

Успехи современной биологии, 2020, T. 140, № 6, стр. 540-554
Методы in vitro, используемые для изучения ДНК-белковых взаимодействий
Л. К. Савинкова 1, *, Е. Б. Шарыпова 1, Н. А. Колчанов 1
1 Федеральный исследовательский центр Институт цитологии и генетики Сибирского отделения РАН
Новосибирск, Россия
* E-mail: savinkl@mail.ru
Поступила в редакцию 29.06.2020
После доработки 29.06.2020
Принята к публикации 29.06.2020
Аннотация
Взаимодействие белков с ДНК лежит в основе всех процессов функционирования клетки. К ним относятся: транскрипция, репликация и репарация ДНК, поддержание структуры хромосомы, активация и ингибирование генов с помощью белков-факторов транскрипции и белков-ферментов, участвующих в модификации хроматина, поддержании структуры нуклеосом, контроле экспрессии генов и т.д. Транскрипционные факторы (ТФ) связываются с определенными наборами последовательностей ДНК, активируя или ингибируя транскрипцию генов, а также участвуют в разнообразных процессах передачи сигнала, включая изменение дифференцировки клеток, развития и влияния окружающей среды. Такая важная роль взаимодействий ДНК с транскрипционными факторами способствовала их активному изучению в течение многих лет и разработке различных методов для определения сайтов связывания на матрице ДНК и белков, взаимодействующих с ними, включая футпринтинг, саузерн- и вестерн-блотт-анализы, систематическую эволюцию лигандов экспоненциальным обогащением (SELEX), иммунопреципитацию хроматина (ChIP), сдвиг электрофоретической подвижности (EMSA), поверхностный плазмонный резонанс (SPR) и т.д. Каждый метод имеет свои преимущества и недостатки. Методы и подходы, используемые для изучения связывания ТФ/ДНК, можно разделить на in vivo и in vitro. Методы in vivo (например, ChIP) используются для изучения взаимодействия ТФ/ДНК, происходящего в конкретных условиях, включающих тип ткани, клетки, момент времени и др., и сканирования сайтов связывания ТФ по всему геному. Методы in vitro используются для изучения взаимодействия ТФ/ДНК, обусловленного только структурой белка и ДНК, для определения последовательностей сайтов связывания, силы взаимодействия и характеристики комплексов ТФ/ДНК. В обзоре будут перечислены наиболее широко используемые для изучения ДНК-белковых взаимодействий методы in vitro, так как по каждому из них есть хорошие обзоры. Подробно будут рассмотрены метод футпринтинга и методы определения сродства ДНК/белок, скоростей связывания и диссоциации комплексов ДНК/белок.
ВВЕДЕНИЕ
ДНК-белковые взаимодействия участвуют в разнообразных биологических процессах, таких как репликация, транскрипция и репарация ДНК, поддержание структуры хромосомы, активация и ингибирование генов с помощью белков-факторов транскрипции и белков-ферментов, участвующих в модификации хроматина, поддержании структуры нуклеосом, контроле экспрессии генов и т.д. Транскрипционные факторы (ТФ) – это белки, контролирующие транскрипцию (синтез мРНК на матричной ДНК) самостоятельно или чаще в комплексе с другими белками, так как считается, что около 80% белков не работают в одиночку (Berggard et al., 2007). Главная характеристика ТФ заключается в том, что в их составе содержится не менее одного ДНК-связывающего домена, который взаимодействует с определенным участком ДНК, расположенным в регуляторном районе гена. В геноме человека 6% всех генов кодируют ТФ – таким образом, ТФ являются вторым по величине семейством белков после ферментов (10%) (Lambert et al., 2018). Транскрипционные факторы связываются с определенными наборами последовательностей ДНК, активируя или ингибируя транскрипцию генов (Wang et al., 2008; Yamazaki et al., 2013; Slattery et al., 2014). Связывание ТФ с ДНК крайне важно для процессов развития и ответов на сигналы окружающей среды. Известно, что аминокислотные замены, изменяющие структуру белка, объясняют довольно небольшую часть (от 0 до 25%) различий в активности ферментов между различными видами, а замены в некодирующих частях гена (промотор, энхансер, 5'- и 3'-нетранслируемые области) обусловливают большинство различий активности, реализуемых через взаимодействие ТФ с этими районами генов (Loehlin et al., 2019; Garcia-Alonso et al., 2019; Castellated et al., 2020). Нарушение взаимодействия ТФ с ДНК может привести к развитию многих фенотипических нарушений, в том числе, различных заболеваний у человека (Bulyk, 2006; Brown, Jackson, 2015; Jackson et al., 2018). Понимание того, каким образом ТФ распознают свои сайты связывания на матрице ДНК, является основой для понимания регуляции транскрипции в норме и при различных нарушениях. Но достижение этого понимания осложняется многочисленными уровнями сложности, которые лежат между активностью TФ и фенотипом, начиная со сложности в связывании TФ/ДНК, вариабельности участков связывания ТФ и локальной структуры ДНК, геномного контекста, посттрансляционных процессов регуляции и т.д. Такая важная роль ДНК-белковых взаимодействий способствовала их активному изучению в течение многих лет и разработке различных методов для определения сайтов связывания на матрице ДНК и белков, взаимодействующих с ними. Связывание и функции TФ регулируются на нескольких уровнях. Первый и наиболее фундаментальный уровень регуляции достигается за счет предпочтительного связывания TФ со специфическими последовательностями ДНК (Luscombe et al., 2000; Badis et al., 2009; Jolma et al., 2010; Rossouw et al., 2012). Следующие уровни регулирования достигаются посттрансляционными модификациями доменов TФ или связыванием корегуляторов (Slattery et al., 2011; Siggers et al., 2011; Xiao et al., 2016). Модификации, в свою очередь, могут влиять на активность и клеточное расположение TФ. Генетические вариации сайтов связывания ТФ часто связаны с различиями в транскрипции между индивидуумами и видами, что подчеркивает необходимость их точной характеристики (Kasowski et al., 2010; Behera et al., 2018; Wissink et al., 2019).
МЕТОДЫ ВЗАИМОДЕЙСТВИЯ БЕЛКОВ С ДНК
Существуют многочисленные методы, как экспериментальные, так и вычислительные, которые позволяют обнаружить и всесторонне охарактеризовать специфичность, с помощью которой ТФ взаимодействуют с сайтами ДНК. Для характеристики взаимодействий TФ с ДНК используются два разных подхода: in vitro и in vivo. Способы in vitro, как правило, нацелены на то, чтобы идентифицировать сайты консенсусного связывания TФ, получить характеристику аффинности и энергии связывания и других параметров, влияющих на взаимодействие. Методы in vivo также дают информацию о сайтах связывания TФ, но в биологическом контексте специфичных взаимодействий, и об их распределении по всему геному.
Экспериментальные методы можно далее подразделить на методы, которые дают качественные или количественные характеристики, причем большинство методов относится к первой категории. Для определения взаимодействия ДНК–белок in vitro и in vivo было разработано множество подходов, которые рассматриваются ниже, включая метод отпечатков (стопы) (footprinting), который использовался для выявления сайтов связывания белка на матрице ДНК (промоторы, энхансеры, сайленсиры) и выявления их последовательности (Galas et al., 1978; Kiyama, Oishi, 1995).
Метод систематической эволюции лигандов методом экспоненциального обогащения (systematic evolution of ligands by exponential enrichment, SELEX) для определения высокоаффинных олигонуклеотидов, связываемых белками, был разработан в 1990 г. Туерком и Голдом (Tuerk, Gold, 1990). Метод основан на использовании чередующихся циклов отбора аптамеров (одноцепочечные ДНК или РНК) из рандомизированных библиотек вариабельных последовательностей и их амплификации (Djordjevic, 2007). Несвязавшиеся олигонуклеотиды отмывались; последовательности, связанные в первом раунде, амплифицировались, переводились в одноцепочечное состояние и снова связывались тем же белком. В результате через несколько раундов связывания получали популяцию из высокоаффинных олигонуклеотидов, которые можно было выделить и секвенировать.
Метод сдвига электрофоретической подвижности (electrophoretic mobility shift assay, EMSA) (Ruscher et al., 2000) основан на торможении в геле ДНК, связанной с белком, относительно свободной ДНК (РНК). Метод широко используется для количественного определения сродства белка и ДНК, для определения кинетических характеристик образования комплексов ДНК как с индивидуальными белками, так и с ядерными и клеточными экстрактами (Henriksson-Peltola et al., 2007).
Метод cаузерн-блоттинга (sousern-blotting assay) включает электрофорез в агарозном геле для фракционирования ДНК, соединенный с методами переноса разделенной по длине ДНК на мембранный фильтр для гибридизации c заданной последовательностью (Stoddard, Howe, 1989; Brown, 2001).
Метод вестерн-блоттинга (western blotting assay) используется для определения в образце интересующих белков, которые после электрофореза в денатурирующих условиях переносятся на нитроцеллюлозную мембрану и детектируются с помощью специфичных антител (Kurien, Scofield, 2006; Mishra et al., 2017).
Метод саузерн-вестерн блоттинга (southwestern assay) используется для определения белков и их специфических сайтов связывания. В этом анализе используется разделение белков в полиакриламидном геле с додецилсульфатом натрия, ренатурация белков, перенос электроблоттингом на иммобилизирующую мембрану и визуализация по их связыванию с радиоактивно меченой ДНК методом авторадиографии (Dooley et al., 1992; Labbe et al., 2009).
Метод иммунопреципитации хроматина (chromatin immunoprecipitation, ChIP) (Kim, Ren, 2006; Kaboord, Perr, 2008; Pawlak et al., 2018) и его вариации (Spencer et al., 2003; Zeng et al., 2006) были разработаны для поиска мест связывания ТФ по всему геному in vivo и для изучения модификаций гистонов. Метод основан на обработке живых клеток формальдегидом, вызывающим образование ковалентных сшивок между ДНК и взаимодействующими с ней белками, выделении, расщеплении ДНК на небольшие фрагменты, добавлении антител, специфических к исследуемому белку и иммунопреципитации. Вместе с белком выделяются районы ДНК, с которыми он связан. Для определения нуклеотидной последовательности районов связывания белка используются высокопроизводительные методы секвенирования (Lee et al., 2006; Hoffman, Jones, 2009).
Метод поверхностного плазмонного резонанса (surface plasmon resonance, SPR) (Stockley, Persson, 2009) – оптический метод обнаружения взаимодействия двух разных молекул, в которых одна подвижна, а другая закреплена на сенсорной поверхности микрочипа, – используется для определения силы взаимодействия между нуклеиновыми кислотами и белком для изучения параметров образующихся комплексов. Метод основан на изменении показателя преломления падающего света определенной длины волны на поверхности биосенсора при образовании комплекса, которое регистрируется в виде сенсограммы – зависимости сигнала от времени и массы (Mariani, Minunni, 2014).
Метод фагового дисплея (phage display assay) разработан для изучения белок/белок-, белок/пептид- и белок/ДНК-взаимодействий в 1985 г. Смитом (Smith, 1985) и В. Петренко одновременно. Смит в 2018 г. получил за разработку этого метода Нобелевскую премию по химии. Метод заключается в том, что ген исследуемого белка или белкового домена встраивается в ген оболочки бактериофага, и фаг синтезирует чужой белок на своей оболочке. Большое разнообразие белков, экспрессируемых таким образом, дает пул вариантов, называемых библиотекой фагового дисплея, из которой белок может быть выделен и почти аффинно очищен с помощью антител или высокоаффинных участков ДНК и может использоваться для изучения его взаимодействия с другими белками, нуклеиновыми кислотами, антителами и т.д. (Choo, Klug, 1995; Isalan, Choo, 2001).
Метод дрожжевого одногибридного анализа (yeast one-hybrid assay, Y1H) (Alexander et al., 2001) разработан для идентификации белков, которые связываются со специфическим фрагментом ДНК.
Метод дрожжевого двугибридного анализа (yeast two-hybrid assay, Y2H) (Vidal et al., 1996) используют для определения сетей взаимодействующих белков. Эти методы основаны на том, что большинство ТФ содержат ДНК-связывающий и активирующий домены. Если эти домены физически разделены, то ТФ не активен и не может привлечь РНК-полимеразу на промотор, чтобы инициировать транскрипцию. При взаимодействии домены сближаются, и ТФ становится активным.
В случае дрожжевого одногибридного анализа к ДНК-связывающему домену присоединяется исследуемая на взаимодействие с белком последовательность ДНК (которую часто называют приманкой), и с активирующим доменом (добыча) трансляционно слит исследуемый белок, а в случае двугибридного анализа к обоим доменам присоединяются белки для выяснения взаимодействия между ними, оба они экспрессируются в одной дрожжевой клетке. Если белок взаимодействует с ДНК-последовательностью (Y1H) или белки взаимодействуют друг с другом (Y2H), то активирующий и ДНК-связывающий домены сближаются, ТФ переходит в активное состояние и в клетке инициируется транскрипция репортерного гена. Если взаимодействия нет между двумя белками или между белком и последовательностью ДНК, то транскрипции репортерного гена не наблюдается.
Метод биохимической экстракции белков (pull- down assay) используется для обнаружения физических взаимодействий между двумя или более белками и для подтверждения предсказанного межбелкового взаимодействия или выявления новых взаимодействующих партнеров (Louche et al., 2017). Метод схож по методологии с экспериментами по коиммунопреципитации с использованием аффинного лиганда для захвата взаимодействующих белков. Разница между этими двумя методами заключается в том, что при коиммунопреципитации используются иммобилизованные антитела для захвата белковых комплексов, а в pull-down используется очищенный и биохимически меченый белок, иммобилизованный на аффинном лиганде – это биотинилированный ДНК-зонд, связанный со стрептавидином в качестве “приманки” для связывания любых взаимодействующих белков, которые могут быть получены из клеточных и ядерных экстрактов, систем экспрессии и др. (Maryas et al., 2018). Неспецифически связанные и с низким сродством белки вымываются, остаются белки, связанные с высокой аффинностью. Затем они идентифицируются с помощью методов EMSA и масс-спектрометрии (Singh, Nath, 2019).
С помощью многочисленных полномасштабных исследований определены ДНК-связывающие мотивы транскрипционных факторов из широкого спектра организмов. В результате выяснился эволюционный консерватизм специфичности связывания ТФ, свидетельствующий о том, что ТФ с близкородственными по аминокислотной последовательности доменами для связывания ДНК (РНК) имеют очень похожие последовательности сайтов связывания (Weirauch et al., 2014), что может использоваться для предсказания ТФ, связывание которых изменено полиморфизмами, ассоциированными с повышенным риском заболеваний человека (Ray et al., 2013).
Каждый метод имеет свои преимущества и недостатки. Например, подходы с высокой пропускной способностью очень эффективны при определении сайтов взаимодействия белок/ДНК в масштабе полного генома, что способствовало разработке успешных биоинформатических инструментов для анализа и прогнозирования сайтов связывания транскрипционных факторов с ДНК. Но эти методы слишком дороги, чтобы использоваться в ежедневной практике. Поэтому классические методы анализа ДНК-белковых взаимодействий, а также их модификации, не утрачивают своей значимости и часто бывают необходимы для уточнения деталей результатов, полученных в полномасштабных исследованиях. Мы рассмотрим более подробно некоторые классические методы, использующиеся in vitro для уточнения результатов общегеномных исследований и получения количественных характеристик комплексов белок/ДНК.
МЕТОД ФУТПРИНТИНГА
Метод футпринтинга (footprinting) с использованием ДНКазы I – один из первых методов для визуализации физических контактов при связывании белка с последовательностью ДНК, который был разработан в 1978 г. Галасом и Шмитцем (Galas, Schmitz, 1978). Метод представляет собой сочетание метода выделения фрагментов ДНК, защищенных белком от гидролиза ДНКазой I и метода секвенирования Максама–Гилберта. С помощью электрофореза в денатурирующем полиакрил-амидном геле (ПААГе) сравнивают связанные белком последовательности ДНК, меченные радиоактивным фосфором по 5'-концу и обработанные ДНКазой I, с контрольными, гидролизованными ДНКазой I, но не связанными белком. Каждая дорожка образует лесенку из бандов ДНК, но на одной дорожке остается “след” – отсутствие бандов на месте связывания белка, – на основании чего расшифровывается последовательность сайта связывания (рис. 1).
Рис. 1.
Схематическое изображение эксперимента футпринтинга. Фрагмент ДНК метится (звездочка) на одном конце одной цепи, связывается с белком и гидролизуется, например ДНКазой I. Белок (лиганд) защищает ДНК от гидролиза в своем сайте связывания, и это проявляется в виде отсутствия бандов – футпринта, когда продукты расщепления разделяются с помощью электрофореза в денатурирующем полиакриламидном геле (по: Hampshire et al., 2007, с модификациями).
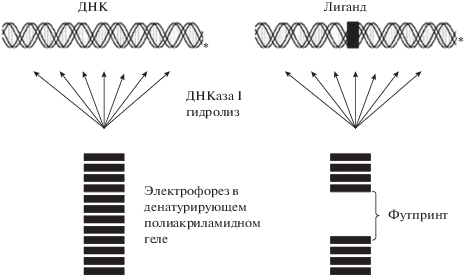
Для экспериментов может использоваться любая нуклеаза, но ДНКаза I является самым простым вариантом нуклеазы – эндонуклеазой, специфичной к двуцепочечной ДНК и не имеющей специфичности к последовательности ДНК, хотя некоторую предпочтительность к основаниям ДНК и ее структуре она все же имеет (Cardew, Fox, 2010). Это выражается в разной интенсивности бандов. Практически любая последовательность ДНК может служить субстратом для ДНКазы I, но предпочтительно использовать относительно короткие фрагменты длиной 80–300 п.н. и последовательности, которые не обогащены GC-парами и не содержат длинных повторов AT-пар, которые плохо гидролизуются (Bailly et al., 2005) и, соответственно, дают плохое разрешение полос при электрофорезе и их не так легко обнаружить. При использовании метода футпринтинга необходимо оптимизировать как количество ДНКазы I, так и минимальную концентрацию ДНК-связывающего белка. Условия надо подбирать так, чтобы в реакционной смеси не было избытка ДНК по отношению к белку. Иначе избыточная ДНК будет гидролизоваться, банды появятся на месте “следа” белка и усложнят или исказят картину футпринта. При инкубации неочищенных образцов белка с фрагментом ДНК необходимо добавлять в реакционную смесь конкурентную ДНК для исключения неспецифического связывания белков. Поскольку при футпринтинге метится одна нить ДНК, это позволяет определить нить, с которой связывается белок. Метод футпринтинга также используется для выявления сайтов связывания лекарств c ДНК (Bailly et al., 2005). Модификации этого метода включают использование автоматического секвенатора ДНК вместо гель-электрофореза и авторадиографии, и флуоресцентное мечение ДНК вместо радиоактивного (Yindeeyoungyeon, Schell, 2000; Wilson et al., 2001), когда не требуется высокая точность.
Может также проводиться химическое расщепление ДНК с использованием гидроксильных радикалов (Tullius, Dombroski, 1987). Гидро-ксильные радикалы производятся в результате реакции между ${\text{Fe}}_{2}^{ + }$ и H2O2 и представляют собой высоко реакционноспособные свободно диффундирующие частицы небольшого размера (Tullius, Dombroski, 1987) в отличие от громоздкой молекулы ДНКазы I. В результате они способны гидролизовать углеродные связи через маленькую бороздку ДНК, проникая более близко к связанному белку, и более точно локализовать положение сайта связывания (отпечаток), но, к сожалению, они также могут гидролизовать более доступные для них основания под белком и искажать футпринт. Гидролиз радикалами также дает четкую лесенку бандов из продуктов расщепления, не зависящую от последовательности.
В настоящее время для общегеномных исследований (genome-wide association studies, GWAS) часто используется метод гидролиза ДНКазой I с последующим высокопроизводительным секвенированием (метод DNAse-seq) (Boyle et al., 2008; Song, Crawford, 2010). Выявление гиперчувствительных сайтов ДНКазы I исторически было ценным инструментом для идентификации различных типов регуляторных элементов (включая промоторы, энхансеры, инсуляторы и др.). Исследования показали, что большинство таких элементов, связанных с распространенными заболеваниями и признаками, сконцентрировано в регуляторной ДНК, обогащенной гиперчувствительными участками ДНКазы I (Maurano et al., 2012).
ДНКазу I используют для расщепления обедненной нуклеосомами ДНК, тогда как участки ДНК, плотно упакованные в нуклеосомные структуры и структуры более высокого порядка, более устойчивы к гидролизу. С помощью метода DNase-seq идентифицируют гиперчувствительные сайты ДНКазы I по всему геному путем захвата расщепленных ДНКaзой фрагментов ДНК и секвенирования их с помощью высокопроизводительного секвенирования следующего поколения (next generation sequencing, NGS). В одном эксперименте можно идентифицировать несколько активных регуляторных областей из потенциально любого типа клеток любых видов, имеющих секвенированный геном (Song, Crawford, 2010).
Разработано несколько методов с высокой пропускной способностью для анализа большого количества гиперчувствительных к ДНКазе сайтов ДНК с использованием чипов (DNКase-chip) или высокопроизводительного секвенирования (DNase-seq) (Crawford et al., 2006; Boyle et al., 2008). Оба метода основаны на традиционном расщеплении ДНК с оптимальными концентрациями ДНКазы для преимущественного гидролиза открытых участков хроматина. Использование твердофазной технологии, включающей иммобилизацию фрагментов ДНК с предполагаемым сайтом связывания исследуемого белка, биотинилированных по одному концу и радиоактивно меченных по-другому, на парамагнитных шариках, покрытых стрептавидином, позволяет быстро и эффективно очищать гидролизованные фрагменты. Этот метод особенно облегчает извлечение интересующего белка из сырого экстракта, так как неспецифические комплексы белок–ДНК можно удалить элюирующим буфером до добавления ДНКазы, и они не будут искажать картину (Sandaltzopoulos, Becker, 1994). Высокая стабильность взаимодействия стрептавидин/биотин обеспечивает иммобилизацию ДНК в широком диапазоне условий реакции, делает метод более удобным и пригодным для одновременной обработки нескольких образцов.
Метод футпринтинга реже использовали для изучения кинетики взаимодействия белок–ДНК – скоростей ассоциации и диссоциации комплексов белков с олигонуклеотидами (Brenowitz et al., 1986). Скорость ассоциации белка можно определить измерением скорости появления “следа” путем гидролиза реакционной смеси после различных временных периодов инкубации. Аналогично, скорость диссоциации комплекса белок–ДНК можно определить путем измерения скорости исчезновения “следа” после добавления подходящего агента, который может изолировать из реакционной среды освободившийся при диссоциации белок. Например, это достигается путем добавления большого избытка ДНК тимуса теленка для белка, а для олигонуклеотидов – добавления избытка цепи, которая комплементарна олигонуклеотиду, образующему триплекс. Сайты связывания белков могут быть легко определены визуальным осмотром, сравнивая интенсивность полос в контрольной и обработанной ДНКазой дорожках. Для количественного определения кинетических характеристик взаимодействия белок–ДНК, высушенный гель экспонируют с помощью рентгеновской пленки в течение ночи при –70°C с усиливающим экраном и затем сканируют с помощью фосфоримиджера Storm, Typhoon, PharosFX Plus (Bio-Rad) и др. (Brenowitz et al., 1986; Fletcher, Fox, 1996; Dhavan et al., 2002). Но гораздо более эффективным и удобным для определения кинетических параметров взаимодействия белка с ДНК является метод “задержки в геле”, о котором будет сказано ниже.
Несмотря на то, что ChIP (иммунопреципитация хроматина) является эффективным способом выявления фрагментов ДНК, специфично связываемых белком in vivo, для точного определения последовательности ДНК, связываемой белком, обычно используют классический метод футпринтинга in vitro из-за его простоты и эффективности. Но следует иметь в виду, что использование этого метода для изучения высокодинамичных ТФ с коротким временем взаимодействия с ДНК, например Sox2 и глюкокортикоидный рецептор (Sung et al., 2014), может не показать адекватных “следов”. Основным недостатком большинства методов отпечатков является трудоемкая и проблемная подготовка фрагментов нуклеиновой кислоты, радиоактивно меченных 32Р. Футпринтинг с использованием флуоресцентно меченных фрагментов лишен этих недостатков, но относительно дорог и менее точен.
МЕТОД СДВИГА ЭЛЕКТРОФОРЕТИЧЕСКОЙ ПОДВИЖНОСТИ
Метод сдвига электрофоретической подвижности (electrophoretic mobility shift assay, EMSA) является очень эффективным для исследования многих аспектов ДНК-белковых взаимодействий in vitro. Метод основан на том, что ассоциация белка с сайтом связывания на фрагменте ДНК приводит к снижению подвижности ДНК в неденатурирующем полиакриламидном геле относительно свободной, не связанной с белком ДНК. Степень снижения подвижности ДНК связана с зарядом, размером и формой молекулы связанного белка. Метод EMSA чаще всего используется для определения термодинамических (сродства белков к конкретным сайтам ДНК) (Powell et al., 1993, 2004) и кинетических параметров взаимодействия белков с ДНК (Henriksson-Peltola, 2007; Dey et al., 2012). Мечение нуклеиновой кислоты по 5'- или 3'-концу 32P-фосфатом является широко используемым подходом, поскольку обеспечивает высокую чувствительность и не создает конформационных изменений, которые могли бы влиять на связывание. При использовании меченных радиоактивным изотопом нуклеиновых кислот, анализ является высокочувствительным, что позволяет использовать небольшие концентрации белка и нуклеиновых кислот (0.1 нМ или менее). Когда высокая чувствительность не нужна, также доступны варианты с использованием флуоресцентных, биотиновых и хемилюминесцентных меток нуклеиновой кислоты и иммуногистохимическое окрашивание (Berger et al., 1993; Mukhopadhyay et al., 2003). Метод может быть использован для оценки влияния других белков на специфическое связывание белок/ДНК (Buratowski, Chodosh, 2001), а также косвенно может указывать на конформационные изменения в белке в результате изгиба ДНК (Buratowski, Chodosh, 2001). Например, взаимодействия с кофакторами могут индуцировать специфическое связывание ДНК с белками за счет изменения конформации остатков белка, контактирующих с ДНК (Powell et al., 1993). Конформационные изменения, вызванные каждым кофактором, могут быть очень слабыми по отдельности, но при этом взаимоусиливающими (Powell et al., 2004).
Белок в комплексе с нуклеиновой кислотой может быть идентифицирован путем добавления антитела. Антитело, специфичное к исследуемому белку, либо блокирует образование комплекса (в случае связывания с ДНК-связывающим доменом белка), либо образует тройной комплекс антитело–белок–ДНК, что приводит к дальнейшему снижению подвижности и образованию “супершифта” при электрофоретическом разделении (Buratowski, Chodosh, 2001). Для доказательства специфичности связывания можно истощить исследуемый белок в неочищенном экстракте специфичным антителом и добавление ДНК с сайтом связывания покажет отсутствие банда белок/ДНК. Специфичность связывания также может быть легко подтверждена экспериментами по конкурентному связыванию с избытком немеченой, “холодной”, ДНК. Только специфичная конкурентная ДНК должна снижать интенсивность замедленной радиоактивной полосы.
Для определения равновесных констант диссоциации комплексов (КD) белка с фрагментами ДНК, несущими сайт связывания, используется традиционный подход, включающий титрование белка в фиксированной концентрации нуклеиновой кислотой в возрастающей концентрации или наоборот. Предварительно определяется время установления равновесного состояния. Для этого снимается кинетическая кривая связывания белка с изучаемым фрагментом ДНК. Эксперименты по равновесному связыванию белка с ДНК проводятся в условиях, оптимальных для взаимодействия, которые близки к условиям in vivo. После электрофоретического разделения гели высушиваются и экспонируются с экраном Imaging ScreenK (Kodak) для фосфоимиджера Molecular Imager PharosFX Plus (Bio-Rad). Как известно, равновесная константа диссоциации комплексов белок/ДНК (КD) равна концентрации лиганда, при которой количество образованных комплексов составляет половину от максимального. Микроденситометрия окрашенных гелей и авторадиограммы гелей позволяют количественно определять содержание белка и ДНК в каждом комплексе. Расчет равновесных констант диссоциации комплексов белок/ДНК может проводиться в программе GraphPad Prism 5. Метод EMSA позволяет определить стехиометрическое соотношение белка, специфически связанного с ДНК, и точно измерить относительную аффинность (сродство) связывания данного белка с ДНК. Используемые при связывании условия низкой ионной силы и гелевая матрица стабилизируют комплекс белок/ДНК от диссоциации. Кинетика диссоциации комплексов белок/ДНК легко измеряется для комплексов с временем жизни более одной минуты (время, необходимое для ручного смешивания компонентов реакции).
Кинетические характеристики – константы скорости ассоциации (ka) и константы скорости диссоциации (kd), отражающие скорости образования и распада комплексов белок/ДНК, определяют, снимая кинетику, то есть определяя количество образующихся комплексов белок/ДНК при постоянной концентрации белка и изменяющейся во времени концентрации ДНК (или наоборот). Кинетические характеристики образования комплексов белок/ДНК описывают важные биомолекулярные процессы, такие как транскрипция ДНК, репарация, модификация хроматина и др. Кроме того, знание кинетических параметров комплексов необходимо при их использовании в биотехнологии и биомедицине. Эксперименты по определению кинетических параметров образования комплексов белок/ДНК проводятся с использованием нескольких концентраций радиоактивно или флуоресцентно меченного фрагмента ДНК, и, как правило, очищенного белка (рис. 2).
Рис. 2.
Определение кинетических параметров взаимодействия белок/ДНК методом EMSA на примере взаимодействия ТАТА-связывающего белка (ТВР) с олигонуклеотидом, идентичным ТАТА-боксу промотора гена LEP человека: ka= (11 ± 0.2) × 103 M–1c–1; kd = (8 ± 1) ×10–4 c–1; KD = kd/ka = 70 ± 20 nM; t1/2 = 14.4 ± 4 мин (по: Arkova et al., 2015, с модификациями).
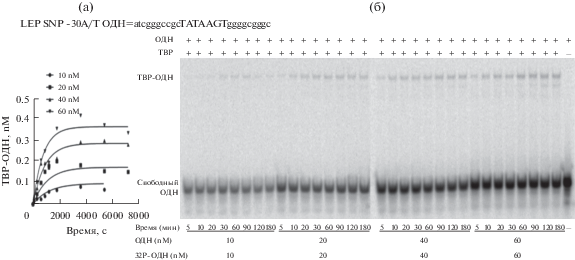
Из измеренных констант ассоциации и диссоциации комплексов можно определить константу равновесной диссоциации комплексов белок/ДНК, которая характеризует сродство белок/ДНК, KD = kd/ka; из кинетических констант можно также определить средний период полураспада (или время жизни) комплексов белок/ДНК (t1/2), который равен ln2/kd. Можно посчитать свободную энергию Гиббса ΔG0 = −RT lnKA, где R – универсальная газовая постоянная, Т – абсолютная температура, а KA (константа равновесия) – ka/kd, отрицательные значения которой характеризуют возможность спонтанного протекания реакции связывания белок/ДНК. Эта информация может быть важной для характеристики взаимодействия двух белков с одной и той же последовательностью или одного белка с разными аллелями (Drachkova et al., 2014).
При использовании метода EMSA нужно иметь в виду, что комплексы белок/ДНК могут быть очень чувствительны к механическому перемешиванию и легко диссоциировать. Для точного измерения термодинамических и кинетических параметров взаимодействия белок/ДНК эксперименты необходимо проводить в максимально стандартизированных условиях, чтобы свести к минимуму разброс числовых значений и получить наиболее точные данные. Кроме того, для получения количественных характеристик комплексов белок/ДНК необходимо учитывать, что в используемом препарате белка не все молекулы могут образовывать сайт-специфические комплексы с ДНК, поэтому перед экспериментом необходимо определить число активных молекул в используемом препарате для внесения поправок в числовые значения.
Анализ с помощью EMSA хорошо работает с высокоочищенными белками, с неочищенными клеточными и ядерными экстрактами, но в последних случаях необходимо использовать конкурентную ДНК, например, poly d(I-C), для предотвращения неспецифического связывания. Метод EMSA также не без ограничений, одно из которых заключается в том, что пробы не находятся в химическом равновесии на этапе электрофореза. Короткоживущие комплексы с быстрой скоростью диссоциации во время электрофореза можно не обнаружить или обнаружить значительно меньшее их количество. Метод может использоваться также для изучения взаимодействия белков с РНК.
EMSA и DAPA – метод ДНК-аффинного осаждения (DNA affinity precipitation assay, DAPA) (Miller et al., 2016) используют для идентификации ТФ в ядерных экстрактах больных и условно здоровых, а также верификации предсказанных ОДН (олигодезоксинуклеотиды) из некодирующих генетических вариантов, ассоциированных с каким-либо заболеванием или фенотипом. Последовательности синтетических ОДН предсказываются из геномных баз данных с помощью подхода in silico. Флуоресцентно меченные ОДН и ТФ, связанные с белками ядерного экстракта, анализируют с помощью метода EMSA. Если для EMSA комплексы олигонуклеотидов, связанных с белком в растворе in vitro, анализируют с помощью неденатурирующего и денатурирующего электрофореза в полиакриламидном геле с последующим использованием антител (наблюдается супершифт или отсутствие банда связывания), то для DAPA олигонуклеотиды иммобилизуют на стреповидиновых гранулах в магнитной колонке, через которую затем пропускают ядерный экстракт. ТФ, которые с высоким сродством связываются с ОДН, элюируют и анализируют с помощью масс-спектрометрии или электрофореза в полиакриламидном геле с додецилсульфатом натрия (SDS-PAGE) и последующим анализом белка с помощью вестерн-блоттинга (Miller et al., 2016). Метод позволяет выделить почти аффинно очищенные белки, связанные с иммобилизованными на колонке ОДН определенной последовательности, и идентифицировать их. Недостатки EMSA- и DAPA-методов при использовании неочищенных ядерных экстрактов заключаются в том, что оба метода не дают информации о способах связывания ТФ с ОДН in vivo: связывается ТФ с ОДН напрямую или привлекается посредством белок-белковых взаимодействий. Для этого нужны дополнительные эксперименты. Недостатком метода DAPA является то, что в результате отмывки остаются только комплексы белков с высоким сродством к ДНК, а слабо связанные комплексы вымываются, хотя они тоже могут содержать важную информацию.
МЕТОД ПОВЕРХНОСТНОГО ПЛАЗМОННОГО РЕЗОНАНСА
Поверхностный плазмонный резонанс (surface plasmon resonance, SPR) – физический эффект, который возникает в случае резонанса частоты падающего на поверхность микрочипа электромагнитного поля (фотонов) с собственной частотой колебаний электронов (плазмонов) поверхностного слоя золотой пленки микрочипа (Navratilova et al., 2006; Bronner et al., 2010).
Изменения диэлектрической проницаемости среды и коэффициента отражения в результате взаимодействия вводимого аналита с иммобилизованным на декстрановой поверхности микрочипа лигандом вызывают сдвиг резонансного сигнала, который линейно пропорционален коэффициенту оптического преломления в зоне измерения и массе аналита (Nahshol et al., 2008; Abbas et al., 2011). Это позволяет в режиме реального времени регистрировать взаимодействие белок/ДНК, которое выражается в резонансных единицах (RU). Общий принцип, который лежит в основе работы SPR – полное внутреннее отражение, возникающее при прохождении поляризованного света через среду с высоким и низким показателями преломления (Brockman et al., 1999; Majka, Speck, 2007; Nguyen et al., 2007).
Со времени появления технологии поверхностного плазмонного резонанса, которая была разработана совместными усилиями физиков, химиков, биологов, программистов и инженеров, ее стали использовать в различных областях: для обнаружения микробного загрязнения (Waswa et al., 2007), тестирования пищевых продуктов (Situ et al., 2010), в лабораторной медицине (Helmerhorst et al., 2012), в протеомике (Yuk, Ha, 2005) при изучении межбелковых взаимодействий (Douzi, 2017), при анализе различных биологических жидкостей организма человека для раннего выявления биомаркеров комплексных заболеваний, таких как рак (Reddy et al., 2012) и т.д. (рис. 3).
Рис. 3.
Различные фазы сенсограммы, наблюдаемые при использовании метода поверхностного плазмонного резонанса на биосенсоре. Исходная базовая линия получается после иммобилизации лиганда. После введения аналита увеличение резонансных единиц (RU) соответствует фазе ассоциации. Равновесие сохраняется, когда число событий ассоциации равно числу событий диссоциации. Фаза диссоциации начинается после прекращения введения аналита: уменьшение RU соответствует смыванию аналита с поверхности лиганда. Этап регенерации направлен на полное удаление аналита и достижение базового уровня (по: Douzi, 2017, с модификациями).
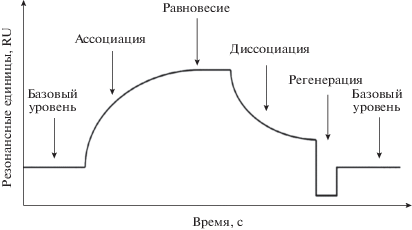
Из сенсограммы можно получить два основных вида информации: о скорости взаимодействия (ассоциация, диссоциация), которая представлена в виде кинетических констант, и об уровне связывания, который представлен в виде констант сродства (Munoz et al., 2013). Кинетические константы дают ценные данные о динамике исследуемой биологической системы, которые могут быть использованы для отбора и конструирования новых молекул, представляющих терапевтический интерес.
Иммобилизация биомолекулы на сенсорной поверхности является ключевым шагом, необходимым для успешного анализа. В частности, необходимо сохранение биологической активности лиганда после иммобилизации, определение оптимального количества лиганда при иммобилизации для избежания стерических помех при взаимодействии, сведение к минимуму неспецифического связывания с поверхностью, необходима воспроизводимость иммобилизации для обеспечения воспроизводимости результатов экспериментов и регенерируемость поверхности биосенсора – диссоциация комплекса лиганд/аналит без изменения биологической активности лиганда (Mariani, Minunni, 2014). Иммобилизацию проводят либо на одномерной матрице, либо на трехмерной, которая обеспечивает больше сайтов связывания, чем иммобилизация на поверхности (Homola, 2008). Иммобилизация на поверхности более проста для выполнения, но количество иммобилизуемого материала ограничено емкостью поверхности матрицы (слишком высокая плотность иммобилизованных элементов может привести к нарушению реакции из-за стерических помех). Иммобилизация на трехмерной матрице обычно обеспечивает больше сайтов связывания, чем иммобилизация на поверхности, что может быть и помехой, так как одновременная иммобилизация через несколько функциональных групп, например, белка, может ограничить конформационную гибкость белка и нарушить его функцию (Homola, 2008). Наиболее широко используемой трехмерной матрицей для иммобилизации молекул является карбоксиметилированный декстран. Основные подходы к иммобилизации молекул на поверхности сенсоров SPR основаны на гидрофобных и электростатических взаимодействиях, на образовании ковалентной связи, образованной между нуклеофильными функциональными группами аминокислот (например, аминогруппами, лизином; тиоловыми группами, цистеином) белка и электрофильными группами (например, активированными карбоксильными группами и альдегидами) на поверхности сенсора (Oshannessy et al., 1992; Lofas et al., 1995). Иммобилизация с образованием ковалентной связи стабильна, но белки могут содержать много функциональных групп, способных к образованию ковалентных связей, и иммобилизация через эти функциональные группы приводит к случайной ориентации иммобилизованных белков. Часто используется подход к иммобилизации белков, основанный на высокоаффинном взаимодействии авидина и биотина (Johnson et al., 2003). Другой подход основан на биоаффинной иммобилизации. Наиболее распространенным примером этого подхода является иммобилизация в результате взаимодействия между белком, содержащим подвижный гистидиновый линкер, и ионами хелатированных металлов (Zhen et al., 2005). С учетом этих подводных камней многие авторы считают, что гораздо предпочтительней иммобилизовать ДНК (Zhen et al., 2007). Нужно также иметь в виду, что поверхности чипов для SPR могут быть микрогетерогенными и состоять из участков с набором разных связывающих свойств. Кроме того, поверхность чипа может представлять собой низкоаффинные неспецифические сайты связывания для анализируемого вещества, даже в отсутствие иммобилизованного партнера связывания она может как способствовать образованию комплексов, так и ингибировать их образование (Nguyen et al., 2007). Подробно методы иммобилизации рассмотрены в очень хороших обзорах (Lofas et al., 1995; Rusmini et al., 2007; Nguyen et al., 2007). Вторым наиболее распространенным источником получения искаженных результатов анализа с использованием SPR, после неоднородности участков поверхности чипа, являются ограничения массопереноса (Schuck, Zhao, 2010). Массоперенос – это доставка реагента из раствора к иммобилизованному на поверхности чипа реагенту для образования комплекса. Если скорость транспорта медленнее, чем реакция связывания аналита с лигандом, то транспорт будет тормозить кинетику связывания. Но если кинетика переноса реагента на иммобилизованную молекулу намного быстрее, то наблюдаемое связывание будет представлять истинную кинетику взаимодействия изучаемых веществ (Myszka et al., 1998a; Karlsson, 1999; Schuck et al., 1999). Таким образом, скорость массопереноса – это критический фактор, который необходимо учитывать при разработке эксперимента с биосенсором и при оценке кинетических констант. Итак, два наиболее распространенных источника отклонения кинетики поверхностного связывания SPR от идеальной кинетики связывания псевдопервого порядка бимолекулярных реакций – это ограничения массопереноса и неоднородность участков поверхности микрочипа (Schuck, Zhao, 2010). Кроме того, иногда необходимо проводить дополнительные эксперименты для определения оптимальной скорости потока. Скорость потока должна быть достаточной для проведения кинетических экспериментов. О ее оптимальности свидетельствует устойчивое плато на сенсограмме (Nguyen et al., 2007) – плато устанавливается там, где происходит ассоциация и диссоциация комплексов с одинаковой скоростью. Высокие скорости потока могут не позволить по времени собрать полностью результаты по образованию комплексов – для длительного мониторинга фазы ассоциации бывает необходимо увеличить объем впрыскиваемого буфера с аналитом (Myszka et al., 1998b).
Следует иметь ввиду, что регенерация поверхности чипа также имеет решающее значение для получения воспроизводимых данных хорошего качества. Некоторые комплексы диссоциируют довольно быстро и для регенерации достаточно поверхность чипа промыть буферным раствором после стадии диссоциации. Для плохо диссоциирующих комплексов требуется регенерационный раствор с высоким содержанием соли или с низким (высоким) рН. Регенерационный раствор должен полностью удалять соединения с поверхности чипа, чтобы не искажать кинетику связывания в следующем цикле концентраций, но не оказывать разрушающего воздействия на поверхность чипа. Поэтому после использования регенерационного раствора необходимо промывание всей системы (инъекционные иглы, пробирки для подачи образца) многократными объемами используемого буфера.
Биосенсоры на основе SPR имеют аналитические характеристики (чувствительность, воспроизводимость), сопоставимые с получаемыми с использованием классических методов электрофореза, хроматографии и др. Преимуществом их является использование немеченых реагирующих веществ, мониторинг взаимодействия в режиме реального времени, высокая производительность и быстрое получение результатов. К преимуществам биосенсоров SPR следует отнести также то обстоятельство, что поверхность биосенсора, по существу, представляет собой систему аффинной очистки в миниатюре, что открывает возможности для изучения мультибелковых комплексов (Pattnaik, 2005; Sundberg et al., 2007). Для небольших анализируемых веществ, элюирование материала с поверхности микрочипа биосенсора позволяет проводить масс-спектрометрическую идентификацию молекул. Наконец, процесс связывания на биосенсоре толерантен к примесям в образце препарата, которые не ингибируют взаимодействие (Schuck, Zhao, 2010). Итак, чтобы использовать все эти преимущества биосенсора, необходима тщательная подготовка образцов и подбор экспериментальных условий.
Следует отметить, что биосенсоры SPR наиболее широко используются в исследовательских фармацевтических лабораториях по всему миру. Наиболее важные направления фармацевтических исследований связаны с изучением переноса лекарственного средства из места введения в системный кровоток, анализа крови (Peungthum et al., 2017), с ранней диагностикой различных заболеваний (Olaru et al., 2015), диагностикой аллергических заболеваний (Yanase et al., 2012) и т.д. Одной из конечных целей использования биосенсоров является разработка тестирования на месте оказания медицинской помощи. Для этого планируется разработать портативные платформы, миниатюрные аналитические SPR-сенсорные приборы и одноразовые чипы (Choi et al., 2020).
ЗАКЛЮЧЕНИЕ
ДНК-белковые взаимодействия являются неотъемлемым компонентом биологических систем, и их изучение важно для всех биологических процессов. ТФ играют важную роль в регуляции экспрессии генов, особенно при развитии организма и в ответ на изменение условий окружающей среды. Одним из механизмов достижения специфичной и сбалансированной экспрессии генов является связывание ТФ с цис-регуляторными элементами ДНК. Эти взаимодействия вносят вклад в регуляцию пространственно-временной специфичности транскрипции. Для более глубокого понимания и создания точных моделей регуляции генов в зависимости от типа клетки и состояния требуется лучшее понимание того, как ТФ привлекаются на ДНК, какую роль играет величина аффинности взаимодействия ТФ/ДНК, какой вклад во взаимодействие делают другие регуляторные белки и т.д. Достаточно много методов доступно для удачного определения этих взаимодействий и их понимания. На уровне in vitro молекулярно-биологические методы, такие как анализы отпечатков стопы, EMSA, southwestern-блоттинг, фаговый дисплей, SELEX, одногибридный и двугибридный дрожжевые анализы и др. позволяют надежно определять ДНК-белковые взаимодействия. Высокодинамичные инструменты in vivo – иммунопреципитация хроматина и ее разнообразные варианты – надежные методы для характеристики ДНК-белковых взаимодействий в клетках. In silico подходы также развивались за эти годы, чтобы дополнить информацию, доступную для исследователей (Matys et al., 2003; Tsuchiya et al., 2004; Schlundt et al., 2017). Появились более сложные модели связывания и более совершенные методы оптимизации для всех моделей (Alipanahi et al., 2015). Однако все еще существует большой разрыв между качеством прогнозирования событий in vivo на основе результатов in vitro. Одной из причин является отсутствие подробных и надежных знаний о комбинаторных взаимодействиях между ТФ, ТФ и белками-кофакторами и конкурирующими белками, влияние которых до сих пор подробно не изучено (Orenstein, Shamir, 2017). Для того, чтобы решить эту проблему, необходимо объединение методов in vitro и in vivo, характеристика косвенных и низкоаффинных взаимодействий и выяснение их роли в процессах транскрипции. Различные относительно недавние биофизические методы, в том числе флуоресцентные методы, SPR, ЯМР, АСМ и другие имеют большой потенциал для обнаружения взаимодействий белка с нуклеиновыми кислотами. Каждая техника по-своему уникальна и служит уникальной цели. Но тем не менее современное состояние методов оставляет желать лучшего. Необходим “идеальный” метод с минимальным количеством клеток, способных обнаруживать редкие взаимодействия, с высокой специфичностью и чувствительностью, легко модифицируемый, сочетающий нескольких методов in vitro и in vivo, чтобы определить количество взаимодействий и предоставить количественную характеристику геномных взаимодействий белок–ДНК в пространстве и времени. Разработанные методы помогут исследователям оценить динамику ДНК-белковых взаимодействий в клеточном развитии и прогрессировании заболевания.
Список литературы
Abbas A., Linman M.J., Cheng C. New trends in instrumental design for surface plasmon resonance-based biosensors // Biosens. Bioelectron. 2011. V. 269. P. 1815–1824.
Alexander M.K., Bourns B.D., Zakian V.A. One-hybrid systems for detecting protein–DNA interactions // Meth. Mol. Biol. 2001. V. 77. P. 241–259.
Alipanahi B., Delong A., Weirauch M.T. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning // Nat. Biotechnol. 2015. V. 33. P. 831–838.
Arkova O.V., Ponomarenko M.P., Rasskazov D.A. et al. Obesity-related known and candidate SNP markers can significantly change affinity of TATA-binding protein for human gene promoters // BMC Genomics. 2015. V. 16 (Suppl. 13). P. S5. https://doi.org/10.1186/1471-2164-16-S13-S5
Badis G., Berger M.F., Philippakis A.A. et al. Diversity and complexity in DNA recognition by transcription factors // Science. 2009. V. 32. P. 1720–1723.
Bailly C., Kluza J., Martin C. et al. DNase I footprinting of small molecule binding sites on DNA // Meth. Mol. Biol. 2005. V. 288. P. 319–342.
Behera V., Evans P., Face C.J. Exploiting genetic variation to uncover rules of transcription factor binding and chromatin accessibility // Nat. Commun. 2018. V. 9. P. 782. https://doi.org/10.1038/s41467-018-03082-6
Berger R., Duncan M.R., Berman B. Nonradioactive gel mobility shift assay using chemiluminescent detection // Biotechniques.1993. V. 15. P. 650–652.
Berggard T., Linse S., James P. Methods for the detection and analysis of protein–protein interactions // Proteomics. 2007. V. 7. 2833–2842.
Boyle A.P., Davis S., Shulha H.P. et al. High-resolution mapping and characterization of open chromatin across the genome // Cell. 2008. V. 132. P. 311–322.
Brenowitz M., Senear D.F., Shea M.A., Ackers G.K. Quantitative DNase footprint titration: a method for studying protein–DNA interactions // Enzyme structure Part K, 9th edn / Eds C.H.W. Hirs, S.N. Timasheff. New York: Academic Press, 1986. P. 132–181.
Brockman J.M., Frutos A.G., Corn R.M. A multistep chemical modification procedure to create DNA arrays on gold surfaces for the study of protein–DNA interactions with surface plasmon resonance imaging // J. Am. Chem. Soc. 1999. V. 121. P. 8044–8051.
Bronner V., Denkberg G., Peled M. et al. Therapeutic antibodies: discovery and development using the ProteOn XPR36 biosensor interaction array system // Anal. Biochem. 2010. V. 406. P. 147–156.
Brown T. Southern blotting // Curr. Protoc. Immunol. 2001.Chapter 10: Unit 10. 6A. https://doi.org/10.1002/0471142735.im1006as06
Brown J.S., Jackson S.P. Ubiquitylation, neddylation and the DNA damage response // Open Biol. 2015. V. 5. P. 150018. https://doi.org/10.1098/rsob.150018
Bulyk M.L. DNA microarray technologies for measuring protein–DNA interactions // Curr. Opin. Biotechnol. 2006. V. 17. P. 422–430.
Buratowski S., Chodosh L.A. Mobility shift DNA-binding assay using gel electrophoresis // Curr. Protoc. Mol. Biol. / Eds F.M. Ausubel et al., Hью-Йopк: John Wiley and Sons Inc., 2001. V. 36. Iss. 1. P. 12.2.1–12.2.11.
Cardew A.S., Fox K.R. DNase I footprinting. drug-DNA interaction protocols // Meth. Mol. Biol. 2010. V. 613. P. 153–172. https://doi.org/10.1007/978-1-60327-418-0_10
Castellated M., Mothi N., Muñoz V. Eukaryotic transcription factors can track and control their target genes using DNA antennas // Nat. Commun. 2020. V. 11. P. 540. https://doi.org/10.1038/s41467-019-14217-8
Choi J.H., Lee J.H., Son J., Choi J. W. Noble metal-assisted surface plasmon resonance immunosensors // Sensors. 2020. V. 20. P. 1003. https://doi.org/10.3390/s20041003
Choo Y., Klug A. Designing DNA-binding proteins on of filamentous phage // Curr. Opin. Biotechnol. 1995. V. 6. P. 431–436.
Crawford G.E., Davis S., Scacheri P.C. et al. DNase-chip: a high-resolution method to identify DNase I hypersensitive sites using tiled microarrays // Nat. Methods. 2006. V. 3. P. 503–509.
Dey B., Thukral S., Krishnan S. et al. DNA–protein interactions: methods for detection and analysis // Mol. Cell Biochem. 2012. V. 365. P. 279–299.
Dhavan G.M., Mollah A.K., Brenowitz M. Equilibrium and kinetic quantitative DNase I footprinting // Advances in DNA sequence-specific agents, 4th edn / Ed. G.B. Jones. New York: Elsevier, 2002. P. 139–155.
Djordjevic M. SELEX experiments: new prospects, applications and data analysis in inferring regulatory pathways // Biomol. Eng. 2007. V. 24. P. 179–189.
Dooley S., Welter C., Blin N. Nonradioactive southwestern analysis using chemiluminescent detection // Biotechniques. 1992. V. 13. P. 540–543.
Douzi B. Protein-protein interactions: surface plasmon resonance // Meth. Mol. Biol. 2017. V. 1615. P. 257–275. https://doi.org/10.1007/978-1-4939-7033-9_21
Drachkova I.A., Savinkova L.K., Arshinova T.V. et al. The mechanism by which TATA-box polymorphisms associated with human hereditary diseases influence interactions with the TATA-binding protein // Hum. Mutat. 2014. V. 35. P. 601–608. https://doi.org/10.1002/humu.22535
Fletcher M.C., Fox K.R. Dissociation kinetics of actinomycin D from individual GpC sites in DNA // Eur. J. Biochem. 1996. V. 237. P. 164–170.
Galas D.J., Schmitz A. DNAase footprinting a simple method for the detection of protein-DNA binding specificity // Nucl. Acids Res. 1978. V. 5. P. 3157–3170.
Garcia-Alonso L., Holland C.H., Ibrahim M.M. et al. Benchmark and integration of resources for the estimation of human transcription factor activities // Gen. Res. 2019. V. 29. P. 1363–1375. https://doi.org/10.1101/gr.240663.118
Hampshire A.J., Rusling D.A., Broughton-Head V.J., Fox K.R. Footprinting: a method for determining the sequence selectivity, affinity and kinetics of DNA-binding ligands // Methods. 2007. V. 42. P. 128–140.
Helmerhorst E., Chandler D.J., Nussio M., Mamotte C.D. Realtime and label-free bio-sensing of molecular interactions by surface plasmon resonance: a laboratory medicine perspective // Clin. Biochem. Rev. 2012. V. 33. P. 161–173.
Henriksson-Peltola P., Sehlen W., Haggard-Ljungquist E. Determination of the DNA-binding kinetics of three related but heteroimmune bacteriophage repressors using EMSA and SPR analysis // Nucl. Acids Res. 2007. V. 35. P. 3181–3191.
Hoffman B.G., Jones S. Genome-wide identification of DNA–protein interactions using chromatin immunoprecipitation coupled with flow cell sequencing // J. Endocrinol. 2009. V. 201. P. 1–13. https://doi.org/10.1677/JOE-08-0526
Homola J. Surface plasmon resonance sensors for detection of chemical and biological species // Chem. Rev. 2008. V. 108. P. 462–493.
Isalan M., Choo Y. Engineering nucleic acid-binding proteins by phage display // Meth. Mol. Biol. 2001. V. 148. P. 417–429.
Jackson M., Marks L., May G.H.W., Wilson J.B. The genetic basis of disease // Ess. Biochem. 2018. V. 62. P. 643–723. https://doi.org/10.1042/EBC20170053
Jolma A., Kivioja T., Toivonen J. et al. Multiplexed massively parallel SELEX for characterization of human transcription factor binding specificities // Gen. Res. 2010. V. 20. P. 861–873.
Johnson C.P., Jensen I.E., Prakasam A. et al. Engineered protein A for the orientational control of immobilized proteins // Bioconj. Chem. 2003. V. 14. P. 974–978.
Kasowski M., Grubert F., Heffelfinger C. et al. Variation in transcription factor binding among humans // Science. 2010. V. 328. P. 232–235.
Kim T.H., Ren B. Genome-wide analysis of protein–DNA interactions // Annu. Rev. Genom. Hum. Genet. 2006. V. 7. P. 81–102.
Kiyama R., Oishi M. Protection of DNA sequences by triplex-bridge formation // Nucl. Acids Res. 1995. V. 23. P. 452–458. https://doi.org/10.1093/nar/23.3.452
Kurien B.T., Scofield R.H. Western blotting // Methods. 2006. V. 38. P. 283–293.
Labbe S., Harrisson J.F., Seguin C. Identification of sequence-specific DNA-binding proteins by southwestern blotting // Meth. Mol. Biol. 2009. V. 543. P. 151–161.
Lambert S.A., Jolma A., Campitelli L.F. et al. The human transcription factors // Cell. 2018. V. 172. P. 650–665.
Lee T.I., Johnstone S.E., Young R.A. Chromatin immunoprecipitation and microarray-based analysis of protein location // Nat. Protocols. 2006. V. 1. P. 729–748. https://doi.org/10.1038/nprot.2006.98
Loehlin D.W., Ames J.R., Vaccaro K., Carroll S.B. A major role for noncoding regulatory mutations in the evolution of enzyme activity // PNAS USA. 2019. V. 116. P. 12383–12389.
Lofas S., Johnsson B., Edstrom A. et al. Methods for site controlled coupling to carboxymethyldextran surfaces in surface plasmon resonance sensors // Biosens. Bioelectron. 1995. V. 10. P. 813–822.
Louche A., Salcedo S.P., Bigot S. Protein-protein interactions: pull-down assays // Meth. Mol. Biol. 2017. V. 1615. P. 247–255. https://doi.org/10.1007/978-1-4939-7033-9_20
Luscombe N.M., Austin S.E., Berman H.M. et al. An overview of the structures of protein-DNA complexes // Gen. Biol. 2000. V. 1. Art. Reviews001.
Majka J., Speck C. Analysis of protein–DNA interactions using surface plasmon resonance // Adv. Biochem. Eng. Biotechnol. 2007. V. 104. P. 13–36.
Mariani S., Minunni M. Surface plasmon resonance applications in clinical analysis // Anal. Bioanal. Chem. 2014. V. 406. P. 2303–2323. https://doi.org/10.1007/s00216-014-7647-5
Maryas J., Factor J., Čapkova L. et al. Pull-down assay on streptavidin beads and surface plasmon resonance chips for SWATH-MS-based interactomics // Cancer Gen. Proteom. 2018. V. 15. P. 395–404. https://doi.org/10.21873/cgp.20098
Maurano M.T., Humbert R., Rynes E. et al. Systematic localization of common disease-associated variation in regulatory DNA // Science. 2012. V. 337 (6099). P. 1190–1195.
Matys V., Fricke E., Geffers R. et al. TRANSFAC: transcriptional regulation, from patterns to profiles // Nucl. Acids Res. 2003. V. 31. P. 374–378.
Miller D.E., Patel Z.H., Lu X. et al. Screening for functional non-coding genetic variants using electrophoretic mobility shift assay (EMSA) and DNA-affinity precipitation assay (DAPA) // J. Vis. Exp. 2016. V. 114. P. 54093. https://doi.org/10.3791/54093
Mishra M., Tiwari S., Gomes A.V. Protein purification and analysis: next generation Western blotting techniques // Exp. Rev. Proteom. 2017. V. 14. P. 1037–1053. https://doi.org/10.1080/14789450.2017.13881674
Mukhopadhyay J., Mekler V., Kortkhonjia E. et al. Fluorescence resonance energy transfer (FRET) in analysis of transcription-complex structure and function // Meth. Enzymol. 2003. V. 371. P. 144–159.
Munoz E.M., Correa J., Riguera R., Fernandez-Megia E. Real-time evaluation of binding mechanisms in multivalent interactions: a surface plasmon resonance kinetic approach // J. Am. Chem. Soc. 2013. V. 135. P. 5966–5969. https://doi.org/10.1021/ja9074826
Myszka D.G., Jonsen M.D., Graves B.J. Equilibrium analysis of high affinity interactions using BIACORE // Anal. Biochem. 1998a. V. 2. P. 326–330.
Myszka D.G., He X., Dembo M. et al. Extending the range of rate constants available from BIACORE: interpreting mass transport-influenced binding data // Biophys. J. 1998b. V. 75. P. 583–594.
Nahshol O., Bronner V., Notcovich A. et al. Parallel kinetic analysis and affinity determination of hundreds of monoclonal antibodies using the ProteOn XPR36 // Anal. Biochem. 2008. V. 383. P. 52–60.
Navratilova I., Dioszegi M., Myszka D.G. Analyzing ligand and small molecule binding activity of solubilized GPCRs using biosensor technology // Anal. Biochem. 2006. V. 355. P. 132–139. https://doi.org/10.1016/j.ab.2006.04.021
Nguyen B., Tanious F.A., Wilson W.D. Biosensor-surface plasmon resonance: quantitative analysis of small moleculenucleic acid interactions // Methods. 2007. V. 42. P. 150–161.
Olaru A., Bala C., Jaffrezic-Renault N., Aboul-Enein H. Y. Surface plasmon resonance (SPR) biosensors in pharmaceutical analysis // Crit. Rev. Anal. Chem. 2015. V. 45. P. 97–105. https://doi.org/10.1080/10408347.2014.881250
Orenstein Y., Shamir R. Modeling protein–DNA binding via high-throughput in vitro technologies // Brief Funct. Genomics. 2017. V. 16. P. 171–180.
Oshannessy D.J. Brighamburke M., Peck K. Immobilization chemistries suitable for use in the BIAcore surface plasmon resonance detector // Anal. Biochem. 1992. V. 205. P. 132–136.
Pattnaik P. Surface plasmon resonance: applications in understanding receptor-ligand interaction // Appl. Biochem. Biotechnol. 2005. V. 126. P. 79–92. https://doi.org/10.1385/ABAB:126:2:079
Pawlak M., Niescierowicz K., Winata C.L. Decoding the heart through next generation sequencing approaches // Genes (Basel). 2018. V. 9. pii:E289. https://doi.org/10.3390/genes9060289
Peungthum P., Sudprasert K., Amarit R. et al. Surface plasmon resonance imaging for ABH antigen detection on red blood cells and in saliva: secretor status-related ABO subgroup identification // Analyst. 2017. V. 142. P. 1471–1481. https://doi.org/10.1039/c7an00027h
Powell L.M., Dryden D.T., Willcock D.F. et al. DNA recognition by the EcoK methyltransferase. The influence of DNA methylation and the cofactor S-adenosyl-l-methionine // J. Mol. Biol. 1993. V. 234. P. 60–71. https://doi.org/10.1006/jmbi.1993.1563
Powell L.M., Zur Lage P.I., Prentice D.R.A. et al. The proneural proteins Atonal and Scute regulate neural target genes through different E-box binding sites // Mol. Cell Biol. 2004. V. 24. P. 9517–9526.
Ray D., Kazan H., Cook K. B. et al. A compendium of RNA-binding motifs for decoding gene regulation // Nature. 2013. V. 499. P. 172–177. https://doi.org/10.1038/nature12311
Reddy P.J., Sadhu S., Ray S., Srivastava S. Cancer biomarker detection by surface plasmon resonance biosensors // Clin. Lab. Med. 2012. V. 32. P. 47–72.
Rossouw D., Jacobson D., Bauer F.F. Transcriptional regulation and the diversification of metabolism in wine yeast strains // Genetics. 2012. V. 190. P. 251–261.https://doi.org/10.1534/genetics.111.132720
Ruscher K., Reuter M., Kupper D. et al. A fluorescence based non-radioactive electrophoretic mobility shift assay // J. Biotechnol. 2000. V. 78. P. 163–170.
Rusmini F., Zhong Z., Feijen J. Protein immobilization strategies for protein biochips // Biomacromolecules. 2007. V. 8. P. 1775–1789. https://doi.org/10.1021/bm061197b
Sandaltzopoulos R., Becker P. B. Solid phase DNase I footprinting: quick and versatile // Nucl. Acids Res. 1994. V. 22. P. 1511–1512. https://doi.org/10.1093/nar/22.8.1511
Schlundt A., Tants J. N., Sattler M. Integrated structural biology to unravel molecular mechanisms of protein-RNA recognition // Methods. 2017. V. 15. P. 118–136. https://doi.org/10.1016/j.ymeth.2017.03.015
Schuck P., Zhao H. The role of mass transport limitation and surface heterogeneity in the biophysical characterization of macromolecular binding processes by SPR biosensing // Meth. Mol. Biol. 2010. V. 627. P. 15–54. https://doi.org/10.1007/978-1-60761-670-2_2
Schuck P., Zhao H., Karlsson R. Affinity analysis of non-steady-state data obtained under mass transport limited conditions using BIAcore technology // J. Mol. Recognit. 1999. V. 12. P. 285–292. https://doi.org/10.1002/(SICI)1099-1352(199909/10)12:5<285:AID-JMR469>3.0.CO;2-Y
Siggers T., Duyzend M.H., Reddy J. et al. Non-DNA-binding cofactors enhance DNA-binding specificity of a transcriptional regulatory complex // Mol. Syst. Biol. 2011. V. 7. P. 555. https://doi.org/10.1038/msb.2011.89
Singh B., Nath S.K. Identification of proteins interacting with single nucleotide polymorphisms (SNPs) by DNA pull-down assay // Meth. Mol. Biol. 2019. V. 1855. P. 355–362. https://doi.org/10.1007/978-1-4939-8793-1_30
Situ C., Mooney M.H., Elliott C. T., Buijs J. Advances in surface plasmon resonance biosensor technology towards high-throughput, food-safety analysis // TrAC-Trends. Anal. Chem. 2010. V. 29. P. 1305–1315.
Slattery M., Riley T., Liu P. et al. Cofactor binding evokes latent differences in DNA binding specificity between Hox proteins // Cell. 2011. V. 147. P. 1270–1282. https://doi.org/10.1016/j.cell.2011.10.053
Slattery M., Zhou T., Yang L. et al. Absence of a simple code: how transcription factors read the genome // Tr. Biochem. Sci. 2014. V. 39. P. 381–399. https://doi.org/10.1016/j.tibs.2014.07.002
Smith G.P. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface // Science. 1985. V. 228. P. 1315– 1317. https://doi.org/10.1126/science.4001944
Song L., Crawford G. E. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells // Cold Spring Harb. Protoc. 2010. V. 2010. pdb.prot 5384. https://doi.org/10.1101/pdb.prot5384
Spencer V., Sun J.M., Li L., Davie J.R. Chromatin immunoprecipitation: a tool for studying histone acetylation and transcription factor binding // Methods. 2003. V. 31. P. 67–75. https://doi.org/10.1016/s1046-2023(03)00089-6
Stockley P.G., Persson B. Surface plasmon resonance assays of DNA–protein interactions // Meth. Mol. Biol. 2009. V. 543. P. 653–669. https://doi.org/10.1007/978-1-60327-015-1_38
Stoddard S.F., Howe M.M. Localization and regulation of bacteriophage Mu promoters // J. Bacteriol. 1989. V. 171 (6). P. 3440–3448. https://doi.org/10.1128/jb.171.6.3440-3448.1989
Sundberg E.J., Andersen P.S., Gorshkova I.N., Schuck P. Surface plasmon resonance biosensing in the study of ternary systems of interacting proteins // Protein interactions: biophysical approaches for the study of complex reversible systems / Ed. P. Schuck. V. 5. New York: Springer, 2007. P. 97–141.
Sung M.H., Guertin M.J., Baek S., Hager G.L. DNase footprint signatures are dictated by factor dynamics and DNA sequence // Mol. Cell. 2014. V. 56. P. 275–285. https://doi.org/10.1016/j.molcel.2014.08.016
Tsuchiya Y., Kinoshita K., Nakamura H. PreDs: a server for predicting dsDNA-binding site on protein molecular surfaces // Bioinformatics. 2004. V. 21. P. 1721–1723.
Tuerk C., Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase // Science. 1990. V. 249. P. 505–510. https://doi.org/10.1126/science.2200121
Tullius T.D., Dombroski B.A. Hydroxy radical footprinting: a high-resolution method for mapping protein-DNA contacts // Meth. Enzymol. 1987. V. 155. P. 537–558. https://doi.org/10.1016/0076-6879(87)55035-2
Vidal M., Brachmann R.K., Fattaey A. et al. Reverse two-hybrid and one-hybrid systems to detect dissociation of protein–protein and DNA–protein interaction // PNAS USA. 1996. V. 93. P. 10315–10320. https://doi.org/10.1073/pnas.93.19.10315
Wang L., Xu G., Chen H. et al. DrRRA: a novel response regulator essential for the extreme radioresistance of Deinococcus radiodurans // Mol. Microbiol. 2008. V. 6. P. 1211–1222. https://doi.org/10.1111/j.1365-2958.2008.06113.x
Waswa J., Irudayaraj J., DebRoy C. Direct detection of E. coli O157:H7 in selected food systems by a surface plasmon resonance biosensor // LWT. 2007. V. 40. P. 187–192.
Weirauch M.T., Yang A., Albu M. et al. Determination and inference of eukaryotic transcription factor sequence specificity // Cell. 2014. V. 158. P. 1431–1443. https://doi.org/10.1016/j.cell.2014.08.009
Wilson D.O., Johnson P., McCord B.R. Nonradiochemical DNase I footprinting by capillary electrophoresis // Electrophoresis. 2001. V. 22. P. 1979–1986.
Wissink E.M., Vihervaara A., Tippens N.D. et al. Nascent RNA analyses: tracking transcription and its regulation // Nat. Rev. Genet. 2019. V. 20. P. 705–723. https://doi.org/10.1038/s41576-019-0159-6
Xiao Z., Zou Q., Liu Y., Xuerui Yanga X. Genome-wide assessment of differential translations with ribosome profiling data // Nat. Commun. 2016. V. 7. P. 11194. https://doi.org/10.1038/ncomms11194
Yamazaki S., Hayano M., Masai H. Replication timing regulation of eukaryotic replicons: Rif1 as a global regulator of replication timing // Tr. Genet. 2013. V. 29. P. 449–460. https://doi.org/10.1016/j.tig.2013.05.001
Yanase Y., Hiragun T., Yanase T. et al. Evaluation of peripheral blood basophil activation by means of surface plasmon resonance imaging // Biosens. Bioelectron. 2012. V. 32. P. 62–68. https://doi.org/10.1016/j.bios.2011.11.023
Yuk J.S., Ha K.S. Proteomic applications of surface plasmon resonance biosensors: analysis of protein arrays // Exp. Mol. Med. 2005. V. 37. P. 1–10. https://doi.org/10.1038/emm.2005.1
Zeng P.Y., Vakoc C.R., Chen Z.C. et al. In vivo dual cross-linking for identification of indirect DNA associated proteins by chromatin immunoprecipitation // Biotechniques. 2006. V. 41. P. 694–698.
Zhen G., Zurcher S., Falconnet D. et al. NTA-functionalized poly(l-lysine)-g-poly(ethylene glycol): a polymeric interface for binding and studying 6 his-tagged proteins // Conf. Proc. IEEE Eng. Med. Biol. Soc. 2005. V. 1. P. 1036–1038.
Zhen G., Zurcher S., Falconnet D. et al. Analysis of protein–DNA interactions using surface plasmon resonance // Adv. Biochem. Eng. Biotechnol. 2007. V. 104. P. 13–36.
Дополнительные материалы отсутствуют.
Инструменты
Успехи современной биологии