

Успехи физиологических наук, 2020, T. 51, № 1, стр. 3-17
Роль малых G-белков в регуляции ионных каналов
А. В. Карпушев a, *, В. Б. Михайлова a, **, Д. В. Абрамочкин b, ***
a Национальный медицинский исследовательский центр им. В.А. Алмазова
Москва, Россия
b Московский государственный университет им. М.В. Ломоносова
Москва, Россия
* E-mail: akarpushev@yandex.ru
** E-mail: hastar.teerend@yandex.ru
*** E-mail: abram340@mail.ru
Поступила в редакцию 08.08.2019
После доработки 05.09.2019
Принята к публикации 20.10.2019
Аннотация
Малые G-белки играют важную роль в основных клеточных процессах, включая дифференцировку, клеточное деление, динамику цитоскелета, мембранный и белковый транспорт. Присутствуя во всех эукариотических клетках и являясь ключевыми ферментами сигнальных каскадов, малые G-белки связывают рецепторы различных регуляторных факторов с множеством эффекторных молекул, среди которых важное место занимают ионные каналы. В обзоре приведены данные по функциональному взаимодействию малых G-белков Ras, Rho и Rab семейств с различными ионными каналами.
ВВЕДЕНИЕ
Малые G-белки, также известные как малые или мономерные ГТФазы (гуанозин трифосфатазы), составляют суперсемейство, носящее название Ras (Rat sarcoma), данное по первым членам, Ras протоонкогенам, обнаруженным в начале 1980-х [79]. Малые G-белки образуют разветвленные сигнальные сети, активируемые различными внешними экстраклеточными факторами, и могут опосредовать высокоспецифичные клеточные ответы благодаря наличию множества эффекторов. Малые G-белки являются ключевыми регуляторами большинства процессов в клетке, включая дифференцировку, пролиферацию, динамику и транспорт везикул и органелл, ядерную динамику и регуляцию цитоскелета [79]. Как одноклеточные, так и многоклеточные эукариотические организмы экспрессируют гены суперсемейства Ras белков. На сегодняшний день малые G-белки обнаружены во всех эукариотических клетках, а у человека выявлено 167 малых ГТФаз [62, 81]. В соответствии со структурным и функциональным сходством малые G-белки подразделяются на пять основных семейств: Ras (совпадает с названием всего суперсемейства), Rho, Rab, Arf и Ran [122]. Белки семейства Ras в основном регулируют экспрессию генов, белки семейства Rho участвуют как в реорганизации цитоскелета так и в экспрессии генов, члены семейств Rab и Arf регулируют внутриклеточный везикулярный транспорт, а белки семейства Ran регулируют нуклео-цитоплазматический транспорт во время фаз G1, S и G2 клеточного цикла и организацию микротрубочек во время фазы М [109].
Малые G-белки состоят из одной полипептидной цепи молекулярной массой 20-25 кДа, гомологичной альфа-субъединице гетеротримерных G-белков, и демонстрируют высокоаффинное связывание с ГДФ и ГТФ, а также обладают слабой собственной гидролитической ГТФ/ГДФ активностью [13]. Таким образом, малые ГТФазы, функционируя как ГТФ/ГДФ-регулируемые молекулярные “выключатели”, могут находиться в двух состояниях: “включенном” ГТФ-связанном или “выключенном” ГДФ-связанном. ГТФ-связанная форма является активной, т.е. обладающей высоким сродством к эффекторным молекулам, в то время как ГДФ-связанная форма инактивирована [9, 80]. Контроль цикла активации/инактивации малых G-белков требует участия положительных и отрицательных регуляторных факторов. Степень и продолжительность нахождения в активной форме определяется взаимодействием с тремя типами факторов: факторы обмена гуаниновых нуклеотидов (guanine nucleotide exchange factors, GEFs), белки-активаторы ГТФазной активности (GTPase-activating proteins, GAPs) и ингибиторы диссоциации гуаниновых нуклеотидов (guanine nucleotide dissociation inhibitors, GDIs) (рис. 1) [15, 63, 113].
Рис. 1.
Схема активации/инактивации малых G-белков суперсемейства Ras. GEF (guanine nucleotide exchange factor) - фактор обмена гуаниновых нуклеотидов, GAP (GTPase-activating protein) – активатор ГТФазной активности, GDI (guanine nucleotide dissociation inhibitor) – ингибитор диссоциации гуаниновых нуклеотидов, ГДФ – гуанозиндифосфат, ГТФ – гуанозинтрифосфат. GEF активирует малый G-белок, катализируя замену ГДФ на ГТФ. GAP активирует гидролиз ГТФ и приводит малый G-белок в неактивное состояние. GDI поддерживает малый G-белок в неактивном состоянии.
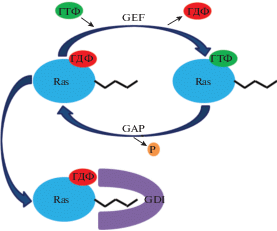
GEFs катализируют замену ГДФ на ГТФ, что приводит к активации малых ГТФаз [95]. GEFs являются связующим звеном между мембранными рецепторами нейротрансмиттеров, цитокинов, различных факторов роста и малыми G-белками [16, 63]. Поскольку малые G-белки имеют низкий уровень собственной каталитической активности, для гидролиза ГТФ необходимо взаимодействие с GAPs. В нативном состоянии GAPs находятся в свернутой, автоингибированной конформации, что ограничивает доступность каталитического сайта. GAPs контролируются несколькими различными механизмами, включая фосфорилирование/дефосфорилирование, липид-белковое или белок-белковое взаимодействия и деградацию/ресинтез [58, 60, 61]. Взаимодействие с GDIs поддерживает малые ГТФазы в неактивном состоянии [22]. Большинство малых G-белков содержат модифицированный липидами С-конец, который локализует их на клеточной мембране. Самый важный эффект GDIs – это их способность контактировать с пренилированным С-концом и отрывать от мембраны ГДФ-связанные малые ГТФазы, сохраняя их в инактивированной форме в цитозоле путем защиты гидрофобного липидного хвоста от водного растворителя [16, 31, 41, 57, 64]. Цитозольная фракция GDI-связанных малых G-белков защищена от замены гуаниновых нуклеотидов ГДФ/ГТФ белками GEFs [20]. Диссоциация комплекса GDI с малой ГТФазой является необходимым условием для GEF- и GAP-опосредованной регуляция активности малых G белков. Сигнальные молекулы, включая Src киназу, протеинкиназу С (РКС) и протеинкиназу А (РКА), являются регуляторами GEFs и влияют на сродство последних к малым G-белкам [98]. Стабильность и субклеточная локализация малых G-белков регулируется также посттрансляционными модификациями, такими как фосфорилирование или убиквитинирование [1].
Малые G-белки локализуются либо в цитозоле, либо на цитоплазматической или внутриклеточных мембранах. Обычно, представители каждого из семейств малых G-белков локализуются на определенном типе мембран [109]. Например, белки Ras семейства локализуются на цитоплазматической поверхности клеточной мембраны. Эта локализация обусловлена посттрансляционным липидированием (пренилирование) и протеолизом [23]. Rho белки, напротив, динамически перемещаются между плазмолеммой и цитозолем. Большинство Rab белков посредством геранилгеранилирования мотивов Cys-X-Cys и Cys-Cys локализуются на цитоплазматических поверхностях клеточной мембраны или на мембранах внутриклеточных компартментов. Белки Arf также взаимодействуют с различными мембранами, а малые G-белки семейства Ran перемещаются между цитозолем и ядром через комплексы ядерных пор [109].
Взаимодействие малых G-белков с эффекторами приводит к изменению в активности последних. За прошедшие два десятилетия накопилось большое количество данных о том, что ионные каналы являются одними из конечных эффекторов малых G-белков. Сигнальные пути с участием малых G-белков могут модулировать активность широкого спектра мембранных ионных каналов. Регуляция токов ионных каналов обусловливается двумя фундаментальными механизмами: изменением вероятности открытого состояния одиночных каналов и изменением общего количества каналов на мембране. Первый механизм связан с контролем воротного аппарата ионного канала. Второй механизм может регулироваться как на уровне экспрессии генов, так и на уровне транспорта и локализации белковых субъединиц на наружной мембране или их интернализации. Малые G-белки могут участвовать на всех этапах контроля активности ионных каналов. Например, для эпителиального натриевого канала ENaC (epithelial sodium channel) показано, что K-Ras повышает вероятность открытого состояния канала, а RhoA увеличивает экспрессию канала на клеточной мембране (рис. 2) [76]. При этом нет данных об эффектах малых G-белков на селективность ионных каналов.
