

Успехи физиологических наук, 2019, T. 50, № 1, стр. 58-74
Магнитно-резонансная спектроскопия: неинвазивные исследования метаболизма мозга человека в норме и патологии
Н. А. Семенова 1, 2, 3, А. В. Манжурцев 1, 3, П. Е. Меньщиков 1, 2, 3, М. В. Ублинский 1, 3, *, Т. А. Ахадов 3
1 Институт биохимической физики им. Н.М. Эмануэля РАН
119334 Москва, Россия
2 Институт химической физики им. Н.Н. Семёнова РАН
119334 Москва, Россия
3 НИИ Неотложной детской хирургии и травматологии
119180 Москва, Россия
* E-mail: maxublinsk@mail.ru
Поступила в редакцию 31.07.2018
Аннотация
Обзор содержит сведения о магнитно-резонансной спектроскопии как методе исследования биохимии in vivo, данные о метаболизме и функции веществ, определяемых методами протонной магнитно – резонансной спектроскопии в мозге человека, результаты собственных исследований и литературные данные о метаболических сдвигах в локусах мозга при функциональных нарушениях, вызванных патологией, данные динамических исследований концентраций метаболитов в периоде нейроактивации в норме и патологии, прижизненные оценки нарушений баланса основных нейромедиаторов.
Сведения о химических процессах, протекающих в клетках органов и тканей in vivo, необходимы для решения фундаментальной задачи связи между метаболизмом и биологической функцией. Возможность получить такие сведения открывает магнитно-резонансная спектроскопия (МРС) – единственный в настоящее время метод, позволяющий в динамике обмена веществ измерять внутриклеточные концентрации ряда важнейших метаболитов и определять скорости некоторых химических реакций в процессе функционирования живой системы. Таким образом, МРС представляет собой уникальный метод надежного наблюдения биохимических процессов in vivo, необходимый в исследованиях физико-химических механизмов биологической функции.
Измеряемые параметры зависят от состояния биологической системы, следовательно, могут различаться в норме и патологии и изменяться под влиянием различных факторов, в том числе, медикаментозных воздействий. Это, наряду с такими важнейшими свойствами МРС, как биологическая безопасность и неинвазивность, открывает широкие возможности для применения МРС в прикладной медицине в качестве метода диагностики, прогноза исхода заболеваний и контроля эффективности терапии.
МРС представляет собой адаптацию хорошо известного в химии метода спектроскопии ядерного магнитного резонанса (ЯМР) к исследованию химических процессов в клетках органов и тканей. В отличие от ЯМР-спектроскопии важнейшей особенностью МРС является необходимость локализовать область интереса – зону организма, в которой такое исследование проводится. Для этого используются методы, разработанные в магнитно-резонансной томографии (МРТ). Оба метода – МРС и МРТ основаны на явлении ЯМР.
ЯДЕРНЫЙ МАГНИТНЫЙ РЕЗОНАНС. МАГНИТНО-РЕЗОНАНСНАЯ СПЕКТРОСКОПИЯ – ЯМР-СПЕКТРОСКОПИЯ IN VIVO
ЯМР – это поглощение энергии атомными ядрами, имеющими магнитные свойства. К “магнитным” ядрам относятся ядра атомов, широко распространенных в биологических системах, например, таких, как атомы водорода, фосфора, углерода, натрия, имеющие магнитные изотопы 1H, 31P, 13C, 23Na. Магнитные свойства характеризуются магнитным моментом или спином, который можно представить как крошечную магнитную стрелку. В объекте, содержащем магнитные ядра, например протоны (ядра 1Н), спины ориентированы хаотично (рис. 1а). Но в постоянном магнитное поле ${{B}_{0}}$ спины ориентируются лишь двумя способами: “по полю” или “против поля” (рис. 1б). Каждой ориентации соответствует строго определенное энергетическое состояние. Иными словами, для ядер 1H разрешены два магнитных энергетических уровня. Разность энергий между этими уровнями пропорциональна величине ${{B}_{0}}{\text{:}}$ $\Delta E = \gamma \hbar {{B}_{0}},$ где $\hbar $ − постоянная Планка, $\Delta E$ − разность энергий между магнитными уровнями, $\gamma $ − коэффициент пропорциональности, (гиромагнитное отношение), определяется строением ядра. Воспользуемся известным соотношением:$\hbar 2\pi \nu = \Delta E,$ где $\nu $ – частота переходов между энергетическими уровнями, или резонансная частота.
Рис 1.
Ядра 1Н в отсутствие постоянного магнитного поля (а); ориентация 1Н в постоянном магнитном поле ${{B}_{0}}$ в соответствии с двумя магнитными энергетическими уровнями с разностью энергий между ними $\Delta E$.

Отсюда:
Из соотношения (1) видно, что при заданном значении ${{B}_{0}}$ резонансные частоты зависят только от $\gamma ,$ т.е. от типа ядра. Следовательно, каждый тип ядер при одной и той же величине постоянного магнитного поля имеет фиксированную частоту ядерного магнитного резонанса, которая является неотъемлемой характеристикой данного типа ядер [37].
Ориентация спинов “по полю” энергетически наиболее выгодна, поэтому на нижнем энергетическом уровне (рис. 1б) оказывается несколько больше ядер, чем на верхнем. Этот избыток формирует макроскопическую намагниченность с вектором, направленным по полю, параллельно ${{B}_{0}}.$ Облучение переменным магнитным полем В1 на резонансной частоте $\nu ,$ приводит к поглощению энергии $\Delta E$ и переходу ядер на верхний энергетический уровень; вектор макроскопической намагниченности отклонится от своего равновесного положения. Если облучение прекратить, ядра вернутся к равновесному распределению по магнитным энергетическим уровням, и вектор макроскопической намагниченности снова окажется ориентированным параллельно ${{B}_{0}}.$ При этом на частоте $\nu $ выделяется энергия $\Delta E,$ которую улавливает специальное устройство.
Процесс возвращения вектора макроскопической намагниченности к равновесному положению называется релаксацией и имеет специфические временные характеристики – величины T1 и T2 (времена восстановления продольной и поперечной составляющей вектора макроскопической намагниченности, соответственно).
Значения Т1 и Т2 являются основными параметрами, использующимися в МРТ, по ним получают изображения внутренних органов и тканей. В МРС решающую роль играет тот факт, что в составе молекулы на ядро оказывает воздействие локальное магнитное поле, возникающее вследствие взаимодействия электронов с полем ${{B}_{0}}.$ Это приводит к небольшому сдвигу резонансной частоты, который определяется электронным окружением ядра, следовательно, химическим строением молекулы, содержащей рассматриваемое ядро. Сдвиг частоты ядра, обусловленный локальным полем электронов, называется химическим сдвигом δ. Ядра, включенные в разные структурные фрагменты молекулы, имеют различающиеся δ, что приводит к образованию спектра, в котором сигналы имеют малые (по сравнению с резонансной частотой $\nu $) частотные сдвиги, их измеряют в миллионных долях (ppm). Значения δ являются специфическими характеристиками структурных фрагментов молекулы.
В молекулах ядерные спины взаимодействуют друг с другом, что приводит к расщеплению некоторых сигналов в спектре и образованию мультиплетов. Спин-спиновое расщепление подчиняется строгим закономерностям, которые определяют число линий и величину их частотного сдвига в мультиплете. Эта величина, называемая константой спин-спинового взаимодействия J, постоянна для данного структурного фрагмента молекулы [37].
Спектральные данные – значения δ и J, мультиплетность, интенсивности сигналов – являются характеристиками химического строения, достаточными для установления структуры молекулы, идентификации соединений, а также для качественного и количественного анализа смесей низкомолекулярных органических соединений, которые присутствуют в живых системах. Такой анализ проведен для тканевых экстрактов на ЯМР-спектрометрах с высокой напряженностью поля ${{B}_{0}}$ (≥9.4 Т). Полученные спектры высокого разрешения позволяют установить химические структуры метаболитов и определить для сигналов величины δ. Затем в спектрах МРС значения δ используются для идентификации отдельных метаболитов. Измерение интенсивностей сигналов в спектрах in vivo дает возможность определить относительные концентрации отдельных метаболитов в зонах интереса.
Разные типы клеток имеют разные метаболические потребности и по-разному протекающие обменные процессы, что делает необходимой локализацию объема интереса (volume of interest, VOI). В современных томографах локализация VOI проводится методами пространственного кодирования, применяющимися в МРТ. Локализация достигается с помощью специальных последовательностей радиочастотных импульсов [88] в присутствии градиентов магнитного поля. Задача градиентов состоит в том, чтобы спины, находящиеся в разных точках объекта, резонировали на уникальных для своего расположения частотах. Это осуществляется за счет добавления пространственно-зависимого магнитного поля (G) к постоянному магнитному полю томографа. Тогда поле в присутствии градиента будет иметь вид B(r) = B0+ rG, где r –расстояние от точки до центра магнита. Резонансная частота, таким образом, тоже становится зависимой от r: ν(r) = γB0 + rG.
