательностей митогенома. Табличные материалы для уточнения подробностей расчетов могут быть представлены авторами для заинтересованных читателей по запросу. Данные о генетических расстояниях внутри видов и в иерархии таксонов убеждают в том, что в большинстве исследованных филумов имеется зависимость между р-расстоянием и рангом таксона, близкая к линейной, с минимальными величинами в группе сравнения внутри видов. Как следует из полученных данных, видовые кластеры на огромном массиве данных ДНК-штрихкодов отчетливо выделяются не только по отдельным генам, но и по митогеномам в целом (рис. 3б) (Stoeckle, Thaler, 2018). Есть также исключения из этого правила, когда разрывы между видовыми кластерами минимальны или отсутствуют вовсе (Turanov et al., 2014; Kartavtsev et al., 2017; Stoeckle, Thaler, 2018). В основном такие случаи относятся к таксонам, не достигшим видового уровня (подвиды, полувиды или молодые виды), но могут быть также обусловлены присутствием видов (форм), не формирующих размножающихся половым путем репродуктивных единиц, то есть видов, не подпадающих под БКВ, которые могут возникать в результате генетической трансформации, не затрагивающей или мало влияющей на структурные гены (Картавцев, 2009, 2013; Kartavtsev, 2009, 2011a,b, 2013a), как это уже было отмечено выше. Известны также примеры значительной вариабельности скорости молекулярной эволюции для таксонов одного ранга в различных филумах, которые могут затруднять применимость генетических расстояний для межвидовых сравнений (Картавцев, 2009, 2013; Avise, Aquadro, 1982; Avise, 2000; Kartavtsev, 2009, 2011a,b, 2013a). Поэтому универсальность ДНК-штрихкода и шкалы генетических расстояний не является абсолютной, что не вполне осознается в некоторых обобщениях по ДНК-штрихкодированию (Stoeckle, Thaler, 2018). Выходом является создание алгоритмического инструментария и программных средств более универсального подхода, учитывающего необходимость использования различных мер генетической дивергенции и изменчивости, с использованием нескольких генотипических дескрипторов, а также фенотипических идентификаторов (Картавцев, 2009, 2013; Kartavtsev, 2009, 2011a, b, 2013a, 2015).
Рис. 3.
Однофакторный дисперсионный анализ, показывающий изменчивость средних значений р-расстояний (ось Y) для групп сравнения сопоставленных последовательностей 16S рРНК и митогеномов животных. Группы сравнения (ось X): 1 – внутри видов; 2 – внутри родов; 3 – внутри подсемейств–семейств; 4 – внутри отрядов; (а) – данные анализа последовательностей 16S рРНК животных второго набора последовательностей (GenBank, 01.05.2018); (б) – данные анализа последовательностей полного митогенома животных (GenBank, 13.02.2006–27.04.2018).

Обобщая все полученные данные, можно сформулировать три главных вывода.
Вывод 1. Диагностика видов является высокоэффективной в силу низкой внутривидовой и значительной межвидовой изменчивости используемых ММ (рис. 3, группы 1 и 2), что обнаруживается как на уровне маркеров отдельных генов мтДНК (Со-1, Cyt-b) и 16S рДНК, так и на уровне полных митогеномов. Это заключение применимо к широкому спектру таксонов животных, для которых есть определенная концептуальная ясность, что такое вид.
Вывод 2. Положительная связь расстояния и ранга таксона с минимумом на внутривидовом уровне доказывает, что видообразование в основном происходит в соответствии с географической моделью D1-типа (Templeton, 1981), а в животном мире преобладает филетическая эволюция. Последние утверждения в полной мере подтверждают базовые принципы БКВ и в целом неодарвинистскую трактовку видообразования и эволюции.
Вывод 3. Из представленных материалов следует, что альтернативные модели видообразования (D3, T2–T4 и др.) редки в природе. В противоположном случае, то есть, если в природе реализуется множество равновероятных способов видообразования, распределение расстояний, ранжированных по таксонам, имело бы равномерный вид или слабый наклон. Очевидно, что представленный анализ и другие доказательства опровергают такую возможность. Однако это не значит, что альтернативных способов нет или они менее важны. Подтверждается справедливость известного положения, что дарвиновская эволюция, дающая наблюдаемое разнообразие живых форм, преобладает во времени, но кардинальные генетические трансформации, хотя и редко возникающие в эволюции, ведут к ароморфозам и дают принципиальные новации живой материи. Хотя это дискуссионный вопрос, который не нашел еще надежного фактического обоснования.
ЗАКЛЮЧЕНИЕ
Идентификация видов на основе молекулярных маркеров, а точнее на основе последовательностей нуклеотидов ДНК или ДНК-штрихкодов, является весьма успешной, как убеждают обширные данные мировой базы данных, BOLD. Однако теория, которая объяснила бы данный факт, пока не создана. В представляемой работе предложен подход, базирующийся на величине индекса ДНК-штрихкодирования, BIN. Используя BIN и массив BOLD, удалось осуществить идентификацию (выделение) таксонов трех рангов (вид, род и семейство) и проделать статистическую оценку состоятельности идентификации, которая достигает более 80%. Кроме того, доказано, что для большинства построенных генных деревьев для маркера Со-1 (ген цитохром оксидазы с, субъединица 1) существует хорошее соответствие их топологии и общепринятой таксономической классификации в иерархии таксонов (вид, род, семейство), по меньшей мере, для проанализированных обширных данных по рыбам. Полученные знания об отклонениях от предсказанных значений для идентификации видов и определения ранга других таксонов позволят улучшить систему описания биоразнообразия на точной молекулярной и биоинформационной основе. Основной вывод из проделанного анализа состоит в том, что успешная идентификация по молекулярным маркерам возникает вследствие преобладания в природе географического способа видообразования. В ходе длительного во времени процесса формирования видов с накоплением мутаций и соответствующих многочисленных нуклеотидных замен в различных дочерних предковых таксонах, могут возникать уникальные последовательности нуклеотидов, ДНК-штрихкоды, которые обнаруживаются посредством молекулярных маркеров. Последующая независимая эволюция возникших филетических линий, включая дальнейшую дивергенцию до уровня рода, семейства и далее, и составляет фактическую базу обнаруженной связи величины BIN и ранга для трех таксонов, выделенного посредством традиционного морфологического подхода. В целом связь генетических расстояний и ранга таксона доказана для различных групп животных по данным для отдельных генов и для полных митогеномов.