Рис. 2.
Схема регуляции активности эпителиального натриевого канала ENaC (epithelial sodium channel) малыми G-белками K-Ras и RhoA. PI3K (phosphoinositide 3-kinase) – фосфоинозитид-3-киназа, PI(4)P5K (phosphatidylinositol-4-phosphate 5-kinases) – фосфатидилинозитол-4-фосфат-5-киназа, ROCK – Rho киназа, PI(3,4,5)P3 – фосфатидилинозитол (3,4,5)-трифосфат, PI(4,5)P2 – фосфатидилинозитол (4,5)-дифосфат, ГТФ – гуанозинтрифосфат. K-Ras повышает вероятность открытого состояния ENaC через PI3K-зависимый сигнальный путь и PI(3,4,5)P3. RhoA увеличивает экспрессию ENaC на клеточной мембране через сигнальный путь, включающий в себя последовательно ROCK, PI(4)P5K и PI(4,5)P2.

В некоторых случаях малые ГТФазы непосредственно взаимодействуют с ионными каналами, в других регуляция опосредуется промежуточными сигнальными белками. Например, Rab11a напрямую связывается с эпителиальными Ca2+ каналами транзиторного рецепторного потенциала ванилоидного подсемейства TRPV5 и TRPV6 (Transient Receptor Potential channel Vanilloid subfamily) [114], а Rem ГТФаза образует регуляторный комплекс со вспомогательной бета-субъединицей Cavβ кальциевого канала Cav1.2 [28]. Точно так же RhoA физически связывается с N-концом калиевого канала Kv1.2 [18]. В то же время K-Ras и RhoA усиливают активность ENaC через сигнальные пути фосфоинозитид-3-киназы PI3K (phosphoinositide 3-kinase) и фосфатидилинозитол-4-фосфат-5-киназы PI(4)P5K (phosphatidy-linositol-4-phosphate 5-kinases) (рис. 2) [75, 76]. Rac1 обеспечивает быстрое встраивание в цитоплазматическую мембрану неселективного катионного канала TRPC5 (Transient Receptor Potential channel Canonical subfamily) посредством стимуляции PI(4)P5K [7].
РЕГУЛЯЦИЯ ИОННЫХ КАНАЛОВ МАЛЫМИ G-БЕЛКАМИ СЕМЕЙСТВА RAS
Гены Ras были впервые идентифицированы как онкогены саркомы при исследовании ретровирусов, выделенных из крыс [24]. Семейство Ras состоит из 36 членов [122]. Белки семейства Ras инициируют целую сеть внутриклеточных сигнальных путей. Наиболее изученный нисходящий сигнальный каскад включает митоген-активируемые протеинкиназы MAPKs (mitogen-activated protein kinases), в частности, регулируемые внеклеточными сигналами киназы ERK 1/2 (extracellular signal-regulated kinase) [130]. Ras ГТФазы могут напрямую связываться и активировать серин/треониновую протеинкиназу Raf [25]. В свою очередь, активированная Raf-киназа фосфорилирует и активирует MEK1/2-киназу, последняя активирует ERK1/2-киназу. Активированная ERK1/2-киназа перемещается в клеточное ядро, где фосфорилирует транскрипционные факторы Ets семейства, что имеет результатом активацию Ets-регулируемых промоторов для индукции экспрессии генов [56, 109]. Rap1 может активировать ERK через b-Raf и p38MAPK [36, 50]. Кроме того Ras ГТФазы активируют Ral GDS, RIN1 и PI3K [10, 44, 96].
Активность белков Ras семейства регулируется факторами GAPs и GEFs. Остатки фосфотирозина рецептора эпидермального фактора роста EGFR (epidermal growth factor receptor) служат в качестве сайтов докинга для SH2 доменов адаптерных белков, таких как Grb2 (growth-factor-receptor-bound protein 2), который, в свою очередь, через SH3 домены рекрутирует Ras-GEF SOS для создания комплекса рецептор–адаптор–GEF, способного активировать Ras ГТФазы [109]. Рецепторы, не обладающие собственной тирозинкиназной активностью, могут также активировать Ras ГТФазы через Src-подобную тирозинкиназу или через лиганд-независимую активацию тирозинкиназных рецепторов [109]. Кроме того, рецепторы, сопряженные с гетеротримерными G-белками GPCRs (G-protein-coupled receptors), такие как мускариновые ацетилхолиновые рецепторы mAChRs (muscarinic acetylcholine receptors), альфа-адренергические рецепторы и рецепторы лизофосфатидной кислоты LPARs (lysophosphatidic acid receptors) могут активировать белки семейства Ras [8, 40, 45, 65].
Передача сигналов, опосредуемая Ras малыми G-белками, может влиять на активность ионных каналов клеточной мембраны. В ряде работ показано, что K-RasA повышает активность ENaC [101, 102]. В эпителии собирательных трубочек почек альдостерон, контролируя экспрессию генов, повышает активность малого G-белка K-Ras [42, 69, 99, 100, 105]. K-Ras усиливает активность ENaC за счет увеличения вероятности открытого состояния канала через PI3K-зависимый сигнальный путь (рис. 2) [69, 101, 102]. Непосредственно с ионным каналом взаимодействует продукт киназы фосфатидилинозитол (3,4,5)-трифосфата PI(3,4,5)P3. Эффект K-Ras на ENaC блокируется ингибитором фермента PI3K вортманнином и имитируется при гиперэкспрессии активной PI3K. Реабсорбция ионов Na+ через канал ENaC в дистальном отделе нефрона почки является ключевым фактором регуляции объема циркулирующей в организме жидкости и давления крови в сосудах [3, 94, 97, 106]. Таким образом, K-Ras-зависимая регуляция канала ENaC определяет системный баланс Na+ и артериального давления [2].
Воздействие малых G-белков на активность ионных каналов может иметь различный характер в зависимости от типа канала. Например, Ras через MAPK-зависимый сигнальный каскад является негативным регулятором K+ канала входящего выпрямления IRK1 (inward-rectifier potassium channel), экспрессированного в клетках линии эмбриональных почек человека HEK-293 (Human Embryonic Kidney). Активный Ras снижает K+ ток IK1, уменьшая количество каналов IRK1 на поверхности клеточной мембраны. Эффект обусловлен нарушением внутриклеточной транспортировки ионного канала без изменений биофизических характеристик и не затрагивает транскрипционного уровня [34]. Однако Ras увеличивает активность Ca2+ канала Т-типа в нейронах спинальных ганглиев куриного эмбриона [38].
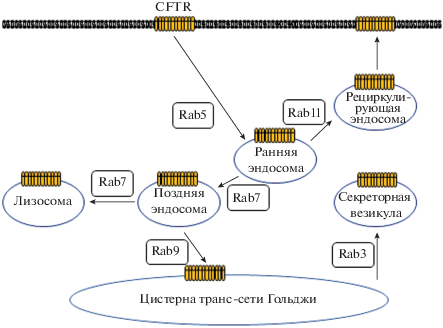
Рис. 3.
Схема модуляции везикулярной транспортировки регулятора трансмембранной проводимости при муковисцидозе CFTR (cystic fibrosis transmembrane conductance regulator) белками Rab.