Это позволяет исследовать анатомическую структуру и обменные процессы в выбранных зонах организма (рис. 2).
Рис. 2.
Выделение чувствительного объема на пересечении ортогональных плоскостей, выбранных градиентами магнитного поля Gx, Gy, Gz (а); расположение VOI в локусе мозга (б).
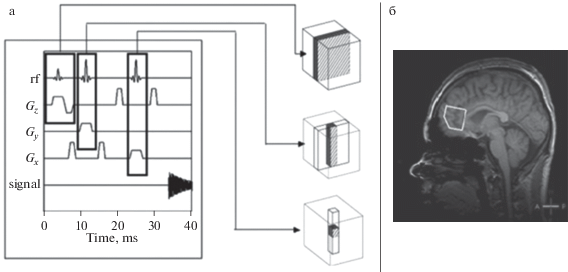
ЯМР частоты находятся в радиодиапазоне – диапазоне низких энергий, что делает метод ЯМР неинвазивным и биологически безопасным. В то же время, низкие энергии означают низкую чувствительность, вследствие которой МРС имеет ряд существенных ограничений. Даже при работе на самых чувствительных ядрах (1Н, гиромагнитное отношение $\gamma $ = 42.57 МГц/Т) в полях с максимальной напряженностью (3 T), разрешенной в медицине, удается наблюдать ЯМР-сигналы соединений, присутствующих в объеме не менее 3 см3 в концентрации не ниже 1 мM. В клетках в таких количествах содержатся конечные продукты метаболических путей (например, многие аминокислоты, лактат, ATP), источники энергии (жиры, гликоген), некоторые биохимические предшественники (холин, инозитол, нейротрансмиттеры). Расширяет возможности МРС применение высокопольных томографов. В настоящее время в мировой науке нарастает количество исследований мозга человека с использованием томографов с величиной ${{B}_{0}}$ = 7 Т. К сожалению, в России такие исследования невозможны из-за отсутствия оборудования.
1Н МР СПЕКТРЫ ГОЛОВНОГО МОЗГА
Головной мозг в норме содержит целый ряд низкомолекулярных соединений, внутриклеточные концентрации которых достаточны для обнаружения их сигналов в 1Н МР спектрах. Мешает наблюдению сигналов метаболитов наличие в ткани большого количества воды (около 95%), сигнал которой оказывается доминирующим и создает проблемы динамического диапазона. От сигнала протонов воды избавляются с помощью специальных методов его подавления, разработанных в ЯМР-спектроскопии [35].
В спектрах нормального мозга человека, зарегистрированных с подавлением сигнала воды, присутствуют сигналы N-ацетиласпартата (NAA), креатина + фосфокреатина (Cr), холинсодержащих соединений (Cho), – холина, фосфохолина, глицерофосфохолина, миоинозитола (mI), суммарный сигнал глутамата (Glu) и глутамина (Gln), Glx. На рис. 3а представлен 1Н МР спектр, зарегистрированный в лобной доле (время регистрации спектра – 7 мин). Видны хорошо разрешенные сигналы NAA, Cho, Cr, mI.
Рис. 3.
Типичные МР спектры, полученные в области головного мозга. а – 1H-МР спектр; б – MEGA-PRESS спектр; в – AspMEGA-PRESS спектр

В клетках мозга в концентрациях, превышающих порог чувствительности метода 1Н МРС, присутствуют не только эти соединения, но и ряд других метаболитов – γ-аминомасляная кислота (GABA), аспартат (Asp), таурин, глицин, глутатион [44]. Однако из-за низких значений ${{B}_{0}}$ (≤3 Т), использующихся с медицинских томографах, перекрываются сигналы соединений, имеющих в своих молекулах сходные структурные фрагменты. Поэтому в представленном спектре не разрешены сигналы GABA, Asp и других перечисленных выше метаболитов.
Для разделения перекрывающихся сигналов в настоящее время наиболее перспективным подходом является упрощение спектров (спектральное редактирование), которое использует наличие мультиплетности в одном из перекрывающихся сигналов и отсутствие такового в другом [70]. Это означает, что спектральное редактирование основано на различии химических структур метаболитов, сигналы которых перекрываются, поскольку мультиплетность определяется J-взаимодействием (см. стр. 5), а оно зависит от химической структуры молекулы.
Использование этого метода значительно расширяет возможности 1Н МРС, позволяя измерять, в дополнение к метаболитам, представленным в спектре на рис. 3a, концентрации GABA (рис. 3б), Asp (рис. 3в), Glu [3, 36, 69].
МЕТАБОЛИЗМ И ФУНКЦИЯ СОЕДИНЕНИЙ, СИГНАЛЫ КОТОРЫХ РЕГИСТРИРУЮТСЯ В 1Н МР СПЕКТРАХ МОЗГА
NAA
NAA – аминокислота, обнаруженная в мозге в высоких концентрациях [111]. Содержание NAA в различных отделах мозга может достигать 20 мМ [73, 82]. Поэтому NAA хорошо детектируется в протонных спектрах мозга [35].
Основное количество NAA синтезируется в митохондриях нейронов из аспартата и ацетил-коэнзима А под действием L-аспартат-N-ацетилтрансферазы [42, 60, 62, 116], фермент локализован в нейрональных митохондриях [42, 60, 124]. Минорным путем образования NAA служит распад нейротрансмиттера N-ацетил аспартилглютамата [89, 106].
Катаболизм NAA происходит вследствие гидролиза под действием аспартоацилазы (ASPA, EC 3.5.1.15) с образованием Asp и ацетата [30, 43]. До недавнего времени предполагалось, что этот процесс локализован исключительно в олигодендроцитах, поскольку ASPA, EC 3.5.1.15 была обнаружена именно в олигодендроцитах [14]. Однако по данным [75] ASPA экспрессируется и в аксонах. Следовательно, и образование, и распад NAA может происходить в одном типе клеток, а именно, в нейронах.
Новые сведения о локализации ферментов синтеза и гидролиза NAA явились основой для возникновения новых гипотез о функциях этой молекулы в головном мозге, которые, несмотря на более, чем полувековую историю изучения NAA, до сих пор остается полностью неустановленными. Ряд данных свидетельствуют об участии NAA в транспорте воды из нейронов в олигодендроциты [16, 28]. NAA может служить источником аспартата и ацетильных групп в синтезе миелина и участвовать в биосинтезе липидов мозга [29, 77]. Предполагается, что NAA выполняет функцию сигнальной молекулы в системе нейроны–глия [17]. Данные [82] указывают на участие NAA в регуляции нейронального энергетического обмена.
Уровень NAA быстро возрастает в периоде развития мозга [55, 110]. Поскольку плотность нейронов в коре уменьшается при созревании мозга, рост NAA может отражать развитие дендритов, синапсов, сомы нейронов, а также функциональное развитие нейронов [112].
NAA обнаруживается в тканях центральной нервной системы и лишь в незначительных количествах присутствует в периферических тканях [35, 56, 73, 78]. Исключение составляют эмбриональные клетки и хрусталик позвоночных [15]. Концентрация NAA варьирует в разных участках мозга [55, 67, 73]. Серое вещество мозга содержит больше NAA, чем белое вещество [73, 111]. Внутриклеточная концентрация NAA в 50–100 раз превышает внеклеточную [94, 114]. Исследования экстрактов культур нейронов и астроцитов [117] и данные иммуногистохимических анализов [76, 104] показали, что NAA локализуется в нейронах, это позволило использовать NAA в качестве нейронального маркера.
МРС является единственным способом прижизненной оценки внутриклеточной концентрации NAA. Корректность этой оценки подтверждается сравнением результатов измерения содержания NAA в тканях мозга, полученных методами 1Н МРС и хроматографии, оба метода дают сходные результаты [95, 96]. При этом, хотя хроматография значительно чувствительнее, чем МРС, прижизненное неинвазивное определение NAA имеет очевидные достоинства.
Снижение интенсивности сигнала NAA в объеме интереса интерпретируется как уменьшение содержания нормально функционирующих нейронов в этой зоне мозга; и наоборот, рост уровня NAA используется как индикатор увеличения уровня нормально функционирующих нейронов в исследуемом локусе.
NAA И ФУНКЦИОНАЛЬНЫЕ РАССТРОЙСТВА ЦЕНТРАЛЬНОЙ НЕРВНОЙ СИСТЕМЫ (ЦНС)
Большинство известных в настоящее время патологических состояний, связанных с нарушениями функций ЦНС, сопровождается уменьшением концентрации NAA и соответствующим уменьшением его сигнала в 1Н МР спектрах мозга [45], рост NAA выявлен только в одном случае – у больных с редкой генетической патологией, болезнью Канаван [45].
