

Успехи современной биологии, 2019, T. 139, № 6, стр. 523-539
Модульность в биологической эволюции и эволюционных вычислениях
А. В. Спиров 1, 2, *, А. В. Еремеев 2, 3, **
1 Институт эволюционной физиологии и биохимии им. И.М. Сеченова РАН
Санкт-Петербург, Россия
2 Институт научной информации по общественным наукам РАН
Москва, Россия
3 Институт математики им. С.Л. Соболева СО РАН, Омский филиал
Омск, Россия
* E-mail: sspirov@yandex.ru
** E-mail: eremeev@ofim.oscsbras.ru
Поступила в редакцию 06.06.2019
После доработки 26.07.2019
Принята к публикации 29.07.2019
Аннотация
В свое время базовые принципы селекционизма были перенесены в упрощенной форме из генетики популяций в область эволюционных вычислений с целью решения прикладных задач оптимизации и адаптации. За почти полувековое развитие этой области компьютерных наук накоплен значительный практический опыт и получены интересные теоретические результаты. Одним из основных свойств биологических систем является модульность, проявляющаяся на всех уровнях их организации, начиная с молекулярно-генетического, заканчивая целыми организмами и их сообществами. В настоящем обзоре сопоставляются явления и закономерности, связанные с модульностью в генетике и эволюционных вычислениях. С точки зрения модульности проводится анализ сходства и различия результатов, полученных в этих областях исследований, обсуждаются возможности обмена знаниями между ними.
ВВЕДЕНИЕ
Одним из факторов, повышающих вероятность благоприятных рекомбинаций и устойчивость приспособленности к локальным изменениям в генотипе, является модульность (Ратнер, 1992; Livnat et al., 2008; Schlosser, Wagner, 2004). В биологической литературе модули понимаются как подсистемы, характеризующиеся высокой степенью интеграции во внутренних связях и значительной автономностью в связях внешних (Schlosser, Wagner, 2004). Выделяются три аспекта модульности: модульность развития, морфологическая модульность и эволюционная модульность (Callebaut, 2005). Несколько неформально модуль развития может определяться как подсистема, проявляющая некоторое относительно автономное поведение (von Dassow, Munro, 1999). Как отмечено в (Muller, Wagner, 1996), по мере расширения познаний о молекулярных механизмах развития у самых разных организмов становятся известны все новые эволюционно-консервативные механизмы. Некоторые из этих примеров показывают, что консервативные молекулярные механизмы могут использоваться в радикально разных контекстах развития, что позволяет предположить, что механизм развития состоит из модульных единиц, по-разному комбинирующихся между собой в процессе эволюции.
На морфологическом уровне модульность характеризует структуру и функцию конкретных частей или элементов организмов, таких как передняя конечность млекопитающих или модульные структуры скелетов животных. Однако, поскольку морфологические модели организации возникают в онтогенезе, морфологическая модульность может рассматриваться также как аспект модульности развития (Eble, 2005). Эволюционный модуль может быть определен на языке отображений генотип–фенотип (genotype–phenotype mapping) как набор фенотипических признаков, высокоинтегрированных фенотипическими эффектами определяющих их генов и относительно изолированных от других подобных множеств признаков за счет незначительности плейотропных эффектов (Wagner, Altenberg, 1996).
Именно эволюционный аспект модульности находится в центре внимания в настоящей статье. С этой точки зрения проводится анализ сходства и различия результатов, полученных в генетике и эволюционных вычислениях с целью выявления возможностей обмена знаниями между этими областями, в каждой из которых накоплен значительный объем знаний и подходов к пониманию эволюционной модульности. Более детальные обзоры по модульности в биологии могут быть найдены, например, в (Schlosser, Wagner, 2004; Callebaut, 2005; Lorenz et al., 2011; Bornberg-Bauer, Albà, 2013).
Эволюционные алгоритмы
Эволюционные алгоритмы (ЭА), к числу которых относятся генетические алгоритмы (ГА), эволюционные стратегии, алгоритмы генетического программирования (ГП) и др., берут начало с монографии Л. Фогеля, А. Оуэнса и М. Уолша 1966 года (Фогель и др., 1969), а также монографий (Ивахненко, 1971; Holland, 1975), где было предложено моделировать процесс биологической эволюции с целью решения задач адаптации, аппроксимации и создания систем искусственного интеллекта. Характерной особенностью ЭА является имитация процесса эволюционной адаптации биологической популяции к условиям окружающей среды, при этом особи соответствуют пробным точкам в пространстве решений задачи оптимизации (называемым фенотипами), а приспособленность особей определяется значениями целевой функции задачи оптимизации11, называемой далее функцией приспособленности. Пробные решения в популяции ЭА кодируются как последовательности символов некоторого алфавита, называемые генотипами (в ГП генотипами являются теоретико-графовые деревья, вершины которых помечены символами).
Принципы наследственности, изменчивости и отбора в эволюционных алгоритмах реализуются при построении новых решений-потомков посредством рандомизированных процедур (операторов), модифицирующих полученные ранее пробные решения подобно процессам мутации и кроссинговера (рекомбинации) в живой природе. Чем большее значение функции приспособленности имеет особь, тем больше для нее вероятность быть выбранной в качестве родительского решения. Например, (μ,λ)-селекция, один из наиболее простых операторов селекции в ЭА, аналогичен массовому отбору в растениеводстве и животноводстве: из популяции численностью λ отбираются μ особей с наибольшими значениями функции приспособленности и родительские генотипы равновероятно выбираются из них для скрещивания и получения потомства. При действии другого широко известного оператора пропорциональной селекции вероятность отбора особи пропорциональна значению функции приспособленности от этой особи. Сравнение понятий приспособленности в генетике популяций и в ГА, использующих оператор пропорциональной селекции, проводится в (Paixão et al., 2015).
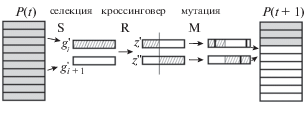
На каждой итерации генетического алгоритма (Holland, 1975; Goldberg, 1989; Vose, 1999) с помощью рандомизированных операторов пропорциональной селекции, мутации и рекомбинации строится новая популяция. Операторы мутации и рекомбинации в упрощенном виде моделируют процессы мутации с заменой нуклеотидов и одиночного мейотического кроссинговера. Численность популяции фиксирована от начала работы алгоритма до конца. Рис. 1 иллюстрирует построение пары потомков в процессе генерации популяции P(t + 1) на основе текущей популяции P(t). Обозначения операторов: S – селекция, R – кроссинговер, M – мутация. Начальная популяция P(1) строится случайным образом. Подробное описание классического генетического алгоритма (Goldberg, 1989) приводится в приложении.
Ввиду простоты адаптации вычислительных схем ЭА, эти методы активно применяются для решения задач оптимизации, возникающих в управлении, планировании, проектировании, распознавании образов и других областях. Эволюционные алгоритмы, и ГА в частности, имеют многочисленные варианты, различающиеся конкретной реализацией операторов селекции, кроссинговера и мутации (De Jong, 2006). В теории ЭА, как правило, рассматриваются вопросы трудоемкости отыскания наилучшего возможного генотипа в процессе работы некоторого ЭА на выбранном классе задач. При этом в первую очередь интересуются средним числом пробных решений до первого получения оптимального генотипа, в зависимости от размерности задачи (Neumann, Witt, 2010). Исследуемые классы задач могут содержать примеры сколь угодно большой размерности, как это принято в теории вычислительной сложности (Гэри, Джонсон, 1982). Для анализа времени первого достижения оптимального генотипа, как правило, используются такие методы теории вероятностей, как цепи Маркова, мартингалы, случайные процессы со сносом, стохастическое доминирование и др.
Постановка вопросов и методы исследования в теории ЭА и в популяционной генетике существенно различаются. В частности, в отношении биологической эволюции, как правило, не так важно достижение оптимума приспособленности, важнее получение достаточно приспособленных генотипов, устойчивых к возможным изменениям окружающей среды. Тем не менее, процессы, исследуемые в обеих областях, имеют много общего. В частности, эти процессы подчиняются принципам наследственности, изменчивости и отбора, соответствующим дарвиновской теории (селекционизм). На основе общности исследуемых процессов авторами (Paixão et al., 2015) предложена общая схема формального описания широкого класса эволюционных процессов, позволяющая сопоставлять аналогичные между собой модели в популяционной генетике и эволюционных вычислениях и дающая основание для переноса результатов между этими областями.
Генетические алгоритмы в вычислительной биологии
Хотя разработки ЭА были вдохновлены идеями дарвиновской эволюции (в самых их общих чертах, так что можно говорить об идеях обобщенного селекционизма; сравни (Dawkins, 1983)), сходства и различия компьютерной и биологической эволюций требуют обсуждения. По совокупности концепций и наработок область исследований ЭА резоннее всего сопоставлять с теорией и практикой эволюции биологических макромолекул (ДНК, РНК и белка). Особенно иллюстративны примеры направленной (искусственной) эволюции небольших по длине молекул ДНК, РНК и полипептидов в экспериментальных подходах типа SELEX (Gopinath, 2007; Chai et al., 2011), как иллюстрируется на рис. 2. Здесь, как и в ГА, в начале эксперимента создается случайная популяция “особей” (обобщение понятия особи существенно для обобщенного селекционизма). В рассматриваемых экспериментальных подходах популяцией является множество биомолекул заданной длины, различающихся своими последовательностями мономеров. С формальной точки зрения такие молекулы описываемы как последовательности букв из 4-буквенного (ДНК, РНК) или 20-буквенного (полипептиды) алфавита, тогда как в теории ЭА чаще всего рассматривают бинарные последовательности, но это не является критичным ограничением.
Рис. 2.
Схема процессов эволюции in vitro (на примере процедур SELEX) весьма сходна со схемой генетических алгоритмов в компьютерных науках. Сходство столь явно, что некоторые авторы рассматривают такие экспериментальные процедуры как реализацию ГА в эксперименте, и наоборот, соответствующие численные эксперименты в ГА можно трактовать как SELEX in silico.
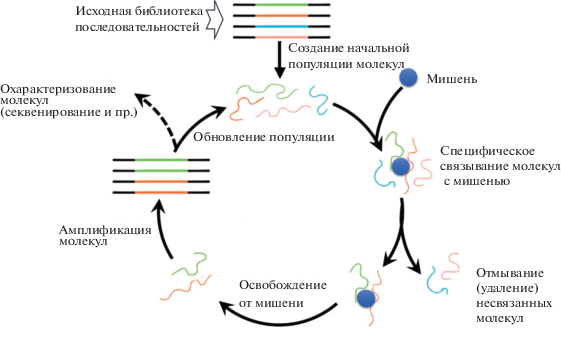
Все молекулы-особи начальной популяции подвергаются процедуре (количественной) оценки их на близость по характеристикам к искомой молекуле (рис. 2). Например, имеет ли она способность узнавать и связываться с заданной молекулой-мишенью (аффинность) и какова специфичность и сила такого связывания. Молекулы, демонстрирующие наибольшую степень аффинности к мишени, отбираются для продолжения эксперимента. Их далее умножают в числе (стадия “размножения”), используя такие экспериментальные процедуры размножения, которые делают копии отобранных молекул с определенным процентом ошибок в воспроизведении их последовательностей. Эта стадия соответствует мутации в ГА. Рекомбинация молекул тоже возможна в экспериментах по направленной эволюции, хотя она технически много сложнее, чем точечные мутации (Lutz, Benkovk, 2008; Stebel et al., 2008). Далее, обновленная популяция вновь подвергается отбору молекул на аффинность к мишени. Отобранные лучшие молекулы вновь умножаются и мутируют. Цикл выполняется, пока не будет найдена молекула требуемых характеристик.
Таким образом, экспериментальные биологические техники такие, как SELEX, вместе с другими подходами направленной эволюции биомолекул вполне резонно трактовать как экспериментальные реализации ЭА. С другой стороны, хорошо развитые современные подходы к анализу ЭА могут служить методической основой для развития теории и практики моделирования биологической эволюции. В последние два десятилетия активно развивалась такая системно-биологическая область, как моделирование эволюции генов и генных сетей (Системная …, 2008, гл. 4,5; Segal et al., 2003). Близка к ней по идеологии и подходам область эволюционного дизайна генов и генных сетей (Jostins, Jaeger, 2010; Spirov, Holloway, 2013). Эти области зачастую относят к приложениям ЭА к современным проблемам биологии (Eremeev, Spirov, 2018). Степень упрощения и формализации организации генов и их функций здесь чрезвычайно высоки. Однако выводы из работ по эволюции генных регуляторных сетей (ГРС) in silico и по эволюционному дизайну генов делаются с биологических и с эволюционно-биологических позиций.
В работах, где в рамках имитационных моделей изучается эволюция ГРС с целью изучения их эволюции в рамках проблематики эволюционной биологии (Spirov, Holloway, 2013, 2016; Payne et al., 2014), процедуры рекомбинации, мутации и селекции, как правило, заимствуются из ГА. Это делается из тех соображений, что перечисленные процедуры в ГА представляют собой достаточно простые абстракции соответствующих реальных биологических процессов. Именно этим обосновывается использование ГА, а не других эвристических алгоритмов, известных в области дискретной оптимизации. ГА для моделирования биологической эволюции ГРС имеют свою специфику. Разными авторами для конкретных целей исследований ГА расширяются так, чтобы в достаточно абстрактной форме описать отображение генотипа в фенотип (Ciliberti et al., 2007), онтогенез (Clune et al., 2012), диплоидность хромосомного набора (Shabash, Wiese, 2015), феномен эпистаза (Sanjuan, Nebot, 2008; Draghi, Plotkin, 2013) и т.д. Некоторые из этих расширений используются и в ГА для решения задач оптимизации, что происходит зачастую параллельно и независимо от реализации расширений в эволюционно-биологических моделях (Spirov, Holloway, 2016).
Более обобщенно можно сказать, что ЭА представляют собой весьма упрощенные и весьма формализованные описания некоторых (возможно ключевых) процессов и механизмов биологической эволюции. Критично то, что степень упрощения и формализации весьма высока и перенос заключений и выводов в областях компьютерной эволюции на реальную биологическую эволюцию приходится делать с осторожностью. Однако значительный опыт количественного анализа разнообразных процессов и механизмов эволюционного поиска в ЭА безусловно заслуживает пристального изучения биологами-эволюционистами, особенно в области молекулярной биологической эволюции.
ЭВОЛЮЦИОННАЯ МОДУЛЬНОСТЬ
При анализе возникновения многих таксономических групп отмечается важная эволюционная роль сальтационных реорганизаций генетического материала (Алтухов, 2003; Carson, 1975). Биологический смысл этих перестроек состоит в том, что они скачком переводят значительную часть генов в мономорфных локусах в гетерозиготное состояние, тем самым, создавая качественно новые возможности адаптации популяции. Однако в схеме сальтационного видообразования присутствует открытая проблема, связанная с тем, что крупные благоприятные мутации (например, полная перестройка гена, кодирующего некоторый белок) являются маловероятными.
В работе (Ратнер, 1992) в развитие идеи (Ohno, 1970) предложен блочно-модульный принцип организации и эволюции молекулярно-генетических систем управления, дающий более правдоподобные сценарии возникновения новых белков и других биомолекул. Согласно этому принципу, эволюция генов, РНК, белков, геномов и молекулярных систем управления на их основе шла путем комбинирования блоков (модулей), причем модулями, из которых составлялись вновь возникающие молекулярно-генетические системы, служили уже функционирующие макромолекулярные компоненты. При этом дублирование генов является основным источником эволюционных инноваций, поскольку позволяет одной копии гена мутировать и исследовать генетическое пространство, в то время как другая копия продолжает выполнять исходную функцию.
Модели процесса эволюции зачастую неявно предполагают, что одна мутация для дублированного гена может дать новое свойство, увеличивающее приспособленность генотипа. Однако некоторые белковые свойства, такие как наличие дисульфидных связей или сайтов связывания лиганда, требуют участия двух или более аминокислотных остатков, и для приобретения таких свойств необходимо мутировать несколько нуклеотидов. Эволюция регуляторных и функциональных сетей (включая сигнальные пути) зачастую также требует более одной благоприятной мутации, причем одновременно в разных генах. В работе (Behe, Snoke, 2004) моделируется простейший вариант эволюции белковых функций в дублированных генах. Авторы заключают, что, хотя генная дупликация и точечная мутация и могут быть эффективным механизмом исследования некоторой “окрестности” в генетическом пространстве (когда даже одиночные мутации приводят к увеличению приспособленности), тем не менее, этот простой путь является проблематичным для развития новых функций, когда требуются множественные мутации. В связи с этим, требуется рассматривать более сложные пути, возможно, связанные со вставкой, делецией, рекомбинацией или другими механизмами перестройки эволюционных модулей.
Модульность в биомолекулах
В биомолекулах (РНК, белок) модули обычно называют доменами. Домен белка определяют как элемент третичной структуры белка, представляющий собой достаточно стабильную и независимую подструктуру, фолдинг (укладка) которой проходит независимо от остальных частей белка (Wetlaufer, 1973). Сходные по структуре домены встречаются не только в родственных белках, но и в совершенно разных. В развитие этой концепции домены определяют как компактные (относительно автономные) единицы структуры и функций, способные укладываться автономно. Имеются основания трактовать домены как единицы эволюции (Bork, 1991; Richardson, 1981). Размеры доменов чрезвычайно варьируют, но средний домен имеет порядка 100 аминокислотных остатков. Число известных доменов составляет много тысяч и продолжает расти (сравни (Neduva, Russell, 2005; Finn et al., 2016)).
Несколько доменов могут формировать мультидоменный и (часто) мультифункциональный белок (Chothia, 1992), поэтому можно говорить о генетической мобильности доменов (Davidson et al., 1993). В мультидоменном белке домены могут выполнять свои функции автономно, а могут функционировать в комплексе с остальными доменами. Соответственно, домены мультидоменного белка могут служить структурными блоками белковых комплексов (как например актомиозины), а могут выполнять специфические каталитические функции (энзимы) или узнавать и специфически связываться с определенными последовательностями нуклеиновых кислот (транскрипционные факторы). Многие домены мультидоменных белков эукариот являются независимыми (однодоменными) белками у прокариот (Davidson et al., 1993).
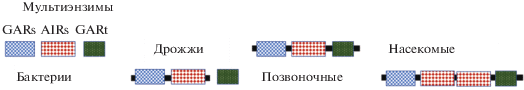
Так, например, у позвоночных имеется фермент GART (GARs-AIRs-GARt – trifunctional purine biosynthetic protein adenosine-3), состоящий из GAR-синтетазы (GARs), AIR-синтетазы (AIRs) и GAR-трансформилазы (GARt) (рис. 3). У насекомых такой мультиэнзим включает не три, а четыре домена: домен AIRs дублирован. У дрожжей домен GARt является отдельным энзимом, тогда как пара GARs-AIRs уже объединена в один мультиэнзим. У бактерий же охарактеризованы три отдельных энзима GARs, AIRs и GARt (Henikoff et al., 1997). Эволюция в данном, достаточно типичном для мультиэнзимов случае, представима как преимущественно последовательное формирование все более сложных мультиэнзимов (ди-, три- и тетра-энзимы) из изначально относительно простых энзимов прокариот. Генетические механизмы, вовлеченные в формирование мультидоменных протеинов, включают такие масштабные реорганизации генетического материала, как инверсии, транслокации, делеции, дупликации, гомологичную рекомбинацию (Bork et al., 1992). Так как домены достаточно “автономны” в формировании своей структуры и выполнении своей функции, с помощью генной инженерии можно “пришить” к одному из белков домен, принадлежащий другому (создавая таким образом белок-химеру). Такая химера при удаче будет совмещать функции обоих белков.
Рис. 3.
GAR-синтетазы, AIR-синтетазы и GAR-трансформилазы в эволюции (по Henikoff et al., 1997, с модификациями).
Так же, как и для протеинов, модульность и мотивы в макромолекулах РНК (и ДНК) привлекают внимание многих исследователей. Особо подчеркивается многоуровневость модульности РНК (Grabow et al., 2013; Grabow, Jaeger, 2013).
Известные, часто встречающиеся модули РНК охарактеризованы в молекулярных деталях (Hendrix et al., 2005; Leontis et al., 2006; Masquida et al., 2010; Grabow et al., 2013; Grabow, Jaeger, 2013). Существенно то, что простые модули могут входить в состав более сложных доменов РНК (Hendrix et al., 2005; Grabow et al., 2013). Тема модульности РНК привлекает особое внимание в связи с гипотезой “Мира РНК” (Gilbert, 1986; Joyce, 2002). Согласно этой гипотезе, случайно появившиеся в “первичном бульоне” небольшие молекулы РНК с разными каталитическими активностями далее комбинировались с образованием составных молекул большей сложности и большим спектром энзиматической активности. Эксперименты с селекцией РНК in vitro с использованием процедур лигации случайных последовательностей и процедур шаффлинга (shuffling), в которых образцы синтетической РНК подвергались направленной селекции для выполнения искомых экспериментаторами биохимических функций, продемонстрировали возможности получения каталитических РНК (Burke, Willis, 1998). Такие экспериментальные процедуры, по мнению исследователей, напоминают гипотетические процессы в Мире РНК. Более того, эксперименты с эволюцией на компьютере, когда простые подходы из области ЭА использовались для реализации модели направленной эволюции популяций небольших молекул РНК, подтвердили вышеприведенные эксперименты in vitro (Manrubia, Briones, 2002). Молекулы РНК из двух желаемых модулей находились быстрее в ходе эволюционного поиска, если сначала две отдельные популяции РНК подвергались селекции на желаемые последовательности (одна популяция – первый модуль, другая – второй), и в итоге найденные мотивы сшивались в составную искомую молекулу (чем если сразу велась селекция молекул с желаемой парой мотивов).
В этом разделе мы вновь сталкиваемся с общей проблемой важности сохранения в эволюции уже найденных мотивов (доменов).
В недавних обзорах (Grabow et al., 2013; Grabow, Jaeger, 2013) особо подчеркивается многоуровневость (иерархичность) модульности РНК. Эти биополимеры уникальны тем, что построены всего из четырех мономеров, а в основе большинства структур более высокого порядка (домены и мотивы вторичной и третичной структуры) – преимущественно комплементарные отношения между основаниями (канонические и неканонические). Эта относительная простота делает возможным формализацию основных механизмов образования и поддержания 2D- и 3D-доменов нуклеиновых кислот и исследование их биоинформационными и компьютерными подходами. Иерархичность модульной организации многих молекул РНК, включающих домены каждого данного уровня, составленные, в свою очередь, из субдоменов предыдущего уровня, навела ряд авторов на лингвистические аналогии (Rivas, Eddy, 2000; Jaeger et al., 2009). А именно, принципы, согласно которым составляется иерархическая структура РНК, могут быть представлены набором правил формальных грамматик, как это делается в лингвистике: части слова (субмотивы) составляются по определенным правилам в слова (мотивы), слова, в свою очередь и по своим правилам, составляются в предложения (мотивы или домены РНК более высокого уровня). С другой стороны, предполагается что когда мы выясним грамматику РНК, то сможем конструировать на компьютере новые молекулы РНК с желаемыми свойствами для их последующего химического синтеза (Jaeger et al., 2009; Geary et al., 2017).
Модульность в генных регуляторных сетях
Модульность генных регуляторных сетей (и более обобщенно – модульность биологических регуляторных сетей) – одна из широко обсуждаемых тем современной системной биологии (Solé et al., 2002; Espinosa-Soto, Wagner, 2010; Clune et al., 2013; Gyorgy, Del Vecchio, 2014). Представление ГРС графами (вершины – гены, дуги – функциональные связи между ними) наглядно иллюстрирует приводимое выше общее определение модульности, когда плотность связей внутри модуля явно выше, чем на его периферии. Каждый модуль как правило включает несколько узловых генов с большим числом связей (гены-хабы, hubs) и множество генов с небольшим количеством связей. ГРС имеют тенденцию аппроксимироваться моделью масштабно-инвариантной сети (scale-free network) (Barabasi, Oltvai, 2004). Такая сеть апроксимируема графом, в котором степени вершин (число ребер, связывающих вершину с другими) распределены по степенному закону, то есть в среднем доля вершин со степенью k пропорциональна k−γ. Параметр γ характеризует различные свойства сети, в частности, роль, которую играют гены-хабы. Одна из простых моделей, объясняющих появление масштабно-инвариантной сети, известна как иерархическая модель сети, которая получается многократным применением определенных правил пошагового добавления новых слоев узлов со все меньшим числом связей к начальному кластеру генов-хабов (Barabasi, Oltvai, 2004).
Еще одна значимая особенность модульности именно в ГРС – это мотивы (Kashtan, Alon, 2005). Мотив характеризуют как типичный, часто встречающийся способ функциональной связи нескольких генов. Если число генов невелико, то несложно перебрать все возможные варианты организации их в сеть. Варианты, которые встречаются много чаще, чем остальные, называют мотивами. Предполагается, что мотивы реализуют собой удачные функциональные решения, пригодные для организации регуляций во многих конкретных случаях, как, например, цепь упреждения (feed-forward loop) (Kashtan, Alon, 2005). Примечательно, что удается наблюдать типичное появление определенных мотивов (например, той же цепи упреждения) в численных экспериментах по эволюции моделей ГРС (Cooper et al., 2008). Иначе говоря, как и в ряде случаев, упомянутых выше, использование подходов из области эволюционных вычислений для теоретических задач эволюционной биологии по проблемам архитектуры ГРС позволило получить согласующиеся результаты. Это пример обратного переноса знаний из кибернетики в биологию, тогда как в свое время эволюционные идеи биологии вдохновили кибернетиков на разработку эволюционных алгоритмов. Вместе с тем, идея “модульного дизайна” была во многом заимствована современной биологической инженерией из обычной инженерии (Hartwell et al., 1999).
Генетическое программирование для генных регуляторных сетей
Одним из широко используемых методов эволюционных вычислений является генетическое программирование (ГП). Джон Коза, автор ГП, неоднократно упоминал, что принцип дублирования с изменением элементов программ, представленных деревьями (Koza et al., 1999), был вдохновлен идеями эволюционной биологии о дупликации и последующей дивергенции дубликатов генов, как изложено в монографии (Ohno, 1970). Возможность представить ГРС как ориентированный граф (при этом каждая дуга имеет знак: плюс – активирующее действие, минус – репрессия) давно используется биологами: это делает наглядными как архитектуру сети, так и “мутации” сети. Активно работающие в этой области лаборатории Сиггиа и Франке обозначили свое направление как эволюция in silico (Francois, Hakim, 2004; Francois et al., 2007; Francois, Siggia, 2010). Пример такого графического представления мутаций модели генной сети приведен на рис. 4.
Рис. 4.
Пример представления ГРС как ориентированного графа, где вершинам соответствуют гены, а дугам соответствует регулирующее действие продукта одного гена на другой. Проиллюстрированы два основных класса мутаций: добавление или удаление генов или их регуляторных связей.

“Мутации” при таком представлении ГРС – это добавление или удаление генов (узлов) и добавление или удаление регуляторных связей (дуг), см. рис. 4. Кроссинговер при таком представлении ГРС может быть реализован как обмен частей графов, как это обычно реализуется в техниках ГП. Такое представление ГРС, в свою очередь, делает возможным непосредственное использование техники ГП для исследований ГРС. Среди довольно широкого круга фундаментальных и прикладных задач в области ГРС нас интересует использование техник ГП для обратной инженерии архитектуры ГП и для исследований эволюции ГРС на компьютере.
В области обратной инженерии организации ГРС, когда исходят из экспериментальных данных по генной экспрессии для вывода архитектуры сети, приложения техник ГП развиваются несколькими группами. Одна из примечательных работ в этой области была опубликована основателем ГП – Козой с соавт. (Koza et al., 2000) и описывала использование ГП для обратной инженерии регуляторной сети лактозного оперона бактерии. В области приложения ГП к анализу биологических и модельных, искусственных регуляторных сетей активно работает группа Банжафа (Banzhaf, 2003; Leier et al., 2006; Hu et al., 2014). Приложение техник ГП для исследования ГРС и их эволюции – это еще один пример обратного трансфера знаний из кибернетики в биологию. Заслуживает упоминания здесь то, что мы не нашли в области приложения техник ГП к проблемам ГРС примеров использования классических схем кроссинговера, используемых в ГП. Соответственно мы можем ожидать дальнейшего прогресса именно в области приложений таких операторов кроссинговера к проблемам ГРС. Заметим здесь также, что простота и естественность кроссинговера как переноса некоторых подграфов графа ГРС должна ускорять эффективность эволюционного поиска архитектуры ГРС по сравнению с другими эволюционными подходами и другими реализациями архитектуры ГРС. В этой связи нам представляется весьма интересным вопрос о том, возможно ли так организовать генетический материал и использовать такие генно-инженерные методы, чтобы они в итоге выполняли кроссинговер согласно схемам ГП.
Возникновение модульности под воздействием рекомбинации
Эффекты рекомбинации могут быть одним из путей возникновения модульности в живой природе, согласно Ливнату с соавт. (Livnat et al., 2008). Эти авторы определяют новую величину, называемую смешиваемостью (mixability) Mi, которая характеризует способность аллели i успешно функционировать в разных комбинациях с другими аллелями и полагается равной средней приспособленности всевозможных генотипов с аллелью i:
Используя имитационное моделирование в рамках классического популяционно-генетического подхода, авторы (Livnat et al., 2008) показывают, что при половом размножении селекция и рекомбинация способствуют преобладанию аллелей с высокой смешиваемостью. Иначе говоря, в таких условиях, согласно (Livnat et al., 2008), селекция благоприятствует тому варианту аллели (или в общем случае варианту последовательности на некотором участке ДНК), который поддерживает в среднем наиболее высокую приспособленность при рекомбинации с различными родительскими генотипами. Эти исследования подтверждают интуитивное предположение Кроу и Кимуры (Crow, Kimura, 1965) о том, что половое размножение благоприятствует преумножению “хорошо смешиваемых” аллелей (good mixers), которые Кроу и Кимура охарактеризовали как аллели, которые вносят большой аддитивный вклад в приспособленность. Данное предположение согласуется с эмпирическими примерами. В частности, Кроу и Кимура отметили, что в случаях, когда у дрозофилы резистентность к лекарственным средствам развивалась в условиях полового размножения, вклад различных хромосом в резистентность имел аддитивный характер (King, Somme, 1958; Crow, Kimura, 1965). С другой стороны, резистентность к лекарственным средствам, развившаяся при бесполом размножении в Escherichia coli, была уменьшена последующей рекомбинацией, что говорит о значительной неаддитивности влияния разных аллелей в этом случае (Cavalli, Maccacaro, 1952). При половом же размножении локусы в моделях (Livnat et al., 2008) приобретают характерные черты эволюционных модулей в смысле Шлоссера (Schlosser, 2004), то есть каждый локус вносит свой вклад в приспособленность, мало зависящий от состояний других локусов.
Сохранение модульности в биологической эволюции
Проблему сохранения модульности в биологической эволюции наиболее естественно начинать рассматривать на уровне последовательности нуклеотидов одного гена. Мультидоменность организации гена включает, как минимум, две весьма различные компоненты: 1) регуляторные модули, рассеянные зачастую на обширных межгенных областях; 2) экзоны, перемежающиеся интронами, которые могут быть длиннее экзонов. В обоих случаях, а особенно в случае регуляторных модулей, кроссинговер должен иметь тенденцию не нарушать целостность функциональных доменов/модулей гена. Вместе с тем, выявленный интенсивный турновер сайтов связывания даже в сходных регуляторных модулях гомологичных генов близкородственных видов (исчезновение одних сайтов и появление других (Kim et al., 2009; Schmidt et al., 2010)) поднимает вопрос, так ли уж важно сохранение регуляторных модулей в сравнении с важностью сохранения целостности экзонов. Далее, мы можем из общих соображений ожидать, что дублирование и умножение числа доменов в пределах одного гена могло бы способствовать эволюции такого гена (Hong et al., 2008; Jeong et al., 2008). Это – открытый вопрос, и мы полагаем, что он заслуживает большего внимания.
Частный случай проблем и механизмов сохранения модулей в эволюции – это эволюция вирусов, в особенности РНК-вирусов. Здесь, как полагают, необходимость высоких темпов эволюционной изменчивости с целью ухода из-под давления иммунитета хозяина приводит к развитию специальных молекулярных механизмов.
Как оказалось (на примере вируса иммунодефицита), области между генами РНК-вируса, содержащие структурные элементы типа шпилек, являются местами преимущественной локализации точек “перекреста” в процессах вирусного кроссинговера (Simon-Loriere et al., 2010; Simon-Loriere, Holmes, 2011). Можно предположить, что наличие структурных элементов в межгенных областях РНК-вирусов обусловлено их ролью как сайтов кроссинговера, делающих более вероятным обмен интактными генами.
МОДУЛЬНОСТЬ В ТЕОРИИ ЭВОЛЮЦИОННЫХ АЛГОРИТМОВ
На начальном этапе теоретического исследования ЭА аспекты модульности исследовались в рамках анализа схем (Holland, 1975) и форм (Radcliffe, 1991). В соответствии с общепринятым в эволюционных вычислениях подходом мы на время предположим, что генотипы представляют собой битовые строки фиксированной длины n. Схемой H с q фиксированными позициями j(1), j(2), …, j(q) называют множество генотипов:
Если среднее значение функции приспособленности на особях схемы H в поколении t в достаточной мере превышает среднюю приспособленность особей текущей популяции, то среднее число представителей схемы H среди особей нового поколения классического генетического алгоритма оценивается снизу известной теоремой о схемах (Holland, 1975; Goldberg, 1989) – см. приложение. Теорема о схемах показывает, что при выборе кодировки решений разработчик ГА должен стремиться к тому, чтобы перспективные свойства решений были бы представлены в генотипе в виде коротких участков (то есть схем с достаточно малыми длиной l и порядком q), называемых строительными блоками (СБ).
В качестве примера функции приспособленности, где каждый СБ является отдельным модулем, в работах Митчелл с соавт. рассматриваются функции Royal Road (Mitchell et al., 1994). Пусть дано некоторое множество схем S и каждой схеме si из S приписан положительный вес ci. Значение Royal Road-функции на генотипе g определяется как w(g): = $\sum\nolimits_{i:g \in {{s}_{i}}} {{{c}_{i}}} $, то есть равно сумме весов всех схем из S, которым принадлежит генотип g. Наиболее часто используемый в анализе ЭА вариант Royal Road-функции обозначается через RRr(g) и имеет схемы равной длины и равного веса. Здесь предполагается, что задан целочисленный параметр r, множество S состоит из n/r схем с непересекающимися наборами фиксированных позиций, длина каждой схемы равна r – 1, а порядок каждой схемы равен r. В таком случае
Рис. 5.
Набор схем, задающих функцию RR8 при n = 56, веса схем и оптимальное решение sopt для этой функции приспособленности.

Процесс поиска оптимума функции RRr при работе ЭА может рассматриваться как упрощенная модель адаптации популяции при наличии нейтральных областей в “ландшафте” функции приспособленности. Каждая такая область соответствует фиксации некоторого количества блоков, которые должны быть заполнены исключительно единичными битами при всевозможных неоптимальных назначениях битов в остальных блоках. В частности, естественное обобщение функций RRr на 4-буквенный алфавит нуклеотидов позволяет с некоторыми упрощениями моделировать процедуры SELEX, направленные на получение кластерных энхансеров (Eremeev, Spirov, 2018). В этой работе применение результатов из теории ЭА к эволюции биомолекул показало, что оценки в терминах численности генотипов с заданным уровнем функции приспособленности более адекватны, чем оценки в терминах среднего времени первого достижения оптимального значения функции приспособленности.
В работе (Jansen, Wegener, 2005) замечено, что простой эволюционной стратегии (1 + 1) EA с популяцией из одной особи требуется построить в среднем порядка 2r(n/r) ln(n/r) особей для достижения оптимума функции RRr . Как ни странно, вычислительные эксперименты (Mitchell et al., 1994) показали, что классический ГА с оператором одноточечного кроссинговера и популяцией большего размера уступают (1 + 1) EA на функциях RRr . Для нескольких вариантов ГА в (Corus et al., 2018) недавно было доказано, что при подходящих настройках параметров алгоритма среднее время поиска оптимума функции RRr ограничено сверху величиной порядка nr ln(n/r). Данная оценка значительно превосходит упомянутое выше время поиска оптимума в (1 + 1) EA, что согласуется с экспериментами (Mitchell et al., 1994).
Отдельного рассмотрения заслуживает один из интересных наблюдаемых положительных эффектов от скрещивания, на основе которого построен недавно предложенный генетический алгоритм (1 + (λ,λ)) (Doerr et al., 2015). В данном алгоритме скрещивание является процедурой “починки” особи после масштабных мутаций, которые чаще всего ухудшают приспособленность, но в силу своего масштаба имеют больший шанс найти некую часть генотипа, улучшающую приспособленность. Эксперименты с модифицированными генетическими алгоритмами в (Doerr et al., 2015) показали способность таких алгоритмов превосходить (1 + 1) EA на функциях RRr именно за счет успешного использования кроссинговера. Другой интересный пример успешного использования модульности в кроссинговере может быть найден в работе (Watson, Jansen, 2007).
Вопросы эффективности использования оператора кроссинговера в ЭА при наличии модульности в структуре функции приспособленности остаются актуальными до сих пор. Теоретический анализ и результаты вычислительных экспериментов указывают на то, что при работе ЭА в процессе оптимизации функции RRr популяция проходит через последовательность эпох, каждая из которых соответствует преодолению очередной области нейтральности (van Nimwegen et al., 1999). Аналогичные эффекты ступенчатой эволюции используются при теоретическом анализе ЭА (Corus et al., 2018), а также наблюдаются в экспериментах in vitro, в частности, в случае кишечной палочки (Elena et al., 1996). В наибольшей мере ступенчатая динамика характерна для “сальтационных процессов”, возникающих в режимах слабой мутации и сильной селекции (SSWM-режимы), и исследуемых как в генетике популяций, так и в ЭА (Paixão et al., 2015). Наиболее детально в теории ЭА исследован частный случай RRr при r = 1, известный как функция приспособленности OneMax. Для этого случая найдены очень точные оценки времени первого достижения оптимума для различных ЭА.
Другая модульная функция приспособленности, определяемая в терминах Royal Road-функций, обозначается через RSr (от англ. Royal Staircase) и задается формулой:
Таким образом, в эволюционных вычислениях, как и в популяционной генетике, актуальны проблемы сальтационных перестроек генотипа и роли модульной структуры и вклада рекомбинации в таких перестройках. В частности, наблюдается затратность по времени образования крупных эволюционных модулей (например, доменов белка из нескольких десятков аминокислот), тогда как сайты связывания в регуляторных областях генов невелики и быстро находятся заново. Аналогичные выводы следуют из теоретических оценок, где длина блока входит в показатель степени. Упрощенные модели эволюционной модульности, такие как RRr- и RSr-функции приспособленности достаточно детально исследованы в теории ЭА, однако для применения этих результатов в биологии требуются дальнейшие исследования.
ЦЕЛЕНАПРАВЛЕННОЕ СОХРАНЕНИЕ МОДУЛЬНОСТИ
Потребность в операторах кроссинговера, сохраняющих СБ, была осознана в ходе развития теории и практики применения ЭА, где разработан целый ряд операторов кроссинговера, способных сохранять СБ (Eremeev, Kolokolov, 1996; Skinner, Riddle, 2004; Zaritsky, Sipper, 2004; El-Mihoub et al., 2006; Li et al., 2006; Kameya, Prayoonsri, 2011; Doerr et al, 2013; Umbarkar, Sheth, 2015). В большинстве из этих алгоритмов кроссинговер проводится на стыках некоторых “смысловых единиц”, аналогичных эволюционным модулям. Так, например, в работе (Eremeev, Kolokolov, 1996) любое допустимое решение z задачи целочисленного линейного программирования (ЦЛП) является вектором с d целочисленными компонентами и для каждой компоненты zi , i = 1, …, d, известно число битов ni, кодирующих ее в двоичной системе счисления. В таком случае естественно использовать строку генотипа из $n = \sum\nolimits_{i = 1}^d {{{n}_{i}}} $ битов и выбирать точку кроссинговера между подстроками, кодирующими координаты вектора z. Доказано, что пара решений-потомков, получаемых под действием такого кроссинговера, получается некоторым случайным поворотом пары родительских решений в пространстве решений задачи ЦЛП. При этом поворот совершается вокруг середины отрезка, соединяющего родительские решения, что позволяет надеяться на получение новых допустимых решений в окрестности имеющейся популяции, возможно с более высоким значением функции приспособленности, чем известные ранее.
Значительного прогресса удалось достичь в решении классической задачи коммивояжера22 с помощью операторов кроссинговера, сохраняющих блоки (Sanchez et al., 2017). В качестве СБ в данном случае выступают альтернативные фрагменты решений. Несмотря на то, что число комбинаций таких блоков растет экспоненциально от их числа, использование “жадных” эвристик или других методов комбинаторной оптимизации позволяет получить практически эффективные процедуры рекомбинации (Eremeev, Kovalenko, 2016; Sanchez et al., 2017).
Одной из традиционных областей применения генетических алгоритмов является оптимизация архитектуры искусственных нейронных сетей в машинном обучении. Исходной мотивацией такого применения генетических алгоритмов послужил эволюционный сценарий возникновения мозга в живой природе. Впоследствии и практика решения прикладных задач подтвердила перспективность такого использования ГА – см., например, монографию Д. Рутковской с соавт. (Рутковская и др., 2006) и приведенные там ссылки. Последние исследования (Lu et al., 2019) показывают, что применение генетического алгоритма NSGA для оптимизации сверточных нейронных сетей оказывается особенно эффективным, если в качестве СБ рассматривать, так называемые, вычислительные блоки и проводить рекомбинацию с сохранением таких блоков. В связи с обсуждением проблем оптимизации нейронных сетей с помощью ЭА необходимо обратить внимание и на широкое обсуждение проблем взаимосвязи модульности и функциональной эволюции центральной нервной системы (Colombo, 2013).
В биологии в то же самое время параллельно и независимо от эволюционных вычислений развиваются представления о важности сохранения модулей в эволюции и о конкретных механизмах такого сохранения.
В биоинженерии развиваются молекулярно-инженерные подходы для манипулирования с доменами в направленной эволюции биологических макромолекул (Voigt et al., 2002). В начале 90-х гг. Стеммер разработал метод “перетасовки” ДНК (DNA shuffling) (Stemmer, 1994a,b). Метод предполагает высокую гомологию рекомбинируемых последовательностей. Это была, по-видимому, первая эффективная реализация генно-инженерных процедур, сходных с гомологичным кроссинговером. В обеих своих первых публикациях по этому подходу (Stemmer, 1994a,b) Стеммер отмечает, что именно в области ГА была продемонстрирована высокая эффективность кроссинговера (в сравнении с точечными мутациями) для решения сложных задач эволюционным поиском. Новые экспериментальные молекулярно-биологические процедуры, рекомбинирующие целые модули, разработаны в области sexual methods в белковой инженерии (Lutz, Benkovk, 2008). Эти биотехнологические подходы предполагают выявление, “вырезание” и “перетасовку экзонов” (exon shuffling) “сшиванием” их в новые химерные молекулы, сходно с тем, как, полагают, это происходило в биологической эволюции (Stebel et al., 2008). Отметим, что методы негомологической рекомбинации не входят в набор стандартных ГА, хотя некоторые авторы развивают процедуры кроссинговера, напоминающие методы негомологической рекомбинации в биотехнологии (Hu, Banzhaf, 2010). Поэтому теоретический анализ эффективности таких методов был бы важен и для ЭА, и для направленной эволюции. Примечательно также то, что принцип экзонно-интронной организации гена вдохновил целую серию публикаций по новым техникам ЭА. Эти новые методы в ЭА были вдохновлены ожиданиями биологов, что обширные интронные вставки между экзонами, кодирующими домены, способствуют сохранению целостности доменов при рекомбинации (Nordin et al., 1997; Kouchakpour et al., 2009; Rohlfshagen, Bullinaria, 2010). Таким образом, для сохранения модульности требуются специальные кроссинговерные механизмы. Такие механизмы разработаны в области направленной эволюции макромолекул. В этих подходах используют ферменты и особенности организации генов. Есть ли подобные механизмы в живой природе – открытый вопрос.
ЗАКЛЮЧЕНИЕ
Таким образом, и в биологии, и в эволюционных вычислениях все больше внимания уделяется проблемам сохранения модулей в эволюции и механизмам такого сохранения.
Перенос идей биологического селекционизма в область прикладной математики привел в итоге к появлению обширной современной области ЭА. Примечательно, что многие проблемы теперь рассматриваются параллельно и (практически) независимо и в эволюционной биологии, и в ЭА. Вместе с тем, подходы современных ЭА все больше используются в современной системной биологии. Мы наблюдаем явную тенденцию к взаимному обмену идеями и подходами в этой области пересечения математики и биологии. В нашем обзоре мы сосредоточились на проблеме модульности и сохранения модулей в эволюции, как это видится математикам и биологам. Мы приходим к заключению, что одна из явно намечающихся точек междисциплинарного роста – это проблема механизмов сохранения доменов/модулей в эволюции, как компьютерной, так и биологической. Значительные перспективы имеет перенос накопленных знаний и методов из области эволюционных вычислений в биоинформатику – как для моделирования известных процессов и анализа имеющихся данных, так и для синтеза новых структур.
Список литературы
Алтухов Ю.П. Генетические процессы в популяциях. М.: Академкнига, 2003. 431 с.
Гэри М., Джонсон Д. Вычислительные машины и труднорешаемые задачи. М.: Мир, 1982. 416 с.
Ивахненко А.Г. Системы эвристической самоорганизации в технической кибернетике. Киев: Техника, 1971. 371с.
Ратнер В.А. Блочно-модульный принцип организации и эволюции молекулярно-генетических систем управления (МГСУ) // Генетика. 1992. Т. 28. № 2. С. 5–23.
Рутковская Д., Пилиньский М., Рутковский Л. Нейронные сети, генетические алгоритмы и нечеткие системы. М.: Горячая линия - Телеком, 2006. 452 с.
Системная компьютерная биология / Ред. Н.А. Колчанов, С.С. Гончаров, В.А. Лихошвай, В.А. Иванисенко. Новосибирск: Изд-во СО РАН, 2008. 769 с.
Фогель Л., Оуэнс А., Уолш М. Искусственный интеллект и эволюционное моделирование / Ред. А. Г. Ивахненко. М.: Мир, 1969. 230 с.
Banzhaf W. Artificial regulatory networks and genetic programming // Proc. Genetic Programming Theory and Practice / Eds R.L. Riolo, B. Worzel. Dordrecht: Kluwer, 2003. P. 43–62.
Barabasi A., Oltvai Z.N. Network biology: understanding the cells' functional organization // Nat. Rev. Genetics. 2004. V. 5. № 2. P. 101–113.
Behe M., Snoke D. Simulating evolution by gene duplication of protein features that require multiple amino acid residues // Protein Science. 2004. V. 13. № 10. P. 2651–2664.
Bork P. Shuffled domains in extracellular proteins // FEBS Lett. 1991. V. 286 № 1–2. P. 47–54.
Bork P., Sander C., Valencia A. An ATPase domain common to prokaryotic cell cycle proteins, sugar kinases, actin, and hsp70 heat shock proteins // PNAS USA. 1992. V. 89 № 16. P. 7290–7294. https://doi.org/10.1073/pnas.89.16.7290
Bornberg-Bauer E., Albà M.M. Dynamics and adaptive benefits of modular protein evolution // Curr. Opin. Struct. Biol. 2013. V. 23. P. 459–466.
Burke D.H., Willis J.H. Recombination, RNA evolution, and bifunctional RNA molecules isolated through chimeric SELEX // RNA. 1998. V. 4. P. 1165–1175.
Callebaut W. The ubiquity of modularity // Modularity: understanding the development and evolution of natural complex systems / Eds W. Callebaut, D. Rasskin-Gutman. Cambridge, MA: The MIT Press, 2005. P. 3–28.
Carson H.L. The genetics of speciation at the diploid level // Am. Nat. 1975. V. 109. № 965. P. 83–92.
Cavalli L.L., Maccacaro G.A. Polygenic inheritance of drug-resistance in the bacterium Escherichia coli // Heredity. 1952. V. 6. P. 311–331.
Chai C., Xie Z., Grotewold E. SELEX (Systematic Evolution of Ligands by EXponential Enrichment), as a powerful tool for deciphering the protein-DNA interaction space // Methods Mol. Biol. 2011. V. 754. P. 249–258.
Chothia C. Proteins. One thousand families for the molecular biologist // Nature. 1992. V. 357. № 6379. P. 543–544.
Ciliberti S., Martin O.C., Wagner A. Innovation and robustness in complex regulatory gene networks // PNAS USA. 2007. V. 104. P. 13591–13596.
Clune J., Mouret J.-B., Lipson H. The evolutionary origins of modularity // Proc. R. Soc. B. Biol. Sci. 2013. V. 280. № 1755. P. 20122863–20122863.
Clune J., Pennock R.T., Ofria C., Lenski R.E. Ontogeny tends to recapitulate phylogeny in digital organisms // Am. Nat. 2012. V. 180. P. E54–E63.
Colombo M. Moving forward (and beyond) the modularity debate: a network perspective // Phil. Sci. 2013. V. 80. P. 356–377.
Cooper M.B., Brookfield J.F.Y., Loose M. Evolutionary modelling of feed forward loops in gene regulatory networks // Biosystems. 2008. V. 91. P. 231–244.
Corus D., Dang D.-C., Eremeev A.V., Lehre P.K. Level-based analysis of genetic algorithms and other search processes // IEEE Trans. Evol. Comp. 2018. V. 22. № 5. P. 707–719. https://doi.org/10.1109/TEVC.2017.2753538
Crow J.F., Kimura M. Evolution in sexual and asexual populations // Am. Nat. 1965. V. 99. P. 439–450.
Davidson J.N., Chen K.C., Jamison R.S. et al. The evolutionary history of the first three enzymes in pyrimidine biosynthesis // BioEssays. 1993. V. 15. № 3. P. 157–164.
Dawkins R. Universal Darwinism // Evolution from molecules to man / Ed. D.S. Bendall. Cambridge: Cambridge Univ. Press, 1983. P. 403–428.
De Jong K.A. Evolutionary computation: a unified approach. Cambridge, MA: MIT Press, 2006. 256 p.
Doerr B., Doerr C., Ebel F. From black-box complexity to designing new genetic algorithms // Theor. Comp. Sci. 2015. V. 567. P. 87–104.
Doerr B., Johannsen D., Kötzing T. et al. More effective crossover operators for the all-pairs shortest path problem // Theor. Comput. Sci. 2013. V. 471. P. 12–26.
Draghi J.A., Plotkin J.B. Selection biases the prevalence and type of epistasis among beneficial substitutions // Evolution. 2013. V. 67. P. 3120–3131.
Eble G.J. Morphological modularity and macroevolution: conceptual and empirical aspects // Modularity: understanding the development and evolution of natural complex systems / Eds W. Callebaut, D. Rasskin-Gutman. Cambridge, MA: The MIT Press, 2005. P. 221–238.
El-Mihoub T.A., Hopgood A.A., Nolle L., Battersby A. Hybrid genetic algorithms: a review // Engin. Lett. 2006. V. 13. № 2. P. 2–11.
Elena S.F., Cooper V.S., Lenski R.E. Punctuated evolution caused by selection of rare beneficial mutations // Science. 1996. V. 272. P. 1802–1804.
Eremeev A.V., Kolokolov A.A. On some genetic and L-class enumeration algorithms in integer programming // Proc. of the First International conference on evolutionary computation and its applications. Moscow, 1996. P. 297–303.
Eremeev A.V., Kovalenko J.V. Experimental evaluation of two approaches to optimal recombination for permutation problems // Proc. of evolutionary computation in combinatorial optimization. LNCS / Ed. F. Chicano, B. Hu, P. Garcia-Sanchez. V. 9595. 2016. P. 138–153.
Eremeev A., Spirov A. Estimates from evolutionary algorithms theory applied to gene design // Proc. 11th International multiconference bioinformatics of genome regulation and structure/systems biology (BGRS\SB). Novosibirsk, Russia, 2018. P. 33–38. https://doi.org/10.1109/CSGB.2018.8544837
Espinosa-Soto C., Wagner A. Specialization can drive the evolution of modularity // PLoS Comput. Biol. 2010. V.6. P. e1000719. https://doi.org/10.1371/journal.pcbi.1000719
Finn R.D., Coggill P., Eberhardt R.Y. et al. The Pfam protein families database: towards a more sustainable future // Bat. Nucl. Acids Res. 2016. Database Issue 44. D. 279–285.
Francois P., Hakim V. Design of genetic networks with specified functions by evolution in silico // PNAS USA. 2004. V. 101. № 2. P. 580–585.
Francois P., Hakim V., Siggia E.D. Deriving structure from evolution: metazoan segmentation // Mol. Syst. Biol. 2007. V. 3. № 1. https://doi.org/10.1038/msb4100192
Francois P., Siggia E.D. Predicting embryonic patterning using mutual entropy fitness and in silico evolution // Development. 2010. V. 137. № 14. P. 2385–2395.
Geary C., Chworos A., Verzemnieks E. et al. Composing RNA nanostructures from a syntax of RNA structural modules // Nano Letters. 2017. V. 17. № 11. P. 7095–7101.
Gilbert W. Origin of life: the RNA world // Nature. 1986. V. 319. P. 618.
Goldberg D. E. Genetic algorithms in search, optimization and machine learning. Reading, MA: Addison-Wesley, 1989. 412 p.
Gopinath S.C. Methods developed for SELEX // Anal. Bioanal. Chem. 2007. V. 387. № 1. P. 171–182.
Grabow W., Jaeger L. RNA modularity for synthetic biology // F1000 Prime Rep. 2013. V. 5. P. 46.
Grabow W.W., Zhuang Z., Shea J.E., Jaeger L. The GA-minor submotif as a case study of RNA modularity, prediction, and design // Wiley Interdiscip. Rev. RNA. 2013. V. 4. № 2. P. 181–203.
Gyorgy A., Del Vecchio D. Modular composition of gene transcription networks // PLoS Comput. Biol. 2014. V. 10. № 3. P. e1003486.
Hartwell L.H., Hopfield J.J., Leibler S., Murray A.W. From molecular to modular cell biology // Nature. 1999. V. 402. (6761 Suppl.). P. C47–C52.
Hendrix D.K., Brenner S.E., Holbrook S.R., RNA structural motifs: building blocks of a modular biomolecule // Q Rev. Biophys. 2005. V. 38. № 3. P.221–243.
Henikoff S., Greene E.A., Pietrokovski S. et al. Gene families: the taxonomy of protein paralogs and chimeras // Science. 1997. V. 278. № 5338. P. 609–614.
Holland J.H. Adaptation in natural and artificial systems. Ann Arbor, MI: Univ. of Michigan Press, 1975. 183p.
Hong J.W., Hendrix D.A., Levine M.S. Shadow enhancers as a source of evolutionary novelty // Science. 2008. V. 321. P. 1314.
Hu T., Banzhaf W. Evolvability and speed of evolutionary algorithms in light of recent developments in biology // J. Artif. Evol. Appl. 2010. V. 2010. Article ID 568375. 28 p.
Hu T., Banzhaf W., Moore J.H. Population exploration on genotype networks in genetic programming // Proc. Parallel Problem Solving from Nature. LNCS / Eds T. Bartz-Beielstein, J. Branke, B. Filipic, J. Smith. V. 8672. Cham: Springer, 2014. P. 424–433.
Jaeger L., Verzemnieks E.J., Geary C. The UA_handle: a versatile submotif in stable RNA architectures // Nucl. Acids Res. 2009. V. 37. P. 215–230.
Jansen T., Wegener I. Real royal road functions – where crossover provably is essential // Dis. Appl. Math. 2005. V. 149. № 1–3. P. 111–125.
Jeong S., Rebeiz M., Andolfatto P. et al. The evolution of gene regulation underlies a morphological difference between two Drosophila sister species // Cell. 2008. V. 132. P. 783–793.
Jostins L., Jaeger J. Reverse engineering a gene network using an asynchronous parallel evolution strategy // BMC Syst. Biol. 2010. V. 4. P. 17. https://doi.org/10.1038/ng1165
Joyce G.F. The antiquity of RNA-based evolution // Nature. 2002. V. 418. P. 214-221.
Kameya Y., Prayoonsri Ch. Pattern-based preservation of building blocks in genetic algorithms // Proc. of IEEE congress on evolutionary computation, 2011. P. 2578–2585.
Kashtan N., Alon U. Spontaneous evolution of modularity and network motifs // PNAS USA. 2005. V. 102. P. 13773–13778.
Kim J., He X., Sinha S. Evolution of regulatory sequences in 12 Drosophila species // PLoS Genet. 2009. V. 5. P. e1000330.
King J.C., Somme L. Chromosomal analysis of the genetic factors for resistance to DDT in two resistant lines of Drosophila melanogaster // Genetics. 1958. V. 43. P. 577–593.
Koza J.R., Bennett F.H., Andre D., Keane M.A. Genetic programming III: Darwinian invention and problem solving. San Francisco, CA: Morgan Kaufmann, 1999. 1116 p.
Koza J.R., Lanza G., Mydlowec W. et al. Automated reverse engineering of metabolic pathways from observed data using genetic programming // Foundations of systems biology / Ed. H. Kitano. Cambridge, MA: MIT Press, 2001. P. 95–117.
Kouchakpour P., Zaknich A., Braunl T. A survey and taxonomy of performance improvement of canonical genetic programming // Knowl. Inf. Syst. 2009. V. 21. № 1. P. 1–39. https://doi.org/10.1007/s10115-008-0184-9
Leontis N.B., Lescoute A., Westhof E. The building blocks and motifs of RNA architecture // Curr. Opin. Struct. Biol. 2006. V. 16. № 3. P. 279–287.
Leier A., Kuo P.D., Banzhaf W., Burrage K. Evolving noisy oscillatory dynamics in genetic regulatory networks // Genetic Programming. EuroGP 2006. LNCS / Eds P. Collet, M. Tomassini, M. Ebner, S. Gustafson, A. Ekart. V. 3905. Berlin, Heidelberg: Springer, 2006. P. 290–299. https://doi.org/10.1007/11729976_26
Li F., Liu Q.H., Min F., Yang G.W. A new adaptive crossover operator for the preservation of useful schemata // Advances in Machine Learning and Cybernetics. LNCS / Eds D.S. Yeung, Z.Q. Liu, X.Z. Wang, H. Yan. V. 3930. Berlin, Heidelberg: Springer, 2006. P. 507–516. https://doi.org/10.1007/11739685_53
Livnat A., Papadimitriou C., Dusho J., Feldman M.W. A mixability theory of the role of sex in evolution // PNAS USA. 2008. V. 105. № 50. P. 19803–19808.
Lorenz D.M., Jeng A., Deem M.W. The emergence of modularity in biological systems // Physics Life Rev. 2011. V. 8. P. 129–160. https://doi.org/0.1016/j.plrev.2011.02.003
Lu Z., Whalen I., Boddeti V. et al. NSGA-net: neural architecture search using multi-objective genetic algorithm // Proc. of the genetic and evolutionary computation conference (GECCO 2019). ACM 2019. P. 419–427. https://doi.org/10.1145/3321707.3321729
Lutz S., Benkovk S. J. Protein engineering by evolutionary methods // Directed molecular evolution of proteins: or how to improve enzymes for biocatalysis / Eds S. Brakmann, K. Johnsson. Weinheim: Wiley-VCH Verlag, 2002. P. 177–213.
Manrubia S.C., Briones C. Modular evolution and increase of functional complexity in replicating RNA molecules // RNA. 2007. V. 13. P. 97–107.
Masquida B., Beckert B., Jossinet F. Exploring RNA structure by integrative molecular modeling // N. Biotechnol. 2010. V. 27. P. 170–183.
Mitchell M., Forrest S., Holland J.H. When will a genetic algorithm outperform hill climbing? // Advances in Neural Information Processing Systems (NIPS 6). San Mateo, CA: Morgan Kaufmann, 1994. P. 51–58.
Müller G.B., Wagner G.P. Homolgy, Hox genes, and developmental integration // Am. Zool. 1996. V. 36. P. 4–13.
Neumann F., Witt C. Bioinspired computation in combinatorial optimization – algorithms and their computational complexity. Berlin: Springer-Verlag, 2010. 216 p.
Neduva V., Russell R.B. Linear motifs: evolutionary interaction switches // FEBS Lett. 2005. V. 579. № 15. P. 3342–3345.
van Nimwegen E., Crutchfield J.P. Optimizing epochal evolutionary search population-size dependent theory // Machine Learn. J. 2001. V. 45. P. 77–114.
van Nimwegen E., Crutchfield J.P., Mitchell M. Statistical dynamics of the Royal Road genetic algorithm // Theor. Comp. Sci. 1999. V. 229. № 1. P. 41–102.
Nordin P., Banzhaf W., Francone F. Introns in nature and in simulated structure evolution // Proc. Bio-Computation and Emergent Computation / Eds D. Lundh, B. Olsson, A. Narayanan. Singapore: World Sci. Publishing, 1997. P. 22–35.
Ohno S. Evolution by gene duplication. N.Y.: Springer-Verlag, 1970. 160 p.
Paixão T., Badkobeh G., Barton N. et al. Toward a unifying framework for evolutionary processes // J. Theor. Biol. 2015. V. 383. P. 28–43.
Payne J.L., Moore J.H., Wagner A. Robustness, evolvability, and the logic of genetic regulation // Artificial Life. 2014. V. 20. P. 111–126.
Radcliffe N.J. Forma analysis and random respectful recombination // Proc. of the Fourth International conference on genetic algorithms. San Diego: Morgan Kaufmann, 1991. P. 222–229.
Richardson J.S. The anatomy and taxonomy of protein structure // Adv. Protein Chem. 1981. V. 34. P. 167–339.
Rivas E., Eddy S.R. The language of RNA: a formal grammar that includes pseudoknots // Bioinformatics. 2000. V. 16. P. 334–340.
Rohlfshagen P., Bullinaria J. Nature inspired genetic algorithms for hard packing problems // Ann. Oper. Res. 2010. V. 179. P. 393–419.
Sanchez D., Whitley D., Tinós R. Building a better heuristic for the traveling salesman problem: combining edge assembly crossover and partition crossover // Genetic and evolutionary computation conference (GECCO), New York. N.Y.: ACM, 2017. P. 329–336.
Sanjuan R., Nebot M.R. A network model for the correlation between epistasis and genomic complexity // PLoS One. 2008. V. 3. P. e2663.
Schlosser G. The role of modules in development and evolution // Modularity in development and evolution / Eds G. Schlosser, G.P. Wagner. Chicago: The Univ. of Chicago Press, 2004. P. 519–582.
Schlosser G., Wagner G.P. Introduction: the modularity concept in development and evolutionary biology // Modularity in development and evolution / Eds G. Schlosser, G.P. Wagner. Chicago: The Univ. of Chicago Press, 2004. P. 1–16.
Schmidt D., Wilson M.D., Ballester B. et al. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding // Science. 2010. V. 328. P. 1036–1040.
Shabash B., Wiese K.C. Diploidy in evolutionary algorithms for dynamic optimization problems: a best-chromosome-wins dominance mechanism // Int. J. Intell. Comp. Cybernet. 2015. V. 8 Iss. 4. P. 312–329.
Segal E., Shapira M., Regev A. et al. Module networks: identifying regulatory modules and their conditionspecific regulators from gene expression data // Nat. Genet. 2003. V. 34. P. 166–176. https://doi.org/10.1038/ng1165
Simon-Loriere E., Martin D.P., Weeks K.M., Negroni M. RNA structures facilitate recombination-mediated gene swapping in HIV-1 // J. Virol. 2010. V. 84. № 24. P. 12675–12682.
Simon-Loriere E., Holmes E.C. Why do RNA viruses recombine? // Nat. Rev. Microbiol. 2011. V. 9. № 8. P.617–626.
Skinner C., Riddle P. Expected rates of building block discovery, retention and combination under 1-point and uniform crossover // Proc. of Parallel Problem Solving from Nature. LNCS. V. 3242. Berlin: Springer-Verlag, 2004. P. 121–130.
Solé R.V., Salazar I., Garcia-Fernandez J. Common pattern formation, modularity and phase transitions in a gene network model of morphogenesis // Physica A. 2002. V. 305. P. 640–647.
Spirov A., Holloway D. New approaches to designing genes by evolution in the computer // Real-world applications of genetic algorithms / Ed. O. Roeva. London, U K: IntechOpen, 2012. P. 235–260. https://doi.org/10.5772/2674
Spirov A., Holloway D. Using evolutionary computations to understand the design and evolution of gene and cell regulatory networks // Methods. 2013. V. 62. № 1. P. 39–55.
Spirov A., Holloway D. Using evolutionary algorithms to study the evolution of gene regulatory networks controlling biological development // Evolutionary computation in gene regulatory network research / Eds H. Iba, N. Noman. Hoboken, NJ, USA: John Wiley & Sons, Inc., 2016. https://doi.org/10.1002/9781119079453.ch10
Stebel S.C., Gaida A., Arndt K.M., Muller K.M. Directed protein evolution // Molecular biomethods handbook / Eds J.M. Walker, R. Rapley. Totowa, NJ: Humana Press, 2008. P. 631–656.
Stemmer W.P. Rapid evolution of a protein in vitro by DNA shuffling // Nature. 1994a. V. 370. P. 389–391.
Stemmer W.P.C. DNA shuffling by random fragmentation and reassembly – in vitro recombination for molecular evolution // PNAS USA. 1994b. V. 91. № 22. P. 10747–10751.
Umbarkar A.J., Sheth P.D. Crossover operators in genetic algorithms: a review // ICTACT J. Soft Comp. 2015. V. 6. № 1. P. 1083–1092.
Voigt C.A., Martinez C., Wang Z.G. et al. Protein building blocks preserved by recombination // Nat. Struct. Biol. 2002. V. 9. P. 553–558.
von Dassow G., Munro E. Modularity in animal development and evolution: elements of a conceptual framework for EvoDevo // J. Exp. Zool (Mol. Dev. Evol). 1999. V. 285. P. 307–325.
Vose M.D. The simple genetic algorithm: foundations and theory. Cambridge, MA: The MIT Press, 1999. 251 p.
Wagner G.P., Altenberg L. Perspective: complex adaptations and the evolution of evolvability // Evolution. 1996. V. 50. P. 967–976.
Watson R.A., Jansen T. A building-block royal road where crossover is provably essential // Proc. of the 9th annual conference on genetic and evolutionary computation (GECCO 07). ACM, 2007. P. 1452–1459.
Wetlaufer D.B. Nucleation, rapid folding, and globular intrachain regions in proteins // PNAS USA. 1973. V. 70. № 3. P. 697–701.
Zaritsky A., Sipper M. The preservation of favoured building blocks in the struggle for fitness: the puzzle algorithm // IEEE Trans. Evol. Comp. 2004. V. 8. № 5. P. 443–455.
Дополнительные материалы отсутствуют.
Инструменты
Успехи современной биологии