Точная доставка каналов в соответствующий мембранный домен является важным аспектом регуляции активности ионных каналов. Поддержание адекватного уровня экспрессии на клеточной мембране функционально активных ионных каналов представляет собой динамический процесс, определяемый балансом антероградного (экзоцитоз, рециркуляция) и ретроградного (интернализация) транспортов. Локализация и стабилизация ионных каналов на мембране регулируется белками-партнерами и вспомогательными субъединицами каналов. Малые G-белки подсемейства RGK (Rad, Rem, Rem2, kir/Gem, Ras-related protein) напрямую связываются с бета-субъединицей Ca2+ канала L-типа и, регулируя экспрессию ионного канала на клеточной поверхности, снижают активность Cav1.2 [5, 28, 29]. Beguin и соавторы обнаружили, что трафик Ca2+ канала L-типа и, следовательно, характеристика Ca2+ тока регулируются малым G-белком kir/Gem подсемейства RGK, который был идентифицирован как партнер вспомогательной бета-субъединицы канала [5]. Взаимодействие kir/Gem с бета-субъединицей ингибировало сборку последней с альфа-субъединицей канала, и как следствие препятствовало экспрессии канала на клеточной поверхности. Прямое взаимодействие белков продемонстрировала коиммунопреципитация Rem и Rad с Cavβ2a экспрессированных в клетках HEK-293 [29]. Также экспрессия Rem в клетках линии мышиных миобластов C2C12 ингибировала активность Ca2+ канала L-типа [29]. При этом, Rem не влияет на активность Ca2+ канала Т-типа Cav3.2, не требующего вспомогательной бета-субъединицы для генерации ионного тока. Кроме того Rem2 подавляет активность Ca2+ канала N-типа Cav2.2 в нейронах спинальных ганглиев и симпатических нейронах паравертебральных ганглиев крысы, не влияя на плотность каналов на клеточной мембране [19].
Немаловажным значением Ras белков является их участие в регуляции сократительной функции миокарда и сердечного ритма. Было показано, что конститутивно-активный H-Ras-V12 уменьшает амплитуду тока Ca2+ канала L-типа в неонатальных желудочковых кардиомиоцитах крысы путем уменьшения экспрессии порообразующей субъединицы α1С Cav1.2 через активацию Raf-MEK-ERK-зависимого сигнального пути [43]. Поскольку общей чертой всех членов подсемейства RGK является их способность мощно ингибировать Ca2+ каналы L-типа, активация RGK ГТФаз уменьшает систолическую функцию левого желудочка [70, 116, 125]. Кроме того, недавно было показано, что Rem предотвращает стимулирующее действие цАМФ-активируемой протеинкиназы (PKA) на Ca2+ канал L-типа, подтверждая физиологическую и/или патофизиологическую роль белков RGK в бета-адренергической регуляции сердечной функции [124]. Эта гипотеза также подтверждается наблюдением, что избыточная экспрессия Rad элиминирует бета-адренергические эффекты на Ca2+ ток L-типа [116]. Одним из преобладающих объяснений этого эффекта является то, что белки RGK препятствуют переносу порообразующей субъединицы α1С на сарколемму, возможно, путем буферизации эндогенных β-субъединиц (β1, β2 и β3), тем самым снижая поверхностную плотность сердечных Ca2+ каналов L-типа [5, 77]. Однако последние данные свидетельствуют, что Rem преимущественно подавляет ток Ca2+ канала L-типа, Cav1.2, снижая вероятность открытого состояния находящихся на клеточной поверхности каналов [124]. Таким образом, белки RGK используют различные механизмы и детерминанты для ингибирования Ca2+ тока канала L-типа.
В 1991 году была установлена связь между геном H-ras и клиническим фенотипом аритмогенного синдрома удлиненного интервала QT (LQTS) [52]. Поскольку Ras является негативным регулятором K+ канала входящего выпрямления Kir2.1 через MAPK-зависимый сигнальный каскад [34], Ras рассматривается как ген-кандидат для этой аутосомно-доминантной аритмии сердца. Трансгенные мыши с тамоксифен-индуцированной кардиоспецифичной экспрессией конститутивно-активного H-Ras-V12 демонстрировали постепенное развитие аритмии [82]. Записи электрокардиограммы указывали на остановку синоатриального узла, идиовентрикулярный ритм, желудочковую тахикардию, нарушение проводимости и ФП. Роль подсемейства RGK в регуляции сердечного ритма напрямую связана с их действием на Ca2+ канал L-типа. In vivo трансдукция белка Gem в левый желудочек сердца морской свинки приводила к значительному сокращению интервала QT вследствие снижения экспрессии Ca2+ канала L-типа и уменьшения длительности ПД [70]. Гиперэкспрессия Gem в атриовентрикулярном узле свиньи удлиняла интервал PR, что указывает на замедление атриовентрикулярной проводимости, а также снижала на 20% частоту сокращений желудочков. В противоположность этому гиперэкспрессия доминантно-негативного мутанта S105N Rad у мышей приводила к удлинению интервала QT. Это было связано с повышенной экспрессией α1с-субъединицы Ca2+ канала L-типа и, как следствие увеличенной длительностью ПД [125]. Также у мышей, экспрессирующих S105N Rad, наблюдались различные аритмии, такие как дисфункция синусового узла, атриовентрикулярная блокада и желудочковая экстрасистолия. Белки Rap и Ras оказывают противоположные действия на K+ токи предсердий. Введение в субнаномолярных концентрациях экзогенных Ras и Ras-GAP в изолированные клетки предсердий блокировало K+ ток IK,ACh, активируемый через М2-мускариновые рецепторы [127]. Этот эффект скорее обусловлен разобщением М2-рецептора и гетеротримерного G-белка, а не изоляцией G-белка от ионного канала [68]. Скрининг ДНК-микрочипов у пациентов с ФП выявил Rap1a как ген, потенциально участвующий в патологическом процессе. Экспрессия Rap1a отрицательно коррелирует с клиническим фенотипом ФП [53]. Интересно, что Rap1а противодействует способности Ras и Ras-GAP ингибировать ацетилхолин-зависимый K+ ток IK,ACh [128]. Также Ras и Rap1 являются антагонистами в регуляции потенциал-зависимых Na+ каналов в клетках гибрида нейробластомы и глиомы NG108-15 [46], НМДА- (N-methyl-D-aspartic acid, NMDA) и АМРА-рецепторов (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor, AMPA) глутаматергической синаптической передачи в органотипической культуре гипокампальных нейронов крыс [47]. Ras опосредует синаптическую потенциацию, регулируя доставку AMPA-рецепторов. Напротив, Rap опосредует синаптическую депрессию, удаляя AMPA-рецептры [134].
Таким образом, Ras ГТФазы модулируя активность ионных каналов, оказываются вовлеченными в регуляцию различных физиологических и патофизиологических процессов в сердечной, нервной и выделительной системах организма.
РЕГУЛЯЦИЯ ИОННЫХ КАНАЛОВ МАЛЫМИ G-БЕЛКАМИ RHO СЕМЕЙСТВА
В середине 1980-х Madaule и Axel [66] идентифицировали первый гомолог Ras в морском моллюске Aplysia и назвали его Rho (Ras homologous). В настоящее время известно 23 члена Rho семейства [39, 71]. Наиболее изученными членами Rho семейства являются RhoА, Rac1 и Cdc42. Rho ГТФазы контролируя внутриклеточную динамику полимеризации F-актина, оказываются задействованными в широком спектре клеточных процессов, включая пролиферацию клеток, дифференцировку, адгезию, секрецию, транскрипцию генов и клеточный цикл [12, 21, 35, 73, 118, 131]. К настоящему времени идентифицировано более 30 потенциальных эффекторов Rho, Rac и Cdc42 [9]. Показано, что передача сигналов Rho ГТФаз модулирует активность нескольких классов ионных каналов, включая K+ каналы Kv1.2, Kir2.1 и ERG, Ca2+ каналы и эпителиальные Na+ каналы, а также неселективный катионный канал TRPC5, Ca2+ каналы TRPV5/6 и потенциал-зависимый Na+ канал Nav1.5 [18, 27, 32, 48, 59, 75, 103, 107, 123].
Подобно другим малым G-белкам, Rho ГТФазы действуют как ГТФ/ГДФ-зависимые молекулярные переключатели. В ГТФ-связанном состоянии Rho ГТФазы взаимодействуют с эффекторными молекулами, такими как Rho киназа (ROCK), WASP, PI(4)P5K и PI3K. RhoA, Rac1, а также ROCK, активируя PI(4)P5K, увеличивают синтез фосфатидилинозитол (4,5)-дифосфата PI(4,5)P2 [87, 119, 120, 126]. Rac и Cdc42 также взаимодействуют с PI3K и увеличивают образование PI(3,4,5)P3 [11, 112, 133]. Посредством фосфолипидных вторичных мессенджеров и адаптерных белков Rho ГТФазы контролируют везикулярный транспорт, влияя как на эндоцитоз, так и на экзоцитоз [54, 78, 83, 110].