Связь между уровнем NAA и когнитивными функциями обнаружена в эксперименте и целом ряде клинических исследований. Так, оценка нейропротекторного эффекта соли янтарной кислоты и холина (ЯХ) на модели хронической ишемии мозга у крыс показала, что данные тестирования когнитивных функций в лабиринте Морриса и результаты измерения уровня NAA хорошо совпадают: под действием ЯХ у ишемизированных крыс достоверно улучшается память и способность к обучению, одновременно повышается уровень NAA, падение которого выявлено у крыс, не получавших препарат [6]. Аналогичное соответствие между содержанием NAA в мозге крыс и сохранностью когнитивных функций наблюдается в модели хронической ишемии после введения животным другого нейропротекторного вещества, пептида HLDF-6 [1].
В клинических исследованиях особое внимание уделяется поиску связей между уровнем NAA и функциональными расстройствами при различных заболеваниях ЦНС. Показано, что суммарное содержание NAA в мозге больных рассеянным склерозом коррелирует с показателями нейропсихологического статуса [66]. Ухудшение когнитивных функций и одновременное уменьшение уровня NAA обнаружено в мозге пациентов на ранней стадии болезни Альцгеймера [41]. Уровень NAA в медиальной височной доле, первичной моторной и сенсорной коре коррелирует с результатами тестов когнитивных функций, использующихся при оценках степени тяжести болезни Альцгеймера [49]. По мере развития этого заболевания возникают региональные изменения NAA. Например, уровень NAA снижается в зонах височной коры и задней поясной извилины лишь в случае глубоких когнитивных нарушений; тогда как малые когнитивные изменения не затрагивает эти области и не сопровождается снижением NAA [51]. В зоне задней поясной извилины снижение NAA наблюдается у пациентов с когнитивными расстройствами при болезни Паркинсона [79]. У пациентов с болезнью Паркинсона между содержанием NAA в области височной коры и показателями когнитивных нарушений найдена корреляция [81]. У больных, когнитивные функции мозга которых не восстановились после инсульта, выявлено снижение NAA в зонах гиппокампов обоих полушарий; тогда как у пациентов с отсутствием постинсультных когнитивных расстройств содержание NAA в гиппокампах не отличается от нормы [122].
Гиппокампы являются зоной нейрогенеза [72]. Показано, активация нейрогенеза введением клеток пуповинной/плацентарной крови вызывает увеличение NAA в височной доле мозга детей с последствиями тяжелой черепно-мозговой травмы, одновременной улучшается неврологического статус этих пациентов [8].
Таким образом, приведенные выше данные демонстрируют, что когнитивные расстройства связаны не только с общим церебральным уровнем NAA, но и со снижением содержания NAA в специфических локусах мозга. Это позволяет получить данные о топографии зон мозга, ответственных за определенные когнитивные функции, и выявить локальные нарушения обменных процессов, вызванные конкретным заболеванием, даже при отсутствии структурных и микроструктурных изменений нервной ткани.
NAA И ЭНЕРГЕТИЧЕСКИЙ ОБМЕН
Механизмы снижения NAA до сих пор не установлены. Имеются данные, указывающие на связь NAA с энергетическим обменом. Так, на моделях фокальной и диффузной черепно-мозговой травмы измерение ATP и NAA в экстрактах мозга крыс показало, что оба показателя имеют одинаковый вид временных зависимостей концентраций в посттравматическом периоде [34]. В [13] предполагается, что концентрация ацетил-коэнзима А (АсСoА), предшественника NAA, определяет уровень NAA и связывает его с энергетикой. Мы показали, что тяжелая травма вызывает не только снижение NAA, но и снижение Asp, другого предшественника NAA; наши данные о концентрациях NAA, Asp и Glu в разные сроки после травмы свидетельствуют о нарушении работы нейронального аспартат-малатного шаттла, регулирующего синтез ATP и NAA [7].
Основной энергозависимый процесс в мозге – передача импульса возбуждения. По оценкам [102] на него приходится 80% всех энергозатрат мозга. Показано, что нейростимуляция изменяет содержание NAA в активированных зонах мозга. Так, по данным [24] видеостимуляция в течение 10 мин вызывает снижение уровня NAA в области зрительной коры.
Зоны нейрональной активности могут быть локализованы методами функциональной магнитно-резонансной томографии (фМРТ). Повышение активности нейронов, вызванное нагрузкой (выполнением как-либо задачи), сопровождается активацией мозгового кровотока в зоне нейростимуляции, что создает в этой зоне избыток оксигемоглобина (Hb) относительно дезоксигемоглобина (dHb) [39]. Парамагнетик dHb снижает время спин-спиновой релаксации Т2 протонов воды и вызывает отрицательный контраст, т.е. снижает яркость в фМРТ – изображениях. Диамагнетик Hb, напротив, не влияет на процесс релаксации. Поэтому при снижении dHb/Hb отрицательный контраст уменьшается, за счет этого возникает положительный BOLD (blood oxygen level dependent) – контраст, поскольку он зависит от насыщения крови кислородом. На рис. 4 представлены зоны BOLD-ответа на предъявление слухового стимула в парадигме ODDBALL и активацию моторной коры (нажатие кнопки пальцем правой руки в ответ на слуховой стимул). Видно, что предъявление слухового стимула активирует слуховую кору, наибольшая активация наблюдается в моторной коре (пациент нажимает кнопку), активируется и префронтальная кора, поскольку требуется принять решение – нажимать или не нажимать кнопку.

BOLD косвенно характеризует локальную нейрональную активность и зависит от изменения скоростей потребления кислорода и венозного кровотока [53]. Вклад венозного кровотока в BOLD можно минимизировать, если для активации использовать короткие единичные стимулы [32]. Тогда BOLD будет зависеть только от изменения скорости потребления кислорода, которое определяется соотношением скорости артериального кровотока и скорости потребления кислорода митохондриями. Отсюда следует, что динамика BOLD-ответа на короткий единичный стимул должна сопровождаться динамикой NAA и определять ее, поскольку синтез ATP зависит от митохондриального окисления, а NAA связан с нейрональным энергетическим обменом.
Мы первыми обнаружили и описали временную зависимость NAA в премоторной коре в динамике BOLD-ответа на короткий единичный стимул [9]. По нашим данным в мозге здорового человека в зоне активации возникает обратимое снижение NAA с минимумом, запаздывающим относительно максимума BOLD на 6 с (рис. 5).
Рис. 5.
Кривая 1 – средние значения (± ошибка среднего) BOLD, кривая 2 – средние значения (± ошибка среднего) относительных интенсивностей сигнала NAA в динамике BOLD.
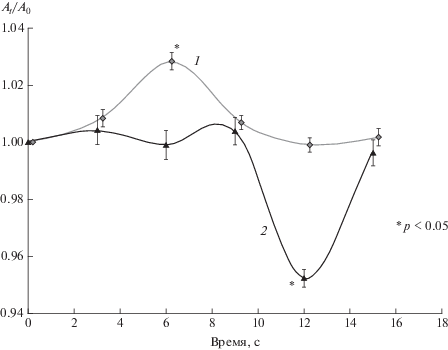
Кинетические ответы на стимул для BOLD и NAA имеют сложный характер. Для BOLD можно идентифицировать четыре, а для NAA шесть кинетически дискриминируемых стадий. На основе экспериментальных данных и их кинетического анализа мы предположили следующие молекулярные механизмы процесса: BOLD-ответ возникает после активации нейронов и инициируется выбросом Glu из везикул; Glu вызывает вазодилатацию и связывает BOLD-ответ и динамику NAA: в энергозависимый возврат Glu в везикулы вовлекается гидролиз NAA, образующиеся при гидролизе NAA ацетильны группы участвуют в синтезе АсСоА, необходимом для активации цикла Кребса и цепи электронного транспорта, что обеспечивает восстановление уровня ATP.
В отличие от нормы у больных на ранней стадии шизофрении отсутствует вызванное такой же нагрузкой падение уровня NAA, а BOLD-ответ достоверно снижен по сравнению с нормой (рис. 6). Если выдвинутая нами гипотеза о связи BOLD и NAA через энергозависимый ретранспорт Glu верна, при шизофрении должно иметь место нарушение транспорта Glu в везикулы и снижение энергозатрат при стимуляции. Действительно, показано, что в мозге больных шизофренией снижено содержание везикулярного транспортера Glu VGLUT1 [87], этот факт свидетельствует о расстройстве везикулярного транспорта Glu. На снижение энергозатрат в зоне нейроактивации у больных шизофренией указывают полученные нами данные анализа фосфатного обмена в зрительной коре при видеостимуляции [2]. Используя функциональную МР спектроскопию на ядрах 31Р мы показали, что предъявление нормальным испытуемым в качестве стимула мигающей шахматной доски вызывает в зрительной коре достоверное снижение макроэрга фосфокреатина (PCr); такая же активация не вносит изменений в показатели фосфатного обмена зрительной коры больных шизофренией.
Рис. 6.
Средние значения (± ошибка среднего) BOLD в области премоторной коры; кривая 1 – группа нормы, кривая 2 – группа патологии

PCr представляет собой депо для быстрого восполнения затрат АТР, являясь участником реакции: PCr + ADP = ATP + Cr, протекающей под действием креатинкиназы. Реакция равновесная; когда необходимо быстро восстановить концентрацию ATP, как это происходит при нагрузке, равновесие смещается вправо, уровень PCr падает, а АТР поддерживается постоянным. Чем больше энергозатраты, тем больше снижается PCr. Отсутствие эффекта нагрузки при шизофрении указывает на уменьшение затрат ATP в зрительной коре мозга больных в условиях видеостимуляции и подтверждает нашу гипотезу.