ФИНАНСИРОВАНИЕ
Исследование поддержано грантом Российского научного фонда (соглашение № 14-50-00034) в области молекулярной систематики морских организмов, а также грантом Российского фонда фундаментальных исследований 15-29-02456-офи по направлению исследования генетических основ биоразнообразия, а также грантом ДВО РАН по программе “Дальний Восток” № 18-4-040 по тематике Комплексное исследование биоразнообразия рыб и беспозвоночных животных на основе ДНК-штрихкодирования, разработки и поддержки баз данных и биобанкинга.
Список литературы
Алтухов Ю.П., Салменкова Е.А., Омельченко В.Т. Популяционная генетика лососевых рыб. М.: Наука, 1997. 288 с.
Антонов А.С., Белозерский А.Н. Сравнительное изучение нуклеотидного состава дезоксирибонуклеиновых кислот некоторых позвоночных и беспозвоночных животных // Докл. АН СССР. 1961. Т. 138. С. 1216–1220.
Антонов А.С., Мирошниченко Г.П., Слюсаренко А.Г. Данные о первичной структуре ДНК в систематике растений // Успехи соврем. биол. 1971. Т. 74. С. 247–261.
Бейли Н. Математика в биологии и медицине. М.: Мир, 1970. 326 с.
Боркин Л.Я., Литвинчук С.Н. Гибридизация, видообразование и систематика животных // Труды ЗИН РАН. 2013. Прилож. 2. С. 83–139.
Васильев В.П. Эволюционная кариология рыб. М.: Наука, 1985. 209 с.
Жохова Е.В., Родионов А.В., Повыдыш М.Н. и др. Современное состояние и перспективы использования ДНК-штрихкодирования и ДНК-фингерпринтинга для анализа качества лекарственного растительного сырья и лекарственных растительных препаратов // Успехи соврем. биол. 2019. № 1. В печати.
Картавцев Ю.Ф. Молекулярная эволюция и популяционная генетика. Владивосток: ДВГУ, 2005. 234 с.
Картавцев Ю.Ф. Молекулярная эволюция и популяционная генетика. Владивосток: ДВГУ, 2009. 280 с.
Картавцев Ю.Ф. Генетическая дивергенция видов и других таксонов. Географическое видообразование и генетическая парадигма Неодарвинизма в действии // Успехи соврем. биол. 2013. № 5. С. 419–451.
Картавцев Ю.Ф., Ли Д.С. Анализ нуклеотидного разнообразия по генам цитохрома b и цитохромоксидазы 1 на популяционном, видовом и родовом уровнях // Генетика. 2006. Т. 42. № 4. С. 437–461.
Картавцев Ю.Ф., Шарина С.Н., Чичвархин А.Ю. и др. Генетическая дивергенция мидий (Mollusca, Mytilidae) по нуклеотидным последовательностям ядерных генов 28S рРНК, 18S pPHК и H3 // Генетика. 2018. V. 54. № 6. P. 639–660. (Kartavtsev Yu.Ph., Sharina S.N., Chichvarkhin A.Yu. et al. Genetic divergence of mussels (Mollusca, Mytilidae) based on the 28S rRNA, 18S rRNA, and H3 nuclear gene sequences // Genetika. 2018. V. 54. № 6. P. 639–660.)
Кафанов А.И. Подсемейство Mytilinae Rafinesque, 1815 (Bivalvia: Mytilidae) в кайнозое северной Пацифики // Фауна и распределение моллюсков: северная Пацифика и Полярный бассейн. Владивосток: ДВНЦ АН СССР, 1987. С. 65–103.
Кирпичников В.С. Генетические основы селекции рыб. Л.: Наука, 1979. 392 с.
Майр Э. Зоологический вид и эволюция. М.: Мир, 1968. 398 с.
Олейник А.Г. Молекулярная эволюция гольцов рода Salvelinus: филогенетические и филогеографические аспекты: Автореф. дис. … д.б.н. Владивосток: ИБМ, 2013. 47 с.
Панов Е.Н. Гибридизация и этологическая изоляция у птиц. М.: Наука, 1989. 510 с.
Тимофеев-Ресовский М.В., Воронцов Н.Н., Яблоков А.В. Краткий очерк теории эволюции. М.: Наука, 1977. 301 с.
Фролов С.В. Изменчивость и эволюция кариотипа лососевых рыб. Владивосток: Дальнаука, 2000. 229 с.
Шнеер В.С., Родионов А.В. ДНК-штрихкоды растений // Успехи соврем. биол. 2018. Т. 138. № 6. С. 531–537.
Aliabadian M., Nijman V. Avian hybrids: incidence and geographic distribution of hybridization in birds // Contrib. Zoology. 2007. V. 76 (1). P. 59–61.