Эффект RhoA на ENaC аналогичен эффекту K-Ras, в том смысле, что они оба увеличивают активность канала. Однако пути передачи сигнала и механизмы активации значительно различаются [76]. K-Ras повышает вероятность открытого состояния канала, тогда как RhoA увеличивает экспрессию канала на клеточной мембране. RhoA активирует ENaC, инициируя быструю транслокацию и встраивание канала в цитоплазматическую мембрану через сигнальный каскад, включающий в себя последовательно ROCK, PI(4)P5K и PI(4,5)P2 (рис. 2) [75, 103]. Интересно, что фосфолипиды служат сигнальными молекулами в обоих случаях: PI(3,4,5)P3 при K-Ras-зависимой активации и PI(4,5)P2 при RhoA-зависимой активации. Другой малый G-белок Rho семейства Rac1, также как и RhoA, значительно увеличивает активность ENaC как в эпителии собирательных трубочек почки, так и в клетках линии СНО (Chinese hamster ovary), экспрессирующих экзогенный канал [55]. Причем белки WAVE1/2 также являются необходимыми компонентами Rac1-зависимого сигнального пути активации ENaC. Ни дикий тип Cdc42, ни постоянно активный мутант (G12V) не оказывают влияния на активность ENaC. Rac1 способствует встраиванию TRPC5 в плазматическую мембрану в клетках HEK-293 [7]. Этот эффект на TRPC5 является ROCK- и PI(4)P5K-зависимым, что еще раз указывает на то, что PI(4,5)P2 играет роль ключевой сигнальной молекулы. RhoA подавляет цАМФ-зависимую транслокацию водного канала аквапорин-2 (AQP2) на апикальную мембрану эпителиальной выстилки почечных канальцев при помощи контроля организации актинового цитоскелета [110, 111].
Rho ГТФазы участвуют в регуляции сердечной деятельности. В исследованиях in vivo на трансгенных мышах были проанализированы эффекты Rho белков на ионные каналы кардиомиоцитов. Гиперэкспрессия RhoA индуцировала выраженную дисфункцию синусового и атриовентрикулярного узлов, характеризующуюся снижением частоты сердечных сокращений, удлиненным интервалом PR, атриовентрикулярной блокадой второй степени и ФП [84]. В то время как у контрольных мышей атропин вызывал положительный хронотропный эффект вследствие блокады мускариновых рецепторов, у трансгенных мышей этот эффект не наблюдался, т.е. атропин не устранял брадикардию, что можно объяснить тормозящим воздействием RhoA на Kir2.1. Изолированные кардиомиоциты желудочка трансгенных мышей, гиперэкспрессирующих Rho GDIα демонстрировали 40% снижение плотности Ca2+ тока L-типа [129]. Эффект не был связан с изменением в количестве мРНК и белка Ca2+ канала. Экспрессия доминантно-негативной формы RhoA, но не доминантно-негативных Rac1 или Cdc42, имитировала эффект Rho GDIα, указывая на то, что ионный канал является конечным эффектором RhoA в миоцитах сердца. У мышей с гиперэкспрессией Rho GDIα также наблюдался прогрессирующий дефект атриовентрикулярной проводимости [121]. Однако, что удивительно, у трансгенных животных с гиперэкспрессией Rho GDIα желудочковая сократительная функция сохранялась.
RhoA также известен как ингибитор K+ тока входящего выпрямления IK1. В гетерологической экспрессионной системе клеток линии tsA201 конститутивно-активный RhoA ингибировал Kir2.1, а доминантно-негативная форма RhoA устраняла эффект подавления Kir2.1, опосредованный активацией мускариновых рецепторов [48]. Это предполагает участие RhoA в регуляции Kir2.1 при активации мускариновых рецепторов. В другом случае конститутивно-активный Rac1 не оказывал влияния на K+ ток канала Kir2.1 реконструированного в клетках НЕК-293, а доминантно-негативный Rac1 увеличивал K+ ток [14]. Иммуногистохимичекое окрашивание и блокада эффекта Rac1 мутантной формой динамина свидетельствуют о снижении уровня интернализации Kir2.1 в ответ на экспрессию доминантно-негативного малого G-белка.
Также было показано, что RhoA ингибирует K+ каналы задержанного выпрямления Kv1.2, первоначально клонированные из предсердия крысы и реконструированные в ооцитах Xenopus [18]. Эксперименты по коиммунопреципитации подтвердили, что RhoA взаимодействует с Kv1.2. Кроме того, в клетках НЕК-293, экспрессирующих Kv1.2 и мускариновый рецептор М1, инактивация RhoA Clostridium botulinum C3-экзоэнзимом блокировала эффект М1-рецептор-зависимого ингибирования Kv1.2. Следовательно, RhoA регулирует активность Kv1.2 и является центральным компонентом в механизме рецептор-опосредованной супрессии Kv1.2. Большой интерес представляют данные по снижению экспресии РНК и белков, а также пиковой амплитуды Na+ тока сердечной изоформы потенциал-зависимого канала Nav1.5, в культурах клеток рака груди MDA-MB-231 и MCF-7 при подавлении экспрессии RhoA [27]. RhoА и ROCK также участвуют в регуляции Ca2+ токов каналов TRPV5/6, индуцированных лизофосфатидилхолином (lysophosphatidylcholine, LPC) в кардиомиоцитах желудочков морской свинки [59]. LPC является амфипатическим метаболитом мембранного фосфатидилхолина. Высокие концентрации LPC были обнаружены в ишемических сердцах. Предполагается, что LPC является одной из причин перегрузки Ca2+ и аритмии во время ишемии и реперфузии сердца.
Исследования, выполненные на линии клеток гипофиза крысы GH4C1 помогли определить молекулярную идентичность каналов, лежащих в основе эффектов Rac1 и RhoA на сердечный ритм. Мишенью малых G-белков оказался потенциал-зависимый K+ канал Kv11.1 или hERG (human Ether-à-go-go-Related Gene), ответственный за быструю составляющую K+ токов задержанного выпрямления IKr [85, 86]. В клетках GH4C1 Rac1 и RhoA опосредуют противоположную регуляцию канала ERG. RhoА ингибирует ионный канал через серин/треониновую протеинкиназу, тогда как Rac активирует канал через серин/треониновую фосфатазу [107]. Было показано, что серин/треониновая фосфатаза PP5 действует как прямой молекулярный эффектор активированного Rac1 [32, 107].
Таким образом, нарушение Rho-опосредованной регуляции ионных каналов может вносить вклад в изменение сердечного ритма и развитие сердечной патологии.
РЕГУЛЯЦИЯ ИОННЫХ КАНАЛОВ МАЛЫМИ G-БЕЛКАМИ RAB СЕМЕЙСТВА
Rab (Ras-like proteins in brain) белки составляют самую большую группу в суперсемействе малых G-белков [26, 122]. В клетках млекопитающих идентифицировано более 60 Rab белков [37, 49, 72]. Это семейство белков в первую очередь связано с различными аспектами регуляции везикулярного транспорта в эндо- и экзоцитозе [20, 30, 35, 122]. Показано участие Rab5 в эндоцитозе, присутствие Rab7 в ранних эндосомах, Rab9, 24 и 27 в поздних эндосомах и лизосомах, Rab4 и 11 в рециркулирующих эндосомах и Rab3 в секреторных пузырьках [51, 67, 132]. Rab ГТФазы используют ГТФ/ГДФ-зависимый механизм переключения для регуляции каждого из четырех основных этапов внутриклеточного транспорта везикул: (i) отпочковывание везикулы от донорной мембраны, (ii) нацеливание везикулы на акцепторную мембрану, (iii) стыковка везикулы и (iv) слияние везикулы с акцепторной мембраной. Процесс почкования в первую очередь регулируется малыми G-белками Arf семейства [109]. Rab белки могут модулировать активность различных ионных каналов, участвуя в распределении каналов между компартментами в клетке и на цитоплазматической мембране [88]. Rab ГТФазы активируются по крайней мере четырьмя различными типами Rab GEFs (подсемейства, имеющие домен Vps9 или домен DENN, комплекс TRAPP и Sec2) и инактивируются Rab GAPs, принадлежащими большому семейству TBC (Tre2/Bub2/Cdc16)-домен содержащих белков [35].
Rab-зависимый везикулярный транспорт представляет собой важный механизм для поддержания надлежащей экспрессии ионных каналов на клеточной поверхности [104]. Van de Graaf и соавторы идентифицировали Rab11a как белок колокализованный с TRPV5 и TRPV6 в Ca2+-транспортирующих эпителиальных клетках почки [114]. Здесь Rab11a и TRPV5 присутствуют в везикулярных структурах, лежащих под апикальной плазматической мембраной [115]. TRPV5 и TRPV6 преимущественно взаимодействуют с Rab11a в ГТФ-связанном состоянии. Коэкспрессия мутантного белка Rab11a, заблокированного в ГДФ-связанном состоянии приводит к значительному снижению входа Ca2+, вызванному уменьшением экспрессии ионных каналов на апикальной поверхности. Это указывает на прямую роль Rab11a в доставке TRPV5 и TRPV6 к цитоплазматической мембране. Было установлено, что сайтом взаимодействия с Rab11a является участок С-концевой спирали TRPV5, содержащий 5 аминокислотных остатков в положении 595–601. Более того, этот участок является консервативным среди всех идентифицированных видов TRPV5 и TRPV6. Другие Rab ГТФазы Rab7 и Rab22b не демонстрируют эффектов на TRPV5 и TRPV6, что указывает на специфичность взаимодействия TRPV5 и TRPV6 с Rab11a [114]. Вероятно, что эпителиальные Ca2+ каналы TRPV5 и TRPV6 доставляются на цитоплазматическую мембрану посредством Rab11а-зависимого везикулярного транспорта.
Rab3, Rab4 и Rab27a ингибируют активность ENaC посредством белок-белковых взаимодействий [89, 90]. Rab27a ингибирует функцию ENaC через сложный механизм, включающий Munc13–4 (первичный фактор экзоцитоза) и синаптотагминподобный белок SLP-5 [91]. Экспрессия доминантно-негативных Rab11a и Rab11b значительно снижают цAMФ-стимулируемый Na+ ток опосредованный каналом ENaC в клетках в линии клеток mpkCCDc14 (cortical collecting duct), выделенных из собирательных трубочек почки мыши [17]. Аналогично, Rab27a и Rab4 снижают активность регулятора трансмембранной проводимости при муковисцидозе CFTR (cystic fibrosis transmembrane conductance regulator), хлоридного канала на апикальной поверхности эпителия дыхательных путей, кишечника и половых путей [92, 93]. Rab5 регулирует перемещение CFTR с цитоплазматической мембраны в ранние эндосомы. Из ранних эндосом CFTR транспортируется обратно в цитоплазматическую мембрану посредством рециркуляции эндосом контролируемой белками Rab11 и Rme1 [74]. Rab7 контролирует перемещение CFTR из ранних эндосом в поздние эндосомы, а также облегчает доставку CFTR в лизосомы. Rab9 направляет CFTR из поздних эндосом в транс-сеть Гольджи, откуда CFTR повторно через секреторный путь доставляется на цитоплазматическую мембрану. Таким образом, белки Rab играют важнейшую роль в рециркуляции CFTR между клеточной поверхностью и аппаратом Гольджи посредством регуляции везикулярного транспорта (рис. 3 ). Наиболее распространенная мутация в гене CFTR, делеция остатка фенилаланина ΔF508, вызывающая муковисцидоз, нарушает правильную укладку и доставку белка в клеточную мемрану [33, 108]. Исследования Rab-зависимой регуляции транспортировки CFTR показали, что ингибирование Rab5-зависимой интернализации, либо активация Rab11-зависимой стадии рециркуляции приводили к увеличению пула ΔF508-CFTR на цитоплазматической мембране, демонстрируя физиологическую важность регуляции с помощью малых G-белков [33].
В сердце Rab11b был идентифицирован как фактор, регулирующий плотность Ca2+ канала L-типа Cav1.2 на клеточной поверхности [6]. Rab11b экспрессируется в миокарде желудочков сердца; экспрессия доминантно-негативной формы Rab11b-S25N увеличивала пиковый Ca2+ ток L-типа на 98% в неонатальных кардиомиоцитах крысы и на 64% в клетках НЕК-293. Для Rab25 была показана колокализация с Ca2+ каналом Cav1.2 в гладкомышечных клетках церебральных артерий крысы. Нокдаун Rab25 снижал плотность плотность Ca2+ тока в результате протеосомальной и лизосомальной деградации [4]. Таким образом Rab белки играют заметную роль в регуляции работы сердца.
ЗАКЛЮЧЕНИЕ
Исследования последних десятилетий значительно расширили наши представления о роли малых G-белков в регуляции активности ионных каналов. В данной ситуации малые G-белки выступают в качестве посредников в передаче сигналов от мембранных клеточных рецепторов различных регуляторных факторов и медиаторов. Дисбаланс в такой передаче может быть связан с развитием патологии. Поэтому малые G-белки оказываются в фокусе внимания исследований различных патологических состояний, ассоциированных с дисфункцией ионных каналов, таких как сердечные аритмии, почечные и церебральные заболевания. Малые G-белки и инициируемые ими сигнальные каскады могут рассматриваться как потенциальные мишени при разработке терапевтических стратегий.
Работа выполнена при финансовой поддержке РФФИ в рамках научного проекта № 18-315-20049, а также Гранта Президента Российской Федерации по государственной поддержке ведущих научных школ Российской Федерации НШ-5508.2018.7 (соглашение № 075-15-2019-161 от 23.05.2019)
Список литературы
Ahearn I.M., Haigis K., Bar-Sagi D., Philips M.R. Regulating the regulator: post-translational modification of RAS // Nat. Rev. Mol. Cell Biol. 2011. V. 13. P. 39–51. https://doi.org/10.1038/nrm3255
Alvarez de la Rosa D., Canessa C.M., Fyfe G.K., Zhang P. Structure and regulation of amiloride-sensitive sodium channels // Annu. Rev. Physiol. 2000. V. 62. P. 573–594. https://doi.org/10.1146/annurev.physiol.62.1.573
Ashcroft F.M. Ion channels and Disease. 2000. San Diego, CA: Academic Press, pp. 1502.
Bannister J.P., Bulley S., Leo M.D. et al. Rab25 influences functional Cav1.2 channel surface expression in arterial smooth muscle cells // Am. J. Physiol. Cell Physiol. 2016. P. 310. № 11. P. 885–93. https://doi.org/10.1152/ajpcell.00345.2015
Beguin P., Nagashima K., Gonoi T. et al. Regulation of Ca2+ channel expression at the cell surface by the small G-protein kir/Gem // Nature. 2001. V. 411. P. 701–706. https://doi.org/10.1038/35079621
Best J.M., Foell J.D., Buss C.R. et al. Small GTPase Rab11b regulates degradation of surface membrane L-type Cav1.2 channels // Am. J. Physiol. Cell Physiol. 2011. V. 300. P. 1023–1033. https://doi.org/10.1152/ajpcell.00288.2010
Bezzerides V.J., Ramsey I.S., Kotecha S. et al. Rapid vesicular translocation and insertion of TRP channels // Nat. Cell Biol. 2004. V. 6. P. 709–720. https://doi.org/10.1038/ncb1150
Bhattacharya M., Babwah A.V., Ferguson S.S. Small GTP-binding protein-coupled receptors // Biochem. Soc. Trans. 2004. V. 32. P. 1040–1044. https://doi.org/10.1042/BST0321040
Bishop A.L., Hall A. Rho GTPases and their effector proteins // Biochem. J. 2000. V. 348. P. 241–255. https://doi.org/10.1042/0264-6021:3480241
Bliss J.M., Venkatesh B., Colicelli J. The RIN family of Ras effectors // Methods Enzymol. 2005. V. 407. P. 335–344. https://doi.org/10.1016/S0076-6879(05)07028-X
Bokoch G.M., Vlahos C.J., Wang Y. et al. Rac GTPase interacts specifically with phosphatidylinositol 3-kinase // Biochem. J. 1996. V. 315. P. 775–779. https://doi.org/10.1042/bj3150775
Boureux A., Vignal E., Faure S., Fort P. Evolution of the Rho family of Ras-like GTPases in eukaryotes // Mol. Biol. Evol. 2007. V. 24. P. 203–216. https://doi.org/10.1093/ molbev/msl145
Bourne H.R., Sanders D.A., Mccormick F. The GTPase superfamily: conserved structure and molecular mechanism // Nature. 1991. V. 349. P. 117–127. doi: 10. 1038/349117a0.
Boyer S.B., Slesinger P.A., Jones S.V. Regulation of Kir2.1 channels by the Rho-GTPase, Rac1 // J. Cell Physiol. 2009. V. 218. P. 385–393. https://doi.org/10.1002/jcp.21610
Buday L., Downward J. Many faces of Ras activation // Biochim. Biophys. Acta. 2008. V. 1786. P. 178–187. https://doi.org/10.1016/j.bbcan.2008.05.001
Bustelo X.R., Sauzeau V., Berenjeno I.M. GTP-binding proteins of the Rho/Rac family: regulation, effectors and functions in vivo // Bioessays. 2007. V. 29. P. 356–370. https://doi.org/10.1002/bies.20558
Butterworth M.B., Edinger R.S., Silvis M.R. et al. Rab11b regulates the trafficking and recycling of the epithelial sodium channel (ENaC) // Am. J. Physiol. Renal. Physiol. 2012. 302(5): F581–F590. https://doi.org/10.1152/ajprenal.00304.2011
Cachero T.G., Morielli A.D., Peralta E.G. The Small GTP-binding protein RhoA regulates a delayed rectifier potassium channel // Cell. 1998. V. 93. P. 1077–1085. https://doi.org/10.1016/S0092-8674(00)81212-X
Chen H., Puhl H.L., Niu S.L. et al. Expression of Rem2, an RGK family small GTPase, reduces N-type calcium current without affecting channel surface density // J. Neurosci. 2005. V. 25. P. 9762–9772. https://doi.org/10.1523/JNEUROSCI.3111-05.2005
Cherfils J., Zeghouf M. Regulation of Small GTPases by GEFs, GAPs, and GDIs // Physiol. Rev. 2013. V. 93. P. 269–309. https://doi.org/10.1152/physrev.00003.2012
Cingolani L.A., Goda Y. Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy // Nat. Rev. Neurosci. 2008. V. 9. P. 344–356. https://doi.org/10.1038/nrn2373
Claing A. Beta-Arrestins: Modulators of small GTPase activation and function // Prog. Mol. Biol. Transl. Sci. 2013. V. 118. P. 149–174. https://doi.org/10.1016/B978-0-12-394440-5.00006-1
Colicelli J. Human RAS superfamily proteins and related GTPases // Sci. STKE. 2004. V. 2004. RE13. https://doi.org/10.1126/stke.2502004re13
Cox A.D., Der C.J. Ras history: the saga continues // Small GTPases. 2010. V. 1. P. 2–27. https://doi.org/10.4161/sgtp.1.1.12178
Dickson B., Sprenger F., Morrison D., Hafen E. Raf functions downstream of Ras1 in the Sevenless signal transduction pathway // Nature. 1992. V. 360. P. 600–603. https://doi.org/10.1038/360600a0
Diekmann Y., Seixas E., Gouw M. et al. Thousands of Rab GTPases for the cell biologist // PLoS Comput. Biol. 2011. V. 7. e1002217. doi: 02217.https://doi.org/10.1371/journal.pcbi.10
Dulong C., Fang Y.J., Gest C. et al. The small GTPase RhoA regulates the expression and function of the sodium channel Nav1.5 in breast cancer cells // Int. J. Oncol. 2014. 44(2): 539–47. https://doi.org/10.3892/ijo.2013.2214
Finlin B.S., Correll R.N., Pang C. et al. Analysis of the complex between Ca2+ channel beta-subunit and the Rem GTPase // J. Biol. Chem. 2006. V. 281. P. 23557–23566. https://doi.org/10.1074/jbc.M604867200
Finlin B.S., Crump S.M., Satin J., Andres D.A. Regulation of voltage-gated calcium channel activity by the Rem and Rad GTPases // Proc. Natl. Acad. Sci. USA. 2003. V. 100. P. 14469–14474. https://doi.org/10.1073/pnas.2437756100
Gao Y., Wilson G.R., Stephenson S.E.M. et al. The emerging role of Rab GTPases in the pathogenesis of Parkinson’s disease // Mov. Disord. 2018. V. 33. P. 196–207. doi: 27270.https://doi.org/10.1002/mds
Garcia-Mata R., Boulter E., Burridge K. The “invisible hand”: regulation of RHO GTPases by RHOGDIs // Nat. Rev. Mol. Cell Biol. 2011. V. 12. P. 493–504. https://doi.org/10.1038/nrm3153
Gentile S., Darden T., Erxleben C. et al. Rac GTPase signaling through the PP5 protein phosphatase // Proc. Natl. Acad. Sci. USA. 2006. V. 103. P. 5202–5206. https://doi.org/10.1073/pnas.0600080103
Gentzsch M., Chang X.B., Cui L. et al. Endocytic trafficking routes of wild type and DeltaF508 cystic fibrosis transmembrane conductance regulator // Mol. Biol. Cell. 2004. V. 15. P. 2684–2696. https://doi.org/10.1091/mbc.e04-03-0176
Giovannardi S., Forlani G., Balestrini M. et al. Modulation of the inward rectifier potassium channel IRK1 by the Ras signaling pathway // J. Biol. Chem. 2002. V. 277. P. 12158–12163. https://doi.org/10.1074/jbc.M110466200
Goitre L., Trapani E., Trabalzini L., Retta S.F. The Ras superfamily of small GTPases: the unlocked secrets // Methods Mol. Biol. 2014. V. 1120. P. 1–18. https://doi.org/10.1007/978-1-62703-791-4_1
Grewal S.S., Horgan A.M., York R.D. et al. Neuronal calcium activates a Rap1 and B-Raf signaling pathway via the cyclic adenosine monophosphate-dependent protein kinase // J. Biol. Chem. 2000. V. 275. P. 3722–3728. https://doi.org/10.1074/jbc.275.5.3722
Grosshans B.L., Ortiz D., Novick P. Rabs and their effectors: achieving specificity in membrane traffic // Proc. Natl. Acad. Sci. USA. 2006. V. 103. P. 11821–11827. https://doi.org/10.1073/pnas.0601617103
Hahnel C., Gottmann K., Wittinghofer A., Lux H.D. p21ras oncogene protein selectively increases low-voltage-activated Ca2+ current density in embryonic chick dorsal root ganglion neurons // Eur. J. Neurosci. 1992. V. 4. P. 361–368. https://doi.org/10.1111/j.1460-9568.1992.tb00883.x
Hall A. Rho family GTPases // Biochem. Soc. Trans. 2012. V. 40. P. 1378–1382. https://doi.org/10.1042/BST20120103
Hawes B.E., van Biesen T., Koch W.J. et al. Distinct pathways of Gi- and Gq-mediated mitogen-activated protein kinase activation // J. Biol. Chem. 1995. V. 270. P. 17148–17153. https://doi.org/10.1074/jbc.270.29.17148
Heasman S.J., Ridley A.J. Mammalian Rho GTPases: new insights into their functions from in vivo studies // Nat. Rev. Mol. Cell Biol. 2008. V. 9. P. 690–701. https://doi.org/10.1038/nrm2476
Hendron E., Stockand J.D. Activation of mitogen-activated protein kinase (mitogen-activated protein kinase/extracellular signal-regulated kinase) cascade by aldosterone // Mol. Biol. Cell. 2002. V. 13. P. 3042–3054. https://doi.org/10.1091/mbc.e02-05-0260
Ho P.D., Fan J.S., Hayes N.L. et al. Ras reduces L-type calcium channel current in cardiac myocytes. Corrective effects of L-channels and SERCA2 on [Ca2+]i regulation and cell morphology // Circ. Res. 2001. V. 88. P. 63–69. https://doi.org/10.1161/01.res.88.1.63
Hofer F., Fields S., Schneider C., Martin G.S. Activated Ras interacts with the Ral guanine nucleotide dissociation stimulator // Proc. Natl. Acad. Sci. USA. 1994. V. 91. P. 11089–11093. https://doi.org/10.1073/pnas.91.23.11089
Howe L.R., Marshall C.J. Lysophosphatidic acid stimulates mitogen activated protein kinase activation via a G-protein-coupled pathway requiring p21ras and p74raf-1 // J. Biol. Chem. 1993. V. 268. P. 20717–20720.
Imamura Y., Matsumoto N., Kondo S. et al. Effects of ras and Rap1 on electrical excitability of differentiated NG108–15 cells // Neuroscience. 2004. V. 127. P. 973–981. https://doi.org/10.1016/j.neuroscience.2004.05.051
Imamura Y., Matsumoto N., Kondo S. et al. Possible involvement of Rap1 and Ras in glutamatergic synaptic transmission // Neuroreport. 2003. V. 14. P. 1203–1207. https://doi.org/10.1097/00001756-200307010-00003
Jones S.V. Role of the small GTPase Rho in modulation of the inwardly rectifying potassium channel Kir2.1 // Mol. Pharmacol. 2003. V. 64. P. 987–993. https://doi.org/10.1124/mol.64.4.987
Jordens I., Marsman M., Kuijl C., Neefjes J. Rab proteins, connecting transport and vesicle fusion // Traffic. 2005. V. 6. P. 1070–1077. https://doi.org/10.1111/j.1600-0854.2005.00336.x
Kanda Y., Watanabe Y. Adrenaline increases glucose transport via a Rap1-p38MAPK pathway in rat vascular smooth muscle cells // Br. J. Pharmacol. 2007. V. 151. P. 476–482. https://doi.org/10.1038/sj.bjp.0707247
Kawasaki M., Nakayama K., Wakatsuki S. Membrane recruitment of effector proteins by Arf and Rab GTPases // Curr. Opin. Struct. Biol. 2005. V. 15. P. 681–689. https://doi.org/10.1016/j.sbi.2005.10.015
Keating M., Atkinson D., Dunn C. et al. Linkage of a cardiac arrhythmia, the long QT syndrome, and the Harvey ras-1 gene // Science. 1991. V. 252. P. 704–706. https://doi.org/10.1126/science.1673802
Kharlap M.S., Goriunova L.E., Timofeeva A.V. et al. Gene expression analysis in myocytes of right atrial appendages in patients with atrial fibrillation using cDNA microarray technique // Kardiologiia. 2008. V. 48. P. 34 – 42.
Klussmann E., Tamma G., Lorenz D. et al. An inhibitory role of Rho in the vasopressin-mediated translocation of aquaporin-2 into cell membranes of renal principal cells // J. Biol. Chem. 2001. V. 276. P. 20451–20457. https://doi.org/10.1074/jbc.M010270200
Karpushev A.V., Levchenko V., Ilatovskaya D.V. et al. Novel role of Rac1/WAVE signaling mechanism in regulation of the epithelial Na+ channel // Hypertension. 2011. V. 57. P. 996–1002. https://doi.org/10.1161/HYPERTENSIONAHA.110.157784
Lange-Carter C.A., Johnson G.L. Ras-dependent growth factor regulation of MEK kinase in PC12 cells // Science. 1994. V. 265. P. 1458–1461. https://doi.org/10.1126/ science.8073291
Lawson C.D., Ridley A.J. Rho GTPase signaling complexes in cell migration and invasion // J. Cell Biol. 2018. V. 217. P. 447–457. https://doi.org/10.1083/jcb.201612069
Levay M., Settleman J., Ligeti E. Regulation of the substrate preference of p190RhoGAP by protein kinase C-mediated phosphorylation of a phospholipid binding site // Biochemistry. 2009. V. 48. P. 8615–8623. https://doi.org/10.1021/bi900667y
Li L., Matsuoka I., Suzuki Y. et al. Inhibitory effect of fluvastatin on lysophosphatidylcholine-induced nonselective cation current in guinea pig ventricular myocytes // Mol. Pharmacol. 2002. V. 62. P. 602–607. https://doi.org/10.1124/mol.62.3.602
Ligeti E., Dagher M.C., Hernandez S.E. et al. Phospholipids can switch the GTPase substrate preference of a GTPase-activating protein // J. Biol. Chem. 2004. V. 279. P. 5055–5058. https://doi.org/10.1074/jbc.C300547200
Ligeti E., Welti S., Scheffzek K. Inhibition and termination of physiological responses by GTPase activating proteins // Physiol. Rev. 2012. V. 92. P. 237–272. https://doi.org/10.1152/physrev.00045.2010
Liu W.N., Yan M., Chan A.M. A thirty-year quest for a role of R-Ras in cancer: from an oncogene to a multitasking GTPase // Cancer Lett. 2017. V. 403. P. 59–65. https://doi.org/10.1016/j.canlet.2017.06.003
Loirand G., Scalbert E., Bril A., Pacaud P. Rho exchange factors in the cardiovascular system // Curr. Opin. Pharmacol. 2008. V. 8. P. 174–180. https://doi.org/10.1016/j.coph.2007.12.006
Loirand G., Pacaud P. The role of Rho protein signaling in hypertension // Nat. Rev. Cardiol. 2010. V. 7. P. 637–647. https://doi.org/10.1038/nrcardio.2010.136
Lopez De J.M., Stope M.B., Oude Weernink P.A. et al. Cyclic AMP-dependent and Epac-mediated activation of R-Ras by G protein-coupled receptors leads to phospholipase D stimulation // J. Biol. Chem. 2006. V. 281. P. 21837–21847. https://doi.org/10.1074/jbc.M604156200
Madaule P., Axel R. A novel Ras-related gene family // Cell. 1985. V. 41. P. 31–40. https://doi.org/10.1016/0092-8674(85)90058-3
Markgraf D.F., Peplowska K., Ungermann C. Rab cascades and tethering factors in the endomembrane system // FEBS Lett. 2007. V. 581. P. 2125–2130. https://doi.org/10.1016/j.febslet.2007.01.090
Martin G.A., Yatani A., Clark R. et al. GAP domains responsible for ras p21-dependent inhibition of muscarinic atrial K channel currents // Science. 1992. V. 255. P. 192–194. https://doi.org/10.1126/science.1553544
Mastroberardino L., Spindler B., Forster I. et al. Ras pathway activates epithelial Na+ channel and decreases its surface expression in Xenopus oocytes // Mol. Biol. Cell. 1998. V. 9. P. 3417–3427. https://doi.org/10.1091/mbc.9.12.3417
Murata M., Cingolani E., McDonald A.D. et al. Creation of a genetic calcium channel blocker by targeted gem gene transfer in the heart // Circ. Res. V. 95. P. 398–405. https://doi.org/10.1161/01.RES.0000138449.85324.c5
Narumiya S., Thumkeo D. Rho signaling research: history, current status and future directions // FEBS Lett. 2018. V. 592. P. 1763–1776. https://doi.org/10.1002/1873-3468.13087
Pereira-Leal J.B., Seabra M.C. Evolution of the Rab family of small GTP-binding proteins // J. Mol. Biol. 2001. V. 313. P. 889–901. https://doi.org/10.1006/jmbi.2001.5072
Peru Y. Colón de Portugal R.L., Acevedo S.F., Rodan A.R. et al. Adult neuronal Arf6 controls ethanol-induced behavior with Arfaptin downstream of Rac1 and RhoGAP18B // J. Neurosci. 2012. V. 32. P. 17706–17713. https://doi.org/10.1523/jneurosci.1944-12
Picciano J.A., Ameen N., Grant B.D., Bradbury N.A. Rme-1 regulates the recycling of the cystic fibrosis transmembrane conductance regulator // Am. J. Physiol. Cell Physiol. 2003. V. 285. P. 1009–1018. https://doi.org/10.1152/ajpcell.00140.2003
Pochynyuk O., Medina J., Gamper N. et al. Rapid translocation and insertion of the epithelial Na+ channel in response to RhoA signaling // J. Biol. Chem. 2006. V. 281. P. 26520–26527. https://doi.org/10.1074/jbc.M603716200
Pochynyuk O.M., Zheleznova N.N., Medina J.L. et al. Regulation of epithelial sodium channels (ENaC) by small G-proteins // Biol. Membr. 2006. V. 23. P. 382–393.
Pochynyuk O., Stockand J.D., Staruschenko A. Ion channel regulation by Ras, Rho, and Rab small GTPases // Exp. Biol. Med. 2007. V. 232. P. 1258–1265. https://doi.org/10.3181/0703-MR-76
Pochynyuk O., Tong Q., Staruschenko A. et al. Regulation of the epithelial Na+ channel (ENaC) by phosphatidylinositides // Am. J. Physiol. Renal. Physiol. 2006. V. 290. P. 949–957. https://doi.org/10.1152/ajprenal.00386.2005
Reiner D.J., Lundquist E.A. Small GTPases // WormBook. 2018. Aug 16. V. 2018. P. 1–65. https://doi.org/10.1895/wormbook.1.67.2
Repasky G.A., Chenette E.J., Der C.J. Renewing the conspiracy theory debate: does Raf function alone to mediate Ras oncogenesis? // Trends Cell Biol. 2004. V. 14. P. 639–647. https://doi.org/10.1016/j.tcb.2004.09.014
Rojas A.M., Fuentes G., Rausell A., Valencia A. The Ras protein superfamily: evolutionary tree and role of conserved amino acids // J. Cell Biol. 2012. V. 196. P. 189–201. https://doi.org/10.1083/jcb.201103008
Ruan H., Mitchell S., Vainoriene M. et al. Gi alpha 1-mediated cardiac electrophysiological remodeling and arrhythmia in hypertrophic cardiomyopathy // Circulation. 2007. V. 116. P. 596–605. https://doi.org/10.1161/CIRCULATIONAHA.106.682773
Runnels L.W., Yue L., Clapham D.E. The TRPM7 channel is inactivated by PIP(2) hydrolysis // Nat. Cell Biol. 2002. V. 4. P. 329–336. https://doi.org/10.1038/ncb781
Sah V.P., Minamisawa S., Tam S.P. et al. Cardiac-specific overexpression of RhoA results in sinus and atrioventricular nodal dysfunction and contractile failure // J. Clin. Invest. 1999. V. 103. P. 1627–1634. https://doi.org/10.1172/JCI6842
Sanguinetti M.C., Jiang C., Curran M.E. et al. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel // Cell. 1995. V. 81. P. 299–307. https://doi.org/10.1016/0092-8674(95)90340-2
Sanguinetti M.C., Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia // Nature. 2006. V. 440. P. 463–469. https://doi.org/10.1038/nature04710
Santarius M., Lee C.H., Anderson R.A. Supervised membrane swimming: small G-protein lifeguards regulate PIPK signalling and monitor intracellular PtdIns(4,5)P2 pools // Biochem. J. 2006. V. 398. P. 1–13. https://doi.org/10.1042/BJ20060565
Saxena S.K., Kaur S. Regulation of epithelial ion channels by Rab GTPases // Biochem. Biophys. Res. Commun. 2006. V. 351. P. 582–587. https://doi.org/10.1016/j.bbrc.2006.10.087
Saxena S.K., Singh M., Shibata H. et al. Rab4 GTP/GDP modulates amiloride-sensitive sodium channel (ENaC) function in colonic epithelia // Biochem. Biophys. Res. Commun.2006. V. 340. P. 726–733. https://doi.org/10.1016/j.bbrc.2005.12.036
Saxena S., Singh M., Engisch K. et al. Rab proteins regulate epithelial sodium channel activity in colonic epithelial HT-29 cells // Biochem. Biophys. Res. Commun. 2005. V. 337. P. 1219–1223. https://doi.org/10.1016/j.bbrc.2005.09.186
Saxena S.K., Horiuchi H., Fukuda M. Rab27a regulates epithelial sodium channel (ENaC) activity through synaptotagmin-like protein (SLP-5) and Munc13–4 effector mechanism // Biochem. Biophys. Res. Commun. 2006. V. 344. P. 651–657. https://doi.org/10.1016/j.bbrc.2006.03.160
Saxena S.K., Kaur S. Rab27a negatively regulates CFTR chloride channel function in colonic epithelia: involvement of the effector proteins in the regulatory mechanism // Biochem. Biophys. Res. Commun. 2006. V. 346. P. 259–267. https://doi.org/10.1016/j.bbrc.2006.05.102
Saxena S.K., Kaur S., George C. Rab4GTPase modulates CFTR function by impairing channel expression at plasma membrane // Biochem. Biophys. Res. Commun. 2006. V. 341. P. 184–191. https://doi.org/10.1016/j.bbrc.2005.12.170
Schild L. The epithelial sodium channel: from molecule to disease // Rev. Physiol. Biochem. Pharmacol. 2004. V. 151. P. 93–107. https://doi.org/10.1007/s10254-004-0023-7
Schmidt A., Hall A. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch // Genes Dev. 2002. V. 16. P. 1587–1609. https://doi.org/10.1101/gad.1003302
Shaw R.J., Cantley L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth // Nature. 2006. V. 441. P. 424–430. https://doi.org/10.1038/nature04869
Snyder P.M. Regulation of epithelial Na+ channel trafficking // Endocrinology. 2005. V. 146. P. 5079–5085. https://doi.org/10.1210/en.2005-0894
Spiering D., Hodgson L. Dynamics of the Rho-family small GTPases in actin regulation and motility // Cell Adhes. Migr. 2011. V. 5. P. 170–180. https://doi.org/10.4161/cam.5.2.14403
Spindler B., Mastroberardino L., Custer M., Verrey F. Characterization of early aldosterone-induced RNAs identified in A6 kidney epithelia // Pflugers. Arch. Eur. J. Physiol. 1997. V. 434. P. 323–331. https://doi.org/10.1007/s004240050403
Spindler B., Verrey F. Aldosterone action: induction of p21ras and fra-2 and transcription-independent decrease in myc, jun, and fos // Am. J. Physiol. Cell Physiol. 1999. V. 276. P. 1154–1161. https://doi.org/10.1152/ajpcell.1999.276.5.C1154
Staruschenko A., Patel P., Tong Q. et al. Ras activates the epithelial Na(+) channel through phosphoinositide 3-OH kinase signaling // J. Biol. Chem. 2004. V. 279. P. 37771–37778. https://doi.org/10.1074/jbc.M402176200
Staruschenko A., Pochynyuk O.M., Tong Q., Stockand J.D. Ras couples phosphoinositide 3-OH kinase to the epithelial Na+ channel // Biochim. Biophys. Acta. 2005. V. 1669. P. 108–115. https://doi.org/10.1016/j.bbamem.2005.01.005
Staruschenko A., Nichols A., Medina J.L. et al. Rho small GTPases activate the epithelial Na(+) channel // J. Biol. Chem. 2004. V. 279. P. 49989–49994. https://doi.org/10.1074/jbc.M409812200
Stenmark H. Rab GTPases as coordinators of vesicle traffic // Nat. Rev. Mol. Cell Biol. 2009. V. 10. P. 513–525. https://doi.org/10.1038/nrm2728
Stockand J.D., Spier B.J., Worrell R.T. et al. Regulation of Na+ reabsorption by the aldosterone-induced small G protein K-Ras2A // J. Biol. Chem. 1999. V. 274. P. 35449–35454. https://doi.org/10.1074/jbc.274.50.35449
Stockand J.D. New ideas about aldosterone signaling in epithelia // Am. J. Physiol. Renal. Physiol. 2002. V. 282. P. 559–576. https://doi.org/10.1152/ajprenal.00320.2001
Storey N.M., O’Bryan J.P., Armstrong D.L. Rac and Rho mediate opposing hormonal regulation of the ether-a-go-go-related potassium channel // Curr. Biol. 2002. V. 12. P. 27–33. https://doi.org/10.1016/s0960-9822(01)00625-x
Swiatecka-Urban A., Brown A., Moreau-Marquis S. et al. The short apical membrane half-life of rescued {Delta}F508-cystic fibrosis transmembrane conductance regulator (CFTR) results from accelerated endocytosis of {Delta}F508-CFTR in polarized human airway epithelial cells // J. Biol. Chem. 2005. V. 280. P. 36762–36772. https://doi.org/10.1074/jbc.M508944200
Takai Y., Sasaki T., Matozaki T. Small GTP-binding proteins // Physiol. Rev. 2001. V. 81. № 1. P. 153–208. https://doi.org/10.1152/physrev.2001.81.1.153
Tamma G., Klussmann E., Maric K. et al. Rho inhibits cAMP-induced translocation of aquaporin-2 into the apical membrane of renal cells // Am. J. Physiol. Renal. Physiol. 2001. V. 281. P. 1092–1101. https://doi.org/10.1152/ajprenal.0091.2001
Tamma G., Klussmann E., Procino G. et al. cAMP-induced AQP2 translocation is associated with RhoA inhibition through RhoA phosphorylation and interaction with RhoGDI // J. Cell. Sci. 