Таким образом, NAA и функциональную активность нейронов, по-видимому, связывают механизмы транспорта Glu в везикулы и в нейрональные митохондрии, где из него образуется предшественник NAA, аспартат (рис. 7).

Glx (Glu + Gln)
Глутамат – аминокислота, которая является основным возбуждающим нейромедиатором мозга [68]. Процесс передачи импульса возбуждения включает в себя накопление Glu в пресинаптических везикулах, выброс нейротрансмиттера в синаптическую щель и активацию постсинаптического рецептора. Постулируется, что везикулярный Glu синтезируется из α-кетоглутарата и аспартата под действием связанной с везикулами аспартат аминотрансферазы [99].
Главная стадия Glu-трансмиссии, накопление Glu, осуществляется с помощью активного транспорта с участием высокоспецифичного транспортера VGLUT; необходимый для этого процесса гидролиз ATP происходит под действием везикулярной ATP-азы, которая создает движущую силу транспорта Glu – электрохимический протонный градиент [99]. Однако возвращающийся в везикулы из межклеточного пространства Glu не покрывает его потери при нейротрансмиссии. [21]. Для поддержания гомеостаза возбуждающего нейротрансмиттера служит глутамин-глутаматный цикл. Цикл начинается с образования Gln в митохондриях астроцитов, где интермедиат цикла Кребса α-кетоглутарат превращается в Glu. Последний покидает митохондрию и в цитозоле астроцитов образует Gln под действием глутаминсинтазы – фермента, локализованного в астроцитах [48]. Синтезированный таким образом Gln выходит в межклеточное пространство, откуда Na+-зависимый транспортер переносит его в глутаматергические нейроны [80]. Там из Gln в результате многоступенчатых превращений образуется Glu, использующийся как нейротрансмиттер [48]. Высвобожденный из везикул нейротрансмиттер переносится глутаматными транспортерами в астроциты [31], где 80% Glu снова превращается в Gln, и цикл Gln–Glu возобновляется [91].
Поглощение Glu везикулами объединяет его метаболическую и нейротрансмиттерную функции: показано, что ингибиторы глутамат дегидрогеназы инактивируют поглощение Glu кортикальными мембранами мозга крыс [123].
Корковые когнитивные функции сильно зависят от регуляции Glu – трансмиссии; гиперактивация Glu рецепторов, вызванная избытком Glu, является причиной эксайтотоксичности, которая приводит к развитию нейродегенерации и функциональным нарушениям [86].
Как отмечалось выше, в протонных спектрах, полученных на медицинских томографах с использованием стандартных методик, сигнал Glu перекрывается с сигналом Gln (пик Glx, рис. 2в). Поэтому подавляющее большинство данных относится к измерению суммарного содержания Glu и Gln, что, несомненно, искажает результаы и затрудняет их интерпретацию. Применение исследовательских томографов с напряженностью поля В0 ≥ 7 Тл позволяет получить сигнал Glu без примеси Gln. С помощью такой техники показано, что при функциональной активации зоны передней поясной извилины в этой области у нормальных испытуемых возрастает [Glu] [115].
GABA
Основной тормозной нейромедиатор γ-амномасляная кислота присутствует в различных зонах мозга в концентрациях порядка 1 мМ. Синтезируется GABA в GABA-ергических нейронах и астроцитах [65] путем декарбоксилирования Glu под дейcтвием глутаматдекарбоксилазы (GAD) [103]. Установлено, что GAD существует в двух изоформах – GAD65 и GAD67 [107], причем GAD65, в основном, присутствует в митохондриях аксонов [23, 65], а GAD67 – в нейрональном цитозоле [65]. Оба фермента кодируются разными генами и по-разному регулируют синтез GABA. GAD65 быстро активируется путем связывания с перидоксалфосфатом, играя ключевую роль при синтезе GABA в синапсах, тогда как GAD67 присутствует в нейрональном цитозоле, связан с коферментом и находится в активной форме постоянно [65]. Таким образом, можно полагать, что содержащаяся в синапсах GABA выполняет нейромедиаторную функцию, а GABA в цитозоле – метаболическую функцию; в покое оба пула находятся в равновесии [120]. Основное количество GAD локализовано в GABA-ергических нейронах, но GАD экспрессируется и в Glu-ергических нейронах; в астроцитах GAD отсутствует [26]. Cинтез GABA регулируется GAD65 [59], in vitro показано, что GABA может быть конкурентным ингибитором GAD [64], внутриклеточная концентрация GABA в норме ниже значений, необходимых для ингибирования этого фермента [64]. Экспрессия GAD67 контролируется уровнем мРНК [101].
Непосредственным предшественником GABA является Glu. Однако ряд данных, полученных на препаратах мозга, свидетельствует о том, что Gln может быть более эффективным по сравнению с Glu экзогенным предшественником GABA [71]. Отсюда следует, что пул GABA поддерживается не только его возвратом в везикулы, но и циклом Gln–Glu [108]. Промежуточной стадией синтеза GABA из Gln является дезаминирование с образованием Glu под действием активированной фосфатом глутаминазы (PAG), распространенной в Glu-эргических и GABA-ергических нейронах [11]. PAG находится на внутренней стороне митохондриальной мембраны, и Glu может после превращения в α-кетоглутарат вступать в цикл Кребса в митохондрии или образовать GABA под действием GAD в цитозоле. По данным 13С МРС установлено, что в культуре нейронов экзогенный 13С-Gln на 60% образует GABA из Glu, поступающего в цикл Кребса [108, 120]. По данным [120] уменьшение метаболического пула GABA в культуре нейронов под действием N-метил D-аспартата (NMDA) и α-амино-3-гидрокси-5-метил-4-изоксазолпропионовой кислоты (AMPA) активирует синтез GABA в цитозоле и не влияет на митохондриальный синтез GABA, тогда как уменьшение везикулярного пула GABA под действием 55 мM K+ в сочетании с ингибитором транспорта GABA тиагабином не изменяет соотношение GABA, синтезирующейся в цитозоле и митохондриях [120].
Катаболизм GABA осуществляется путем трансаминирования c образованием полуальдегида сукцината, который затем подвергается дегидрированию, образуя сукцинат, поступающий в цикл Кребса. Ферменты этих реакций – GABA-трансаминаза (GAT) и сукцинил полуальдегид дегидрогеназа, соответственно.
Вместе с синтезом GABA реакции его распада составляют GABA-шунт, представляющий собой альтернативный путь вступления сукцината в цикл Кребса. Согласно [47] поток метаболитов через этот путь в мозге составляет 10% от общего потока через цикл Кребса; данные получены при инкубации кортикальных срезов в среде, содержащей 1-14С GABA.
Скорость разложения GABA определяется скоростью трансаминирования под действием GAT [57]. Необратимое ингибирование этого фермента γ-винил-γ-аминомасляной кислотой (вигабатрин) приводит к накоплению GABA в цитозоле и везикулах [85]. GAT присутствует в митохондриях нейронов и астроцитов [93]. Показано, что ингибирование GAT сопровождается ростом выброса GABA, обусловленного деполяризацией, в клеточных культурах [26], а также электростимуляцией в церебральных срезах. Полное окисление GABA до СО2 происходит исключительно в астроцитах, что показано методом 13С МРС в культурах нейронов и астроцитов [121].
Дисфункция GABA-нейротрансмиссии включена в патогенез большинства известных в психиатрии и неврологии заболеваний, таких как шизоврения [58], эпилепсия [84], депрессия [97], тревожность [105]. Отсюда возникает колоссальный интерес к 13С и 1Н МРС исследованиям уровня этого нейромедиатора in vivo в норме и патологии. Поскольку природное содержание 13С крайне мало (1.1%), для получения 13С МР спектров необходимо обогащение этим изотопом веществ, присутствующих в объеме образца. С этой целью в кровоток вводят 13С глюкозу (Glc) или 13С ацетат (Ac). Glc является субстратом для нейронов и астроцитов, Ac – только для астроцитов. Таким образом, введение этих субстратов дает возможность изучать обменные процессы в каждом типе клеток мозга: в нейронах и астроцитах.
Рост интенсивности сигнала GABA в 13С МР спектре мозга после инфузии [1-13С]Glc позволил установить, что GABA-ергические нейроны потребляют около 10% от общего количества введенной Glc [118]. На основании этих данных авторы [118] считают, что 10% энергозатрат в коре приходится на GABA-ергические нейроны. При введении [1, 6-13C2]глюкозы и [2-13C] ацетата измерены скорости циклов GABA–Gln и Glu–Gln относительно скоростей соответствующих циклов Кребса; показано, что в нейронах и астроцитах основные энергозатраты связаны с циклом нейротрансмиттеров [83].
Исследование GABA методом 13С МРС в клинике и эксперименте имеет ограниченное применение, поскольку требует инъекцией дорогостоящих веществ, обогащенных магнитным изотопом 13С. Значительно дешевле использовать 1Н МРС. Выше отмечалось, что отсутствие методик для наблюдения GABA, включенных в стандартный набор импульсных последовательностей (ИП) медицинских томографов, затрудняет 1H МРС – исследования GABA. Тем не менее, опубликованные результаты измерений GABA в мозге человека позволили провести мета-анализ 1Н МРС – данных пациентов с психическими расстройствами, который показал, что GABA ниже нормы у пациентов с аутизмом и при депрессии; у больных шизофренией отмечен тренд к уменьшению GABA [100]. Поздние стадии шизофрении характеризуются инактивацией синтеза GABA. Об этом свидетельствует уменьшенное содержание GABA-ергических интернейронов [18] и снижение экспрессии мРНК, кодирующей синтез GAD [10], обнаруженные в постмортальных исследованиях префронтальной коры мозга больных. Эти данные согласуются со снижением содержания GABA на поздних стадиях заболевания, обнаруженном в 1Н МРС – исследовании шизофрении [125].
Для понимания связи GABA-ергической системы с патогенезом шизофрении необходимы данные о содержании GABA в мозге пациентов с ультравысоким риском развития этого заболевания. Нами впервые показано, что уровень GABA у таких пациентов достоверно снижен в медиальной префронтальной коре левой лобной доли, тогда как в том же локусе правого полушария он не отличается от нормы [5]. Обнаружена отрицательная корреляция между уровнем GABA в зоне передней поясной извилины у пациентов с ультравысоким риском развития шизофрении и оценками тяжести негативных продромальных симптомов [74].
У пациентов с диагностированной шизофренией также выявлена связь между когнитивными нарушениями и уровнем GABA: найдена обратная корреляция между содержанием GABA в медиальной префронтальной коре и оценками когнитивных функций [63].
Снижение уровня GABA в той же области мозга коррелирует с возрастным ухудшением внимания у здоровых испытуемых [92]. Приведенные выше данные указывают на связь когнитивных нарушений со снижением активности GABA-ергической системы.
Инактивация GABA-ергической системы является одним из элементов глутаматной гипотезы шизофрении [109]. Согласно этой гипотезе активность тормозных нейронов контролирует активность возбуждающих через сигнал NMDA-рецепторов. Сниженный сигнал NMDA снимает ингибирование возбуждающих нейронов [109]. Таким образом, функциональные нарушения могут быть обусловлены дисбалансом процессов возбуждения–торможения и изменением локальных концентраций основных нейромедиаторов – возбуждающего Glu и тормозного GABA. Баланс возбуждения–торможения определяет кортикальную активность, причем активация возбуждения может реализоваться через угнетение тормозных синапсов [113].
Известно, что дисбаланс уровней Glu и GABA возникает вследствие травмы мозга, в том числе и после сотрясения мозга – травмы, которая не вызывает повреждений нервной ткани, обнаруживающихся современными методами нейровизуализации, но в ряде случаев приводит к ухудшению когнитивных функций [33]. По результатам микродиализа цереброспинальной жидкости установлено, что сотрясение мозга сопровождается ростом внеклеточной концентрации Glu в мозге крыс [52]. В модельных экспериментах на крысах обнаружено увеличение концентрации GAD67 через час после травмы, свидетельствующее об активации синтеза GABA; с предполагаемой избыточной GABA связывают ухудшение рабочей памяти, обнаруженное у травмированных животных [54]. Показано, что травма мозга изменяет экспрессию субъединиц рецепторов GABA в гиппокампе, это приводит к дисбалансу процессов торможения и возбуждения, следствием которого является ухудшение когнитивных функций [12, 27]. Мы показали, что через сутки после сотрясения в лобных долях мозга человека на 36% возрастает GABA, вызывая смещение баланса GABA/Glu [4]. В настоящее время прижизненные исследования связи функционального состояния мозга с балансом GABA–Glu находится лишь в начальной стадии [46, 25 ].
Cho
Сигнал Cho является суперпозицией сигналов холина, моноэфира холина и фосфорной кислоты, фосфохолина (PC) и диэфира, глицерофосфохолина (GPC), наибольший вклад в интенсивность суммарного сигнала вносит PC, хотя относительное содержание холина и его эфиров может варьировать в периоде взросления и, в меньшей степени, как следствие поступления холина с пищей. Холин синтезируется, в основном, в печени и попадает в мозг с кровотоком. Источником увеличения пула холинсодержащих соединений может быть РC, который в мозге, в частности, в нейронах, синтезируется в трех последовательных реакциях переметилирования из фосфатидилэтаноламина [61, 90]. РС синтезируется также при фосфорилировании холина под действием холинкиназы. Холинсодержащие соединения являются предшественниками синтеза и продуктами распада мембран [40, 45].
Для исследований липидного обмена необходимо знать концентрации каждого из холинсодержащих соединений. Неразрешенные в 1Н МР спектрах сигналы PC и GPC разрешаются в спектрах 31Р МР. Таким образом, из сопоставления 1Н МР и 31Р МР спектров можно получить информацию о содержании каждого из этих метаболитов. Значимость такой информации определяется тем, что концентрации холинсодержащих соединений чувствительны к процессам пролиферации, в частности при опухолеобразовании. Во многих опухолях мозга обнаружена повышенная экспрессия холин киназы – фермента синтеза РС [119]. Поэтому следует ожидать, что в опухолях не только повышается интенсивность сигнала Cho, но и меняется соотношение интенсивностей сигналов разных холинсодержащих соединений.
Cr
Это суммарный сигнал креатина и креатинфосфата. Оба метаболита связаны креатинкиназной реакцией, в которой под действием креатинкиназы фосфатная группа обратимо переносится от креатинфосфатана на ADP с образованием ATP. Таким образом, креатинфосфат является запасным источником синтеза ATP. Креатинкиназная реакция активируется, когда необходимо быстро компенсировать затраты ATP. В спектрах 31Р МР фосфокреатин имеет хорошо детектируемый интенсивный сигнал, поэтому, как и в случае холинсодержащих соединений, анализ 1Н МР и 31Р МР спектров, полученных в одной и той же локализации, позволяет оценить концентрации креатина и фосфокреатина раздельно. При острой ишемии интенсивность сигнала Cr в протонных спектрах постоянна, поскольку фосфокреатин в креатинкиназной реакции превращается в креатин в молярном соотношении 1 : 1. В то же время в спектрах 31Р сигнал фосфокреатина падает.
Креатин синтезируется из аргинина в две стадии с участием аргинин-глицин-аминотрансферазы и гуанидиноацетат метилтрансферазы. Показано, что в нейронах и глиальных клетках в большом количестве экспрессируются гены обоих ферментов, поэтому предполагается, что основное количество церебрального креатина эндогенного происхождения [22].
mI
Этот сигнал используют как маркер астроцитов. Известно, что mI выполняет роль осмолита в глиальных клетках [98]. По данным 1Н МРС опухолей мозга рост интенсивности этого сигнала отражает активацию пролиферации глии [38, 50]. Увеличение интенсивности сигнала mI может быть вызвано активацией глии, поскольку mI вовлечен в метаболические процессы, включающие такие соединения как инозитол-1-фосфат, фосфатидилинозитол, инозитол-полифосфатные вторичные мессенджеры, участвующие во внутриклеточной сигнализации [19, 20].
В заключении следует отметить, что магнитно-резонансная спектроскопия в сочетании с функциональной томографией предоставляет широкие возможности для выявления связей биохимических процессов, протекающих в мозге, с биологической функцией. Уже получены интереснейшие данные о химических механизмах функциональных нарушений при патологических состояниях центральной нервной системы. Очевидна перспективность использования в таких исследованиях гетроядерной спектроскопии.
Функциональная МРС, т.е. анализ внутриклеточных нейрохимических процессов в периоде нейроактивации – совсем новая и многообещающая область спектроскопии живых систем – требует серьезных усилий для своего развития и кажется приоритетной в исследованиях связи метаболизма и биологической функции. Несомненно, прогресс будет достигнут использованием высокопольных томографов (В0 ≥ 7 Т), которые позволяют значительно увеличить число наблюдаемых метаболитов.
Работа поддержана грантами РНФ 18-13-00030, РФФИ 17-04-01149 А (разработка методики определения концентрации аспартата в мозге человека in vivo) и РФФИ 18-315-00165 (пилотное исследование легкой черепно-мозговой травмы).
Список литературы
Костанян И.А., Сторожева З.И., Семёнова Н.А. и др. Пептид HLDF-6 уменьшает когнитивные дисфункции и поражения мозга, вызванные хронической ишемизацией мозга крыс // Доклады РАН. 2009. Т. 428. № 4. С. 565–569.