Altukhov Yu.P., Salmenkova E.A. The genetic structure of salmon populations // Aquaculture. 1991. V. 98. P. 11–40.
Alves P.C., Melo-Ferreira J., Freitas H., Boursot P. The ubiquitous mountain hare mitochondria: multiple introgressive hybridization in hares, genus Lepus // Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008. V. 363. P. 2831–2839.
Arnold M.L. Evolution through Genetic Exchange. N.Y.: Oxford Univ. Press, 1997.
Arnold M.L. Evolution through Genetic Exchange. N.Y.: Oxford Univ. Press, 2008.
Arnold M.L., Emms S.K. Paradigm lost: natural hybridization and evolutionary innovation // Endless forms: species and speciation / Eds D.J. Howard, S.H. Berlocher Oxford–N.Y.: Oxford Univ. Press, 1998. P. 379–389.
Arnold M.L., Fogarty N.D. Reticulate evolution and marine organisms: the final frontier? // Int. J. Mol. Sci. 2009. V. 10. P. 3836–3860. DOI: 10.3390/ijms10093836
Asmussen M.A., Arnold J., Avise J.S. Definition and properties of disequilibrium statistics for associations between nuclear and cytoplasmic genotypes // Genetics. 1987. V. 115. P. 755–768.
Avise J.C. Phylogeography. The history and formation of species. Cambridge-London: Harvard Univ. Press, MA, 2000. 447 p.
Avise J.C. Cytonuclear genetic signatures of hybridization phenomena: rationale utility and empirical examples from fishes and other aquatic animals // Rev. Fish Biol. Fisheries. 2001. V. 10. P. 253–263.
Avise J.C., Aquadro C.F. A comparative summary of genetic distances in the vertebrates: pattern and correlations // Evol. Biol. 1982. V. 15. P. 151–185.
Avise J., Saunders N.C. Hybridization and introgression among species of sunfish (Lepomis): analysis by mitochondrial DNA and allozyme markers // Genetics. 1984. V. 108. P. 237–250.
Avise J.C., Wollenberg K. Phylogenetics and origin of species // PNAS USA. 1997. V. 94. P. 7748–7755.
Balakirev E.S., Krupnova T.N., Ayala F.J. DNA variation in the phenotypically-diverse brow alga Sacharina japonica // BMC Plant Biol. 2012. V. 12. P. 108–126.
Balakirev E.S., Romanov N.S., Mikheev P.B., Ayala F.J. Mitochondrial DNA variation and introgression in Siberian taimen Hucho taimen // PLoS One. 2013. V. 8 (8). P. e71147.
Barley D.M., Rana K., Immink A.J. The use of inter-specific hybrids in aquaculture and fisheries // Rev. Fish Biol. Fisheries. 2001. V. 10. P. 325–337.
Barton N.H., Hewitt G.M. Analysis of hybrid zones // Ann. Rev. Ecol. Syst. 1985. V. 16. P. 113–148.
Bayne B.L., Ahrens M., Allen S.K. et al. The proposed dropping of the genus Crassostrea for all Pacific cupped oysters and its replacement by a new genus Magallana: a dissenting view // Shellfish Res. 2017. V. 36 (3). P. 545–547.
Berendzen P.B., Dimmick W.W. Phylogenetic relationships of Pleuronectiformes based on molecular evidence // Copeia. 2002. V. 3. P. 642–652.
Betancur R.R., Orti G. Molecular evidence for the monophyly of flatfishes (Carangimorpharia, Pleuronectiformes) // Mol. Phyl. Evol. 2014. V. 73. P. 18–22.
Birky C.W.Jr. Species detection and identification in sexual organisms using population genetic theory and DNA sequences // PLoS One. 2013. V. 8 (1). P. e52544. DOI: 10.1371/journal.pone.0052544
Bringloe T.T., Cottenie K., Martin G.K., Adamowicz S.J. The importance of taxonomic resolution for additive beta diversity as revealed through DNA barcoding // Genome. 2016. V. 59 (12). P. 1130–1140.
Brower A.V.Z. Delimitation of phylogenetic species with DNA sequences: a critique of Davis and Nixon’s population aggregation analysis // Syst. Biol. 1999. V. 48. P. 199–213.
Busack C.A., Torgaard J.H., Bannon M.P., Gall G.A.E. An electrophoretic karyotypic and meristic characterization of the Eagle Lake trout, Salmo gairdnery aquilarum // Copeia. 1980. V. 3. P. 418–424.
Bush G.L. Modes of animal speciation // Ann. Rev. Ecol. Syst. 1975. V. 6. P. 339–364.
Campton D.E. Natural hybridization and introgression in fishes. Method of detection and genetic interpretation // Population genetics & fishery management / Eds Ryman N., F. Utter. 1987. P. 161–192.
Clark A.G. Natural selection with nuclear and cytoplasmic transmission: I. A deterministic model // Genetics. 1984. V. 107. P. 679–701.
Delaney M.E., Bloom S.E. Replication banding patterns in the chromosomes of rainbow trout // J. Heredity. 1984. V. 5. P. 431–34.
Devlin R.H., Nagahama Y. Sex determination and sex differentiation in fish: an overview of genetic, physiological, and environmental influences // Aquaculture. 2002. V. 208. P. 191–364.
Dobzhansky Th. Evolution, genetics and man. N.Y.: John Wiley & Sons Inc., London: Chapman & Hall, Limited, 1955. 398 p.
Drummond A.K., Bouckaert R.R. Bayesian evolutionary analysis with BEAST. Cambridge: Cambridge University, 2015. 240 p.
Ferris S.D., Sage R.D., Huang C.-M. et al. Flow of mitochondrial DNA across a species boundary // PNAS USA. 1983. V. 80. P. 2290–2294.