Манжурцев А.В., Семенова Н.А., Ублинский М.В. и др. Влияние нейростимуляции на внутриклеточные концентрации протонсодержащих метаболитов и макроэргических фосфатов в коре мозга при шизофрении по данным методов 1H и 31P магнитно-резонансной спектроскопии // Известия РАН. Сер хим. 2016. № 6. С. 160–1636.
Меньщиков П.Е., Семенова Н.А., Манжурцев А.В. и др. Определение уровней N-ацетиласпартата, аспартата и глутамата в локальных структурах головного мозга человека методом J-редактирования спектров протонного магнитного резонанса in vivo // Известия РАН. Сер. хим. 2018. № 4. С. 1–8.
Меньщиков П.Е., Cеменова Н.А., Аxадов Т.А. и др. Рост церебральной концентрации γ-аминомасляной кислоты у детей с легкой черепно-мозговой травмой в остром периоде по данным протонной магнитно-резонансной спектроскопии // Биофизика. 2017. Т. 62. № 6. С. 1221–1231.
Меньщиков П.Е., Семенова Н.А., Ублинский М.В. и др. 1Н МРС и импульсная последовательность MEGA-PRESS в исследовании баланса возбуждающего и тормозного нейромедиаторов в мозге пациентов с ультравысоким риском развития шизофрении // ДАН. 2016. Т. 468. № 1. С. 1–5.
Помыткин И.А., Сторожева 3.И., Семенова Н.А. и др. Нейропротективный эффект янтарнокислого холина по данным тестов когнитивной функции и ЯМР 1Н in vivo в экспериментальной модели хронической ишемии головного мозга // Известия РАН. Сер биол. 2007. № 2. С. 183–187.
Семенова Н.А., Меньщиков П.Е., Манжурцев М.В. и др. ДАН. 2018. в печати.
Семенова Н.А., Сидорин С.В., Ахадов Т.А. и др. Влияние клеточной терапии на уровни метаболитов в структурах мозга детей с последствиями тяжелой черепно-мозговой травмы: исследование методом 1Н магнитно-резонансной спектроскопии // Клеточн. Технол. Биол. Мед. 2011. № 2. С. 99–103.
Ублинский М.В., Семенова Н.А., Ахадов Т.А. и др. Релаксационная кинетика в исследовании нейробиологических процессов методами функциональной магнитно-резонансной томографии и спектроскопии // Известия РАН. Сер хим. 2015. № 2. С. 451–457.
Akbarian S., Kim J. Decreased regional cortical gray matter volume in schizophrenia // Arch. Gen. Psychiat. 1995. V. 52. P. 258–266.
Akiyama H., Kaneko T., Mizuno N. et al. Distribution of phosphate-activated glutaminase in the human cerebral cortex // J. Compat. Nuorol. 1990. V. 297. № 2. P. 239–252.
Almeida-Suhett P., Prager E., Pidoplichko V. et al. GABAergic interneuronal loss and reduced inhibitory synaptic transmission in the hippocampal CA1 region after mild traumatic brain injury // Exp. Neurol. 2015. V. 273. P. 11–23.
Ariyannur P.S., Moffett J.R., Manickam P. et al. Methamphetamine-induced neuronal protein NAT8L is the NAA biosynthetic enzyme: implications for specialized acetyl coenzyme A metabolism in the CNS // Brain Res. 2010. V. 1335. P. 1–13.
Baslow M. Evidence that tri-cellular metabolism of N-acetylaspartate functions as barain’s “operating system”: how NAA metabolism supports meaningful intercellular frequency-encoded communications // Amino Acids. 2010. V. 39. P. 1139–145.
Baslow M.H., Yamada S. Identification of N-acetylaspartate in the lens of the vertebrate eye: a new model for the investigation of the function of N-acetylated amino acids in vertebrates // Exp. Eye Res. 1997. V. 64. P. 283–286.
Baslow M. N-acetylaspartate as a molecular water pump and its role in the etiology of Canavan disease: a mechanistic explanation // J. Mol. Neurosci. 2003.V. 21. № 3. P. 185–190.
Bates T.E, Williams S.R., Gadian D.G. et al. 1H NMR study of cerebral development in the rat // NMR Biomed. 1989. V. 2. P. 225–229.
Beasley C., Zhang Z, Patten I. et al. Selective deficits in prefrontal cortical GABAergic neurons in schizophrenia defined by the presence of calcium-binding proteins // Biol. Psychiatry. 2002. V. 52. P. 708–715.
Berridge M.R. Inositol trisphosphate and diacylglycerol as second messengers // Biochem. J. 1984. V. 22. P. 345–360.
Berridge M.R., Irvin R.F. Inositol phosphates and cell signaling // Nature. 1989. V. 341. P. 197–205.
Bjornsen L.P., Hadera M.G., Zhou Y. et al. The GLT-1 (EAAT2; slc1a2) Glutamate transporter is essential for glutamate homeostasis in the neocortex of the mouse // J. Neurochem 2014. V. 128. № 5. P. 641–649.
Braissant O., Bachmann C., Henry H. Expression and function of AGAT, GAMT and CT1 in the mammalian brain // Subcell Biochem. 2007. V. 46. 67–81.
Buddhala C., Hsu C.C., Wu J.Y.A. Novel mechanism for GABA synthesis and packaging into synaptic vesicles // Neurochem Int. 2009. V. 55. № 1–3. P. 9–12.
Castellano G., Dias C.S.B., Foerster B. et al. NAA and NAAG variation in neuronal activation during visual stimulation // J. Med. Biol. Res. 2012. V. 45. P. 1031–3136.
Chen C., Sigurdsson H.P., Pépés S.E. et al. Activation induced changes in GABA: Functional MRS at 7T with MEGA-sLASER // Neuroimage.2017. V. 156. P. 207–213.
Choi Y., Gruetter R. Neural Metabolism in vivo. Eds. Springer Science + Business Media, LLC 2012. 1169 p.
Cirgis F., Pace J., Sweet J., Miller J. Hippocampal Neurophysiologic Changes after Mild Traumatic Brain Injury and Potential Neuromodulation Treatment Approaches // Front. Syst. Neuroscie. 2016. V. 10. P. 1–10.
D’Adamo A.F.J., Yatsu F.M. Acetate metabolism in the nervous system. N-Acetyl-L-aspartic acid and the biosynthesis of brain lipids // J. Neurochem. 1966. V. 13. P. 961–965.
D'Adamo A.F.Jr., Gidez L.I., Yatsu F.M. Acetyl transport mechanisms. Involvement of N-acetyl aspartic acid in de novo fatty acid biosynthesis in the developing rat brain // Exp. Brain Res. 1968. V. 5. P. 267–273.
D'Adamo A.F., Peisach J., Manner G. et al. N-acetyl-aspartate amidohydrolase: purification and properties // J. Neurochem. 1977. V. 28. P. 739–744.
Danbolt N.C. Glutamate uptake // Prog Neurobiol. 2001. V. 65. № 1. P. 1–105.
Davis T., Kwong K., Weisskoff R. Calibrated functional MRI: Mapping the dynamics of oxidative metabolism // Proc. Natl. Acad. Sci.USA. 1998. V. 95. P. 1834–1839.
Dean P., Sterr A. Long-term effects of mild traumatic brain injury on cognitive performance // Front. Hum. Neurosci. 2013. V. 7. P. 30–41.
Di Pietro V., Amorini A., Tavazzi B. et al. The molecular mechanisms affecting N-acetylaspartate homeostasis following experimental graded traumatic brain injury // Mol. Med. 2014. V. 20. P. 147–157.
Diehl P., Fluck E., Gunther H. et al. NMR. Basic principles and progress. In vivo Magnetic resonance spectroscopy III: potential and limitations. Berlin – Heidelberg – New York: Springer-Verlag. 1992. 190 p.
Edden R., Puts N., Barker P.Macromolecule-suppressed GABA-edited magnetic resonance spectroscopy at 3 T // Magn. Reson. Med. 2012. V. 68. P. 657–661.
Ernst R.R., Bodenhausen G., Wokaun A. Principles of Nuclear Magnetic Resonance in One and Two Dimensions. Oxford: University Press, 1987. 610 p.
Fitzpatrick C.J., Perrine S.A., Ghoddoussi F. et al. Sign-trackers have elevated myo-inositol in the nucleus accumbens and ventral hippocampus following Pavlovian conditioned approach // J. Neurochem. 2016. 136(6). P. 1196–1203.
Fox P., Raichle M. Focal physiological uncoupling of cerebral blood flow and oxidative metabolism during somatosensory activation in human subject // Proc. Natl. Acad. Sci. USA. 1986. V. 83. P. 1140–1144.
Siegel George J., Agranof Bernard W. Basic Neurochemistry: Molecular, Cellular, and Medical Aspects. Lippincott Williams & Wilkins. 1999. 1183 p.
Glodzika L., Sollbergercd M., Gassc A. et al. Global N-Acetylaspartate in Normal Subjects, Mild Cognitive Impairment and Alzheimer’s Disease Patients // J. Alzheimer’s Disease. 2015. V. 4. P. 939–947.
Goldstein F.B. The enzymatic synthesis of N-acetyl-l-aspartatic acid by subcellular preparations of rat brain // J. Biol. Chem. 1969. V. 244. P. 4257–4260.
Goldstein F.B. Amidohydrolases of brain; enzymatic hydrolysis of N-acetyl-l-aspartate and other N-acyl-l-amino acids. // J. Neurochem. 1976. V. 26. P. 45–49.
Govindaraju Y., Young K., Maudsley A. et al. Proton NMR chemical shifts and coupling constants for brain metabolites // NMR Biomed. 2000. V. 13. P. 129–153.
Greenberg J. Neuroimaging. McCraw-Hill Companies Inc., New York. 1999. 774 p.
Haider B., Häusser M., Carandini M. Inhibition dominates sensory responses in the awake cortex // Nature. 2013. V. 493. P. 97–100.
Hammond B.J., Balázs R., Machiyama Y. et al. The operation of the γ-aminobutyrate bypath of the tricarboxylic acid cycle in brain tissue in vitro // Biochem. J. 1970. V. 116. № 3. P. 445–461.
Hertz L. Theglutamate-glutamine (GABA) cycle: importance of late postnatal development and potential reciprocal interactions between biosynthesis and degradation // Front Endocrinol (Lausanne). 2013. V. 4. P. 59–67.
Jessen F., Block W., Traber F. et al. Proton MR spectroscopy detects a relative decrease of N-acetylaspartate in the medial temporal lobe of patients with AD // Neurology. 2000. V. 55. № 5. P. 684–688.
Kaneko J., Harvey J., Bruss M. Clinical Biochemistry of Domestic Animals (Sixth Edition), Academic Press. 928 p.
Kantarci K., Jack C.R.Jr., Xu Y.C. et al. Regional metabolic patterns in mild cognitive impairment and Alzheimer’s disease: A 1H MRS study // Neurology. 2000. V. 55. № 2. P. 210–217.
Katayama Y., Becker D., Tamura T. et al. Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury // J. Neurosurg. 1990. V. 73. P. 889–900.
Kim S., Ogawa S.J. Biophysical and physiological origins of blood oxygenation level-dependent fMRI signals // Cereb. Blood Flow Metab. 2012. V. 32. P. 1188–1196.
Kobori N., Dash P. Reversal of brain injury-induced prefrontal glutamic acid decarboxylase expression and working memory deficits by D1 receptor antagonism // J. Neurosci. 2006. V. 26. P. 4236–4246.
Koller K.J., Coyle J.T. Ontogenis of N-acetyl-aspartate and N-acetyl-aspartyl-glutamate in rat brain // Brain Res. 1984. V. 317. P. 137–140.
Koller K.J., Zaczek R., Coyle J.T. N-Acetyl-aspartyl glutamate: regional levels in rat brain and the effects of brain lesions as determined by a new HPLC method // J. Neurochem. 1984. V. 43. P. 1136–1142.
Larsson O., Schousboe A. Kinetic characterization of GABA-transaminase from cultured neurons and astrocytes // Neurochem. Res. 1990. V. 15. P. 1073–1077.
Lewis D.A., Volk D.W., Hashimoto T. Selective alterations in prefrontal cortical GABA neurotransmission in schizophrenia: a novel target for the treatment of working memory dysfunction // Psychopharmacol. 2004. V. 174. № 1. P. 143–150.
Liden E., Karlsson L., Sellstrom A. Is the concentration of gamma-aminobutyric acid in thenerve terminal regulated via product inhibition of glutamic acid decarboxylase? // Neurochem Res. 1987. V. 12. P. 489–493.
Madhavarao C.N., Chinopoulos C., Chandrasekaran K. et al. Characterization of the N-acetylaspartate biosynthetic enzyme from rat brain // J. Neurochem. 2003. V. 86. P. 824–835.
Magret V., Elkhalil L., Nazih-Sanderson F. et al. Entry of polyunsaturated fatty acids into the brain: evidence that high-density lipoprotein-induced methylation of phosphatidylethanolamine and phospholipase A2 are involved // Biochem. J. 1996. V. 316. P. 805–811.
Margolis R.U., Barkulis S.S., Geiger A. A comparison between the incorporation of 14C from glucose into N-acetyl-l-aspartic acid and aspartic acid in the brain perfusion experiments // J. Neurochem. 1960. V. 5. P. 379–382.
Marsman A., Mandl R., Klomp D. et al. GABA and glutamate in schizophrenia: A 7 T. 1H-MRS study // Neuroimage Clin. 2014. V. 6. P. 398–407.
Martin D.L., Rimvall K. Regulation of gamma-aminobutyric acid synthesis in the brain //J. Neurochem.1993. V. 60. № 2. P. 395–407.
Martin D.L., Rimvall K. Regulation of gamma-aminobutyric acid synthesis in the brain // J. Neurochem.1993. V. 60. № 2. P. 395–407.
Mathiesen H.K., Jonsson A., Tscherning T. et al. Correlation of global N-acetylaspartate with cognitive impairment in multiple sclerosis // Arch. Neurol. 2006. V. 63. № 4. P. 533–536.
Mcelwain H., Bachelard H.S. Biochemistry of the central nervous system. 5th ed., London: Churchill-Livingstone, 1985. 630 p.
McEntee W.J., Crook T.H. Glutamate: Its role in learning, memory, and the aging brain // Psychopharmacology. 1993. V. 111. № 4. P. 391–401.
Men'shchikov P.E., Semenova N.A., Ublinskiy M.V. et al. Spectral editing in proton magnetic resonance spectroscopy. Determination of GABA level in the brains of humans with ultra-high risk for schizophrenia // Dokl. Biochem. & Biophys. 2016. V. 468. № 1. P. 168–172.
Mescher M., Merkle H., Kirsch J. et al. Simultaneous in vivo spectral editing and water suppression // NMR Biomed. 1998. V. 11. P. 266–272.
Meynial-Denis D. Glutamine: Biochemistry, Physiology, and Clinical Applications / Eds. CRC Press NY, 2017. 373 p.
Ming G.L., Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions // Neuron. 2011. V. 70. № 4. P. 687–702.
Miyake M., Kakimoto Y., Sorimachi M. A gas chromatographic method for the determination of N-acetyl-l-aspartic acid, N-acetyl-alpha-aspartylglutamic acid and beta-citryl-l-glutamic acid and their distributions in the brain and other organs of various species of animals // J. Neurochem. 1981. V. 36. P. 804–810.
Modinos G., Şimşek F., Horder J. et al. Cortical GABA in Subjects at Ultra-High Risk of Psychosis: Relationship to Negative Prodromal Symptoms // Int. J. Neuropsychopharmacol. 2018. V. 2. № 2. P. 114–119.
Moffet J., Arun P., Ariyannur P. et al. Extensive aspartoacylase expression in the rat central nervous system // Gli. 2011. V. 59. № 10. P. 1414–1434.
Moffett J.R., Namboodiri M.A., Neale J.H. Immunohistochemical localization of N-acetylaspartate in rat brain // J. Histochem. Cytochem. 1993. V. 41. P. 559–570.
Moffett J., Tieman S.B., Weinberger et al. N-Acetylaspartate. A Unique Neuronal Molecule in the Central Nervous System / Eds. Springer Science + Business Media; New York, NY: 2006. 250 p.
Nadler J.V., Cooper J.R. N-acetyl-l-aspartic acid content of human neural tumors and bovine peripheral nervous tissue // J. Neurochem. 1972. V. 19. P. 313–319.
Nie K., Zhang Y., Huang B. et al. Marked N-acetylaspartate and choline metabolite changes in Parkinson’s disease patients with mild cognitive impairment // Parkinsonism Relat Disord. 2013. V. 19. P. 329–334.
Nissen-Meyer L.S., Chaudhry F.A. Proteinkinase C phosphorylates the system N glutamine transporter SN1 (Slc38a3) and regulates its membrane trafficking and degradation. // Front Endocrinol (Lausanne). 2013. V. 4. P. 138–142.
Pagonabarraga J.,·Gómez-Ansón B., Rotger R. et al. Spectroscopic Changes Associated with Mild Cognitive Impairment and Dementia in Parkinson’s Disease // Dement. Geriatr. Cogn. Disord. 2012. P. 34. № 5–6. P. 312–318.
Pan J.W., Takahashi K. Interdependence of N-acetyl aspartate and high-energy phosphates in healthy human brain // Ann. Neurol. 2005. V. 57. P. 92–97.
Patel A.B., de Graaf R.A., Mason G.F. et al. The contribution o fGABA to glutamate/glutamine cycling and energy metabolism in the rat cortex in vivo // Proc. Natl. Acad. Sci. USA. 2005. V. 102. № 15. P. 5588–5593.
Petroff O.A., Rothman D.L. Measuring human brain GABA in vivo: effects of GABA-transaminase inhibition with vigabatrin // Mol. Neurobiol. 1998. V. 16. № 1. P. 97–121.
Preece N.E., Cerdan S. Metabolic precursors and compartmentation of cerebral GABA in vigabatrin-treated rats // J. Neurochem. 1996. V. 67. P. 1718–1725.
Rahn K.A., Slusher B.S., Kaplin A.I. Glutamate in CNS neurodegeneration and cognition and its regulation by GCPII inhibition // Curr. Med. Chem. 2012. V. 19. № 9. P.1335–1345.
Reynolds G.P., Harte M.K. The neuronal pathology of schizophrenia: molecules and mechanisms // Biochem. Soc. Trans. 2007. V. 35 P. 433–436.
Rink P.A. Introduction into Magnetic Resonance in Medicine. Stuttgart – New York: Theme Medical Publishers Inc., 1990. 228 p.
Robinson M.B., Blakely R.D., Couto R. et al. Hydrolysis of the brain dipeptide N-acetyl-l-aspartyl-l-glutamate: identification and characterization of a novel N-acetylated-alpha-linked acidic dipeptidase activity from rat brain // J. Biol. Chem. 1987. V. 262. P. 14 498–14 502.
Ross B.M., Moszczynska A., Blusztajn J.K. et al. Phospholipid biosynthetic enzymes in human brain // Lipids.1997. V. 32(4). P. 351–358.
Rothman D.L., De Feyter H.M., de Graaf R. et al. 13C MRS studies of neuroenergetics and neurotransmitter cycling in humans // NMR Biomed. 2011. V. 24. № 8. P. 943–957.
Rowland L., Kontson K., West J. et al. In vivo Measurements of glutamate, GABA, and NAAG in schizophrenia // Schizophr. Bull. 2013. V. 39. № 5. P. 1096–1104.
Rowley N.M., Madsen K.K., Schousboe A. et al. Glutamate and GABA synthesis, release, transport and metabolism as targets for seizure control // Neurochem. Int. 2012. l. 61. P. 546–5810.
Sager T.N., Fink-Jensen A., Hansen A.J. Transient elevation of interstitial N-acetylaspartate in reversible global brain ischemia // J. Neurochem. 1997. V. 68. P. 675–682.
Sager T.N., Laursen H., Fink-Jensen A. et al. N-Acetylaspartate distribution in rat brain striatum during acute brain ischemia // J. Cereb. Blood Flow Metab. 1999. V. 19. P. 164–172.
Sager T.N., Topp S., Torup L. et al. Evaluation of CA1 damage using single-voxel 1H-MRS and un-biased stereology: Can non-invasive measures of N-acetyl-aspartate following global ischemia be used as a reliable measure of neuronal damage // Brain Res. 2001. V. 892. P. 166–175.
Sanacora G., Mason G.F., Rothman D.L. et al. Reduced cortical gamma-aminobutyric acid levels in depressed patients determined by proton magnetic resonance spectroscopy. // Archives of general psychiatry. 1999. V. 56. № 11. P. 1043–1047.
Schober A.L., Wilson C.S., Mongin A.A. Molecular composition and heterogeneity of the LRRC8-containing swelling-activated osmolyte channels in primary rat astrocytes // J. Physiol. 2017. V. 15. 595(22). P. 6939–6951.
Schousboe A., Sonnewald U. GABA- glutamine cycle. Amino acid neurotransmitter homeostasis / Eds. Springer International Publishing Switzerland. 2016. 435 p.
Schür R.R., Draisma L.W., Wijnen J.P. et al. Brain GABA levels across psychiatric disorders: A systematic literature review and meta-analysis of (1) H-MRS studies // Hum. Brain. Mapp. 2016. V. 37. P. 3337–3352.
Sheikh S.N., Martin D.L. Elevation of brain GABA levels with vigabatrin (gamma-vinyl-GABA) differentially affects GAD65 and GAD67 expression in various regions of rat brain // J. Neurosci Res. 1998. V. 52. P. 736–741.
Shulman R.G. Energetic basis of brain activity: implications for neuroimaging // Trends Neurosci. 2004. Vol. 27. № 8. P. 489–495.
Siegel G.J., Bernard W., Agranoff B.W. Basic Neurochemistry: Molecular, Cellular, and Medical Aspects. Lippincott Williams & Wilkins, 1999. 1183 p.
Simmons M.L., Frodonza C.G., Coyle J.T. Immunocytochemical localization of N-acetyl-aspartate with monoclonal antibodies // Neuroscience. 1991. V. 45. P. 37–45.
Simpson H.B., Shungu D.C., Bender J. et al. Investigation of cortical glutamate-glutamine and gamma-aminobutyric acid in obsessive-compulsive disorder by proton magnetic resonance spectroscopy // Neuropsychopharmacol. 2012. V. 37. № 12. P. 2684–2692.
Slusher B.S., Robinson M.B., Tsai G. et al. Rat brain N-acetylated alpha-linked acidic dipeptidase activity // J. Biol. Chem. 1990. V. 265. P. 21297–21301.
Soghomonian J.J., Martin D.L. Two isoforms of glutamate decarboxylase: why? // Trends Pharmacol. Sci. 1998.V. 19. № 12. P. 500–505.
Sonnewald H., Schousboe A. Compartmentation of glutamine, glutamate, and GABA metabolism in neurons and astrocytes: functional implications. // Neuroscientist. 2003. V. 9. № 5. P. 398–403.
Sontheimer H. Diseases of the Nervous System. Academic Press, 2015. 520 p.
Takashima S., Chan F., Becker L.E. et al. Morphology of the developing visual cortex of the human infant // J. Neuropathol. Exp. Neurol. 1980. V. 39. P. 487–501.
Tallan H.H. Studies on the distribution of N-acetyl-L-aspartic acid in brain // J. Biol. Chem. 1957. V. 224. № 1. P. 41–45.
Tallan H.H., Moore S., Stein W.H. N-Acetyl-l-Aspartic acid in brain // J. Biol. Chem. 1956. V. 219. P. 257–264.
Taub A., Katz Y., Lampl I. Cortical balance of excitation and inhibition is regulated by the rate of synaptic activity // J. Neurosci.2013. V. 33. № 36. P. 14 359–14 368.
Taylor D.L., Davies S.E.C., Obrenovitch T.P. et al. Extracellular N-acetylaspartate in the rat brain: in vivo determination of basal levels and changes evoked by high K+ // J. Neurochem. 1994. V. 62. P. 2349–2355.
Taylor R., Neufeld R., Schaefer B. et al. Functional magnetic resonance spectroscopy of glutamate in schizophrenia and major depressive disorder: anterior cingulate activity during a color-word Stroop task // Schizophrenia. 2015. P. 15 028–1540.
Truckenmiller M.E., Namboodiri M.A.A., Browstein M.J. et al. N-Acetylation of l-aspartate in the nervous system: differential distribution of a specific enzyme // J. Neurochem. 1985. V. 45. P. 1658–1662.
Urenjak J.1., Williams S.R., Gadian D.G. et al. Specific expression of N-acetylaspartate in neurons, oligodendrocyte-type-2 astrocyte progenitors, and immature oligodendrocytes in vitro // J. Neurochem.1992. V. 59. № 1. P. 55–61.
Van Eijsden P., Behar K.L., Mason G.F. et al. In vivo neurochemical profiling ofrat brain by 1H-[13C] NMR spectroscopy: cerebral energetics and glutamatergic/GABAergic neurotransmission // J. Neurochem. 2010. V. 112. № 1. P. 24–33.
Verma A., Kumar I., Verma N. et al. Magnetic resonance spectroscopy – Revisiting the biochemical and molecular milieu of brain tumors // BBA Clin. 2016. V. 5. P. 170–178.
Waagepetersen H., Sonnewald U., Gegelashvili G. et al. Metabolic Distinction Between Vesicular and Cytosolic GABA in Cultured GABAergic Neurons Using 13C Magnetic Resonance Spectroscopy // J. Neurosci. Res. 2001. V. 63. P. 347–355.
Waagepetersen H.S., Qu H., Hertz L. et al. Demonstration of pyruvate recycling in primary cultures of neocortical astrocytes but not in neurons // Neurochem. Res. 2002. V. 27. № 11. P. 1431–1437. https://www.ncbi.nlm.nih.gov/pubmed/12512946.
Wang S.-Y., Wang M., Wang X.-X. et al. Study on the clinical application of the MRS in the cognitive assessment after stroke // European. Rev. Med. Pharmacol. Sci. 2017. V. 21. P. 2437–2442.
Whitelaw B.S., Robinson M.B. Inhibitors of glutamate dehydrogenase block sodiumdependent glutamate uptake in rat brain membranes // Front Endocrinol (Lausanne). 2013. V. 4. P. 123–128.
Wiame E., Tyteca D., Pierrot N. et al. Molecular identification of aspartateN-acetyltransferase and its mutationin hypoacetylaspartia // Biochem. J. 2010. V. 425. P. 127–136.
Yoon J.H., Maddock R.J., Rokem A. et al. GABA concentration is reduced in visual cortex in schizophrenia and correlates with orientation-specific surround suppression // J. Neurosci. 2010. 30. 3777–27811.
Дополнительные материалы отсутствуют.
Инструменты
Успехи физиологических наук