Fitzpatrick B.M. Rates of evolution of hybrid unviability in birds and mammals // Evolution. 2004. V. 58. P. 1865–1870.
Fong J.J., Chen T.-H. DNA evidence for the hybridization of wild turtles in Taiwan: possible genetic pollution from trade animals // Conserv. Genet. 2010. V. 11 (5). P. 2061–2066.
Fujisawa T., Barraclough T.G. Delimiting species using single-locus data and the generalized mixed Yule coalescent approach: a revised method and evaluation on simulated data sets // Syst. Biol. 2013. V. 62. P. 707–724.
Gardner J.P.H., Skibinski D.O.F. Historical and size-dependent genetic variation in hybrid mussel populations // Heredity. 1988. P. 61. P. 93–105.
Genovart M. Natural hybridization and conservation // Biodivers. Conserv. 2008. V. 18 (6). P. 1435–1439.
Gerber A.S., Tibbets C.A., Dowling T.E. The role of introgressive hybridization in the evolution of the Gila robusta complex (Teleostei: Cyprinidae) // Evolution. 2001. V. 55 (10). P. 2028–2039.
Glemet H., Blier P., Bernatchez L. Geographical extent of Arctic Char (Salvelinus alpinus) mtDNA introgression in brook char populations (S. fontinalis) from Eastern Quebec Canada // Mol. Ecol. 1998. V. 7 (12). P. 1655–1662.
Grant P.R., Grant R.B. Hybridization of bird species // Science. 1992. V. 256. P. 193–197.
Greenfield D.W., Greenfield T. Introgressive hybridization between Gila orcutti and Hesperolecus symmetricus (Pisces: Cyprinidae) in the Guyama River Basin, California. I. Meristics, morphometrics, and breeding // Copeia. 1972. V. 4. P. 849–859.
Greenfield D.W., Abdel-Hameed F., Deckert G.D., Finn R.R. Hybridization between Chrosomus erythrogaster and Notropis cornutus (Pisces: Cyprinidae) // Copeia. 1973. № 1. P. 54–60.
Hanzawa N., Taniguchi N., Shinzawa H. Genetic markers of the artificial hybrids between Tribolodon hakonensis and T. sp. (Ukeguchi-Ugui) // Otsuchi Mar. Res. Cent. Rep. Univ. Tokyo. 1984. V. 10. P. 11–17.
Heath D.A., Rawson P.D., Hilbish T.J. PCR-based nuclear markers identify alien blue mussel (Mytilus spp.) genotypes on the west coast of Canada // Can. J. Fish. Aquat. Sci. 1995. V. 52. P. 2621–2627.
Hebert P.D.N., Cywinska A., Ball S.L. Biological identifications through DNA barcodes // Proc. R. Soc. L. B Biol. Sci. 2003a. V. 270 (1512). P. 313–321.
Hebert P.D.N., Ratnasingham S., De Waard J.R. Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species // Proc. R. Soc. L. B Biol. Sci. 2003b. V. 270 (S1). P. S96–S99.
Hedges S.B., Marin J., Suleski M., Madeline P., Kumar S. Tree of life reveals clock-like speciation and diversification // Mol. Biol. Evol. 2015. V. 32. P. 835–845.
Hewitt G.M. Quaternary phylogeography: the roots of hybrid zones // Genetica. 2011. V. 139 (5). P. 617–638.
Hubbs C., Kuehne R.A., Ball J.C. The fishes of upper Guadelupe River, Texas // Texas J. Sci. 1953. V. 5. P. 216–244.
Hubbs C.L. Hybridization between fish species in nature // Syst. Zoology. 1955. V. 4. P. 1–20.
Hubby J.L., Throckmorton D.H. Protein differences of Drosophila. II. Comparative species genetics and evolutionary problems // Genetics. 1965. V. 52. P. 203–215.
Imoto J.M., Saitoh K., Sasaki T. et al. Phylogeny and biogeography of highly diverged freshwater fish species (Leuciscinae, Cyprinidae, Teleostei) inferred from mitochondrial genome analysis // Gene. 2013. V. 514 (2). P. 112–124.
Inoue K., Takeuchi Y., Miki D., Odo S. Mussel adhesive plaque protein gene is a novel member of epidermal growth factor like gene family // J. Biol. Chem. 1995. V. 270. P. 6698–6701.
Joly S. JML: testing hybridization from species tree // Mol. Ecol. Res. 2012. V. 12. P. 179–184.
Kartavtsev Y.Ph. Analysis of sequence diversity at mitochondrial genes on different taxonomic levels. Applicability of DNA based distance data in genetics of speciation and phylogenetics. Ch. 1 // Genetic diversity / Eds Mahoney C.L., Springer D.A. N.Y.: Nova Science Publishers, Inc., 2009. P. 1–50.
Kartavtsev Y.Ph. Sequence divergence at mitochondrial genes in animals. Applicability of DNA data in genetics of speciation, phylogenetics and molecular ecology // Mar. Genomics. 2011a. V. 49. P. 71–81.
Kartavtsev Y.Ph. Sequence divergence at Co-1 and Cyt-b mtDNA on different taxonomic levels and genetics of speciation in animals // Mitochondrial DNA. 2011b. V. 2 (3). P. 55–65.
Kartavtsev Y.Ph. Sequence diversity at Cyt-b and Co-1 mtDNA genes in animal taxa proved Neo-Darwinism // J. Phyl. Evol. Biol. 2013a. V. 1 (4). P. 1–5. http://dx.org/10.4172/2329-9002.1000120.
Kartavtsev Y.Ph. Some current concerns of Neo-Darwinism: gene introgression throughout a species border // J. Phyl. Evol. Biol. 2013b. V. 1 (5). P. 1–4. http://dx.org/10.4172/2329-9002.1000e107.
Kartavtsev Y.Ph. Molecular evolution and population genetics. A course for marine biology students. N.Y.: CRS Press, Taylor & Francis Publ. Group, 2015. 349 p.
Kartavtsev Y.P. Barcode index number, taxonomic rank and modes of speciation: examples from fish // Mitoch. DNA. Pt A. 2018. V. 29 (4). P. 535–542.
Kartavtsev Y.P., Sviridov V.V., Sasaki T., Hanzawa N. Genetic divergence of far eastern dace belonging to the genus Tribolodon (Pisces, Cyprinidae) and closely related taxa: some insights in taxonomy and speciation // Rus. J. Genetics. 2002. V. 38 (11). P. 1518–1531.
Kartavtsev Y.Ph., Chichvarkhin A.Y., Kijima A. et al. Allozyme and morphometric analysis of two common mussel species of Mytilus genus (Mollusca, Mytilidae) in Korea, Japan and Russia waters // Kor. J. Genetics. 2005. V. 27 (4). P. 289–306.
Kartavtsev Y.P., Park T.-J., Vinnikov K.A. et al. Cytochrome b (Cyt-b) gene sequences analysis in six flatfish species (Pisces, Pleuronectidae), with phylogenetic and taxonomic insights // Mar. Biol. 2007. V. 152 (4). P. 757–773.
Kartavtsev Y.Ph., Katolikova M.V., Sharina S.N. et al. A population genetic study of the hybrid zone of Mytilus trossulus Gould, 1850 and an introduced species, M. galloprovincialis Lamarck, 1819 (Bivalvia: Mytilidae) in Peter the Great Bay in the Sea of Japan // Rus. J. Mar. Biol. 2014. V. 40 (3). P. 208–216.
Kartavtsev Y.Ph., Sharina S.N., Saitoh K. et al. Phylogenetic relationships of Russian Far Eastern Flatfish (Pleuronectiformes, Pleuronectidae) based on two mitochondrial gene sequences, Co-1 and Cyt-b, with inferences in order phylogeny using complete mitogenome data // Mitoch. DNA. 2016. V. 27 (1). P. 667–678. Informa UK Ltd DOI: 10.3109/19401736.2014.913139
Kartavtsev Yu.Ph., Batishcheva N.M., Bogutskaya N.G. et al. Molecular systematics research, DNA barcoding of Altai Osmans, Oreoleuciscus (Pisces, Cyprinidae, Leuciscinae), and nearest relatives, inferred from sequences of cytochrome b (Cyt-b), cytochrome oxidase c (Co-1), and complete mitochondrial genome // Mitoch. DNA. Pt A. 2017. V. 28 (4). P. 502–517.
Kartavtsev Y.Ph., Masalkova N.A., Katolikova M.V. Genetic-and-morphometric variability in the settlements of two mussel species (Mytilus ex. gr. edulis), M. trossulus and M. galloprovincialis, in North-West Japan Sea // J. Shellfish Res. 2018. V. 37 (1). P. 1–17.
Katolikova M., Khaitov V., Väinölä R. Genetic, ecological and morphological distinctness of the blue mussels Mytilus trossulus Gould and M. edulis L. in the White Sea // PLoS One. 2016. V. 11. P. 1–25.
Keck B.P., Near T.J. Geographic and temporal aspects of mitochondrial replacement in Nothonotus darters (Teleostei: Percidae: Etheostomatinae) // Evolution. 2009. V. 64. P. 1410–1428.
Kijewska A., Burzynski A., Wenne R. Molecular identification of european flounder (Platichthys flesus) and its hybrids with european plaice (Pleuronectes platessa) // ICES J. Mar. Sci. 2009. V. 66 (5). P. 902–906.
Kondrashov A.S., Yampolsky L.Y., Shabalina S.A. On the sympatric origin of species by means of natural selection // Endless forms: species and speciation / Eds Howarth D.J., Berlocher S.H. Oxford: Oxford Univ. Press, 1998. P. 90–98.
Mayden R.L., Chen W.-J., Bart H.L. et al. Reconstructing the phylogenetic relationships of the earth’s most diverse clade of freshwater fishes – order Cypriniformes (Actinopterygii: Ostariophysi): a case study using multiple nuclear loci and the mitochondrial genome // Mol. Phyl. Evol. 2009. V. 51 (3). P. 500–514.
Mayer W.E., Tichy H., Klein J. Phylogeny of African cichlid fishes as revealed by molecular markers // Heredity. 1998. V. 80 (6). P. 702–714.
Mayr E. Process of speciation in animals // Mechanisms of speciation / Ed. C. Barigozzi. N.Y.: Alan R. Liss, 1982. P. 1–20.
McCusker M.R., Denti D., van Guelpen L. et al. Barcoding Atlantic Canada’s commonly encountered marine fishes // Mol. Ecol. Res. 2013. V. 13. P. 177–188.
McGuire J.A., Linkem C.W., Koo M.S. et al. Mitochondrial introgression and incomplete lineage sorting through space and time: phylogenetics of crotaphytid lizards // Evolution. 2007. V. 61. P. 2879–2897.
Miya M., Takeshima H., Endo H. et al. Major patterns of higher teleostean phylogenies: a new perspective based on 100 complete mitochondrial DNA sequences // Mol. Phyl. Evol. 2003. V. 26 (1). P. 121–138.
Miya M., Saitoh K., Wood R. et al. New primers for amplifying and sequencing the mitochondrial ND4/ND5 gene region of the Cypriniformes (Actinopterygii: Ostariophysi) // Ichthyol. Res. 2006. V. 53. P. 75–81.
Nanney D.L. Genes and phenes in Tetrahymena // Bioscience. 1982. V. 32. № 10. P. 783–788.
Nedunoori A., Turanov S.V., Kartavtsev Y.Ph. Fish product mislabeling identified in the Russian far east using DNA barcoding // Gene Rep. 2017. V. 8. P. 144–149.
Nei M. Molecular evolutionary genetics. N.Y.: Columbia Univ. Press, 1987. 512 p.
Nei M., Kumar S. Molecular evolution and phylogenetics. N.Y.: Oxford Univ. Press, 2000. 333 p.
Nelson J.S. Hybridization between two cyprinid fishes, Hybopes plumbea and Rhinichthys cataractae, in Alberta // Can. J. Zool. 1966. V. 44. P. 663–968.
Nelson J.S. Occurrence of hybrids between longnose sucker (Catastomus catastomus) and white sucker (C. commersoni) in upper Kananaskis Reservoir, Alberta // J. Fish. Res. Board. Can. 1973. V. 30. P. 557–560.
Nevado B., Koblmüller S., Sturmbauer C. et al. Complete mitochondrial DNA replacement in a lake Tanganyika cichlid fish // Mol. Ecol. 2009. V. 18. P. 4240–4255.
Nevado B., Fazalova V., Backeljau T. et al. Repeated unidirectional introgression of nuclear and mitochondrial DNA between four congeneric Tanganyikan cichlids // Mol. Biol. Evol. 2011. V. 28 (8). P. 2253–2267.
Oleinik A.G., Skurikhina L.A. Mitochondrial DNA diversity and relationships of endemic charrs of the genus Salvelinus from lake Kronotskoye (Kamchatka Penisula) // Hydrobiologia. 2010. V. 650 (1). P. 145–159.
Pardo B.G., Machordom A., Foresti F. et al. Mitochondrial genomics of ostariophysan fishes: perspectives on phylogeny and biogeography // J. Mol. Evol. 2003. V. 56. P. 464–472.
Pardo B.G., Machordom A., Foresti F. et al. Phylogenetic analysis of flatfish (order Pleuronectiformes) based on mitochondrial 16S rDNA sequences // Sci. Mar. 2005. V. 69. P. 531–543. https://doi.org/10.3989/scimar.2005.69n4531.
Penney R.W., Hart M.J. Distribution, genetic structure, and morphometry of Mytilus edulis and M. trossulus within a mixed species zone // J. Shellfish Res. 1999. V. 18 (2). P. 367–374.
Pereira L.H.G., Hanner R., Foresti F., Oliveira C. Can DNA barcoding accurately discriminate megadiverse Neotropical freshwater fish fauna? // BMC Genetics. 2013. V. 14 (20). P. 1–14.
Phillips R.B., Zajicek K.D. Q band chromosomal banding polymorphisms in lake trout (Salvelinus namaycush) // Genetics. 1982. V. 101. P. 227–234.
Phillips R.B., Zajicek K.D., Utter F.M. Q band chromosome polymorphism in Chinook salmon (Oncorhynchus tshawytscha) // Copeia. 1985. № 2. P. 273–278.
Plotner J., Uzzell T., Beerli P. et al. Widespread unidirectional transfer of mitochondrial DNA: a case in western Palaearctic water frogs // J. Evol. Biol. 2008. V. 21. P. 668–681.
Podlesnykh A.V., Brykov V.A., Kukhlevsky A.D. Unstable linkage of molecular markers with sex determination gene in pacific salmon (Oncorhynchus spp.) // J. Heredity. 2017. V. 108 (3). P. 328–333.
Powell J.R. Interspecific cytoplasmic gene flow in the absence of nuclear gene flow: evidence from Drosophila // PNAS USA. 1983. V. 80. P. 492–495.
Prager E.M., Wilson A.C. Slow evolutionary loss of the potential for interspecific hybridization in birds: a manifestation of slow regulatory evolution // PNAS USA. 1975. V. 72. P. 200–204.
Ratnasingham S., Hebert P.D.N. BOLD: the barcode of life data system. Available: www.barcodinglife.org // Mol. Ecol. Notes. 2007. V. 7 (3). P. 355–364.
Rawson P.D., Hilbish T.J. Evolutionary relationships among the male and female mitochondrial DNA lineages in the Mytilus edulis species complex // Mol. Biol. Evol. 1995. V. 12 (5). P. 893–901.
Rawson P.D., Secor C.L., Hilbish T.J. The effect of natural hybridization on the regulation of doubly uniparental mtDNA inheritance in blue mussels (Mytilus spp.) // Genetics. 1996. V. 144. P. 241–248.
Rawson P.D., Argawal V., Hilbish T.J. Hybridization between the blue mussels Mytilus galloprovincialis and M. trossulus along the pacific coast of North America: evidence for limited introgression // Mar. Biol. 1999. V. 134 (1). P. 201–211.
Redin A.D., Kartavtsev Y.Ph. Molecular phylogeny of Russian far eastern flatfish (Pleuronectiformes, Pleuronectidae) based on sequences of mitochondrial genes // Modern achievements in population, evolutionary, and ecological genetics: Int. symp. Vladivostok – Vostok Marine Biological Station, Sept. 3–8. 2017. Vladivostok, 2017. P. 34–35.
Roberts D.G., Gray C.A., West D.J. Marine genetic swamping: hybrids replace an obligately estuarine fish // Mol. Ecol. 2010. V. 19. P. 508–520.
Roje D.M. Incorporating molecular phylogenetics with larval morphology while mitigating the effects of substitution saturation on phylogeny estimation: a new hypothesis of relationships for the flatfish family Pleuronectidae (Percomorpha: Pleuronectiformes) // J. Mol. Phyl. Evol. 2010. V. 56. P. 586–600.
Saarman N.P., Pogson G.H. Introgression between invasive and native blue mussels (genus Mytilus) in the central California hybrid zone // Mol. Ecol. 2015. V. 24 (18). P. 4723–4738.
Saitoh K., Sado T., Mayden R.L. et al. Mitogenomic evolution and interrelationships of the Cypriniformes (Actinopterygii: Ostariophysi): the first evidence toward resolution of higher-level relationships of the world’s largest freshwater fish clade based on 59 whole mitogenome sequences // J. Mol. Evol. 2006. V. 63. P. 826–841.
Saitoh K., Chen W.-J., Mayden R.L. Extensive hybridization and tetrapolyploidy in spined loach fish // Mol. Phyl. Evol. 2010. V. 56. P. 1001–1010.
Sakai H., Ito Y., Shedko S.V. et al. Phylogenetic and taxonomic relationships of northern Far Eastern Phoxinin Minnows and Rhynchocypris (Pisces, Cyprinidae), as inferred from allozyme and mitochondrial 16S rRNA sequence analysis // Zool. Sci. 2006. V. 23. P. 323–331.
Sasaki T., Kartavtsev Y.P., Uematsu T. et al. Phylogenetic independence of Far Eastern Leuciscinae (Pisces: Cyprinidae) inferred from mitochondrial DNA analysis // Gene Gen. Syst. 2007. V. 82. P. 329–340.
Schwartz F.J. World literature to fish hybrids, with an analysis by family, species and hybrid // Publ. Gulf Coast Res. Lab. Museum. 1972. V. 3. 328 p.
Schwartz F.J. World literature on to fish hybrids, with an analysis by family, species and hybrid; Supp. 1 // NOAA Techn. Report NMFS SSRF-750. US Dep. Commerce. 1981. 507 p.
Seehausen O. Hybridization and adaptive radiation // Trends Ecol. Evol. 2004. V. 19. P. 198–207.
Semina A.V., Polyakova N.E., Brykov V.A. Genetic analysis revels a cryptic species in Far Eastern dace of genus Tribolodon // Docl. Russ. Acad. Sci. 2006. V. 4. P. 1–3.
Setzer P.Y.V. An analysis of natural hybrid swarm by means of chromosome morphology // Transac. Am. Fish. Soc. 1970. V. 99. P. 139–146.
Sharina S.N., Kartavtsev Y.P. Phylogenetic analysis of flatfish (Teleostei, Pleuronectiformes) based on the investigation of nucleotide sequences of cytochrome oxidase 1 gene (Co-1) // Russ. J. Genetics. 2010. V. 46 (3). P. 401–407.
Simon R.C., Noble R.E. Hybridization in Oncorhynchus (Salmonidae). I. Viability and inheritance in artificial crosses of chum and pink salmon // Transac. Am. Fish. Soc. 1968. V. 97. P. 109–118.
Sites J.W., Marshall J.C. Operational criteria for delimiting species // Ann. Rev. Ecol. Evol. Syst. 2004. V. 35. P. 199–227.
Skibinski D.O.F., Beardmore J.A., Cross T.F. Aspects of the population genetics of Mytilus (Mytilidae: Mollusca) in the British Isles // Biol. J. Linn. Soc. 1983. V. 19. P. 173–183.
Skurikhina L.A., Kartavtsev Yu.F., Chichvarkhin A.Yu., Pan’kova M.V. Study of two species of mussels, Mytilus trossulus and Mytilus galloprovincialis (Bivalvia, Mytilidae), and their hybrids in Peter the Great Bay of the Sea of Japan with the use of PCR markers // Russ. J. Genetics. 2001. V. 37 (12). P. 1448–1451.
Smietanka B., Burzynski A., Hummel H., Wenne R. Glacial history of the European marine mussels Mytilus, inferred from distribution of mitochondrial DNA lineages // Heredity. 2014. V. 113. P. 250–258.
Smith G.R. Introgression in fishes: significance for paleontology, cladistics, and evolutionary rates // Syst. Biol. 1992. V. 41 (1). P. 41–57.
Sneath P.H., Sokal R.R. Numerical taxonomy. San Francisco: Freeman, 1973. 573 p.
Spolsky C., Uzzell T. Natural interspecies transfer of mitochondrial DNA in amphibians // PNAS USA. 1984. V. 81. P. 5802–5805.
STATISTICA (data analysis software system), version 6. StatSoft Inc. 2001. www.statsoft.com.
Steinke D., Zemlak T.S., Hebert P.D.N., Adamowicz S.A. DNA barcoding of Pacific Canadas fishes // Mar. Biol. 2009. V. 156. P. 2641–2647.
Stevenson M.M., Buchanon T.M. An analysis of hybridization between the cyprinodont fishes, Cyprinodon variegates and C. elegans // Copeia. 1973. № 4. P. 682–692.
Stoeckle M.Y., Thaler D.S. Why should mitochondria define species? // Hum. Evol. 2018. V. 33 (1–2). P. 1–30.
STRUCTURE. Software from http://pritch.bsd.uchicago.edu/structure.html.
Suzuki H., Yasuda S.P., Sakaizumi M. et al. Differential geographic patterns of mitochondrial DNA variation in two sympatric species of Japanese Wood Mice, Apodemus speciosus and A. argenteus // Genes Genet. Syst. 2004. V. 79. P. 165–176.
Suzuki H., Filippucci M.G., Chelomina G.N. et al. A biogeographic view of Apodemus in Asia and Europe inferred from nuclear and mitochondrial gene sequences // Bioch. Genetics. 2008. V. 46 (5–6). P. 329–346.
Takahata N., Slatkin M. Mitochondrial gene flow // PNAS USA. 1984. V. 81. P. 1764–1767.
Tamura K., Stecher G., Peterson D. et al. MEGA6: Molecular evolutionary genetics analysis version 6.0 // Mol. Biol. Evol. 2013. V. 30 (12). P. 2725–2729.
Tateno Y., Nei M., Tajima F. Accuracy of estimated phylogenetic trees from molecular data. I. Distantly related species // J. Mol. Evol. 1982. V. 18. P. 387–404.
Templeton A.R. Mechanisms of speciation – a population genetic approach // Ann. Rev. Ecol. Syst. 1981. V. 12. P. 23–48.
Templeton A.R. Species and speciation: geography, population structure, ecology, and gene trees // Endless forms: species and speciation / Eds D.J. Howard, S.H. Berlocher. N.Y.–Oxford: Oxford Univ. Press, 1998. P. 32–43.
Turanov S.V., Kartavtsev Yu.Ph., Lipinsky V.V. et al. DNA-barcoding of perch-like fishes (Actinopterygii: Perciformes) from far-eastern seas of Russia with taxonomic remarks for some groups // Mitoch. DNA. 2014. V. 40. № 6. P. 447–454. DOI: 10.3109/19401736.2014.945525
Turanov S.V., Kartavtsev Yu.Ph., Lee Y.-H., Jeong D. Molecular phylogenetic reconstruction and taxonomic investigation of eelpouts (Cottoidei: Zoarcales) based on Co-1 and Cyt-b mitochondrial genes // Mitoch. DNA. 2016. V. 28. P. 547–557. DOI: 10.3109/24701394.2016.1155117
Turner G.F., Seehausen O., Knight M.E. et al. How many species of cichlid fishes are there in African lakes? // Mol. Ecol. 2001. V. 10 (3). P. 793–806.
Väinölä R., Strelkov P. Mytilus trossulus in Northern Europe // Mar. Biol. 2011. V. 58. P. 815–833.
Vinnikiov K.A., Thomson R.C., Munroe T.A. Revised classification of the righteye flounders (Teleostei: Pleuronectidae) based on multilocus phylogeny with complete taxon sampling // Mol. Phyl. Evol. 2018. V. 125. P. 147–162. https://doi.org/10.1016/j.ympev.2018. 03.01410.1016/j.ympev.2018.03.014.
Wang J., Kuang Y.Y., Tong G.X., Yin J.H. Genetics analysis on incompatibility of intergeneric hybridizations between Hucho taimen (♂) and Brachymystax lenok (♀) by using 30 polymorphic SSR markers // J. Fish. Sci. China. 2011. V. 3. P. 547–555.
Wiens J.J., Kuczynski C.A., Stephens P.R. Discordant mitochondrial and nuclear gene phylogenies in emydid turtles: implications for speciation and conservation // Biol. J. Lin. Soc. 2010. V. 99. P. 445–461.
Xu L.X., Kuang Y.Y., Tong G.X. et al. An analysis on genetic constitution of Hucho taimen, Brachymystax lenok and hybrids (Hucho taimen [female] × Brachymystax lenok [male]) by SRAP markers // Acta Agric. Univ. Jiangxiensis. 2011. V. 33. P. 1187–1194.
Yonekawa H., Moriwaki K., Gotoh O. et al. Evolutionary relationships among five subspecies of Mus musculus based on restriction enzyme cleavage patterns of mitochondrial DNA // Genetics. 1981. V. 98. P. 801–816.
Yonekawa H., Tsuda K., Tsuchia K. et al. Genetic diversity, geographic distribution and evolutionary relationships of Mus musculus subspecies based on polymorphism of mitochondrial DNA // Problems of evolution / Eds A.P. Kryukov, L.V. Yakimenko Vladivostok: Dalnauka, 2000. P. 90–108.
Yang L., Sado T., Hirt M.V. et al. Phylogeny and polyploidy: resolving the classification of Cyprinine 4 fishes (Teleostei: Cypriniformes) // Mol. Phyl. Evol. 2015. V. 85. P. 97–116.
Zbawicka M., Wenne R., Burzynski A. Mitogenomics of recombinant mitochondrial genomes of Baltic Sea Mytilus mussels // Mol. Genet. Genomics. 2014. V. 289. P. 1275–1287.
Zbawicka M., Trucco M.I., Wenne R. Single nucleotide polymorphisms in native South American Atlantic coast populations of smooth shelled mussels: hybridization with invasive European Mytilus galloprovincialis // Genet. Sel. Evol. 2018. V. 50. P. 5. https://org/doi 10.1186/s12711-018-0376-z.10.1186/s12711-018-0376-z.
Zhang J., Kapli P., Pavlidis P., Stamatakis A. A general species delimitation method with applications to phylogenetic placements // Bioinformatics. 2013. V. 29 (22). P. 2869–2876.
Zhuravlev Y.N., Avetisov V.A. The definition of life in the context of its origin // Biogeosciences. 2006. V. 3. P. 281–291.
Zuckerkandl E., Pauling L. Molecules as documents of evolutionary history // J. Theor. Biol. 1965. V. 8 (2). P. 357–366.
Дополнительные материалы отсутствуют.
Инструменты
Успехи современной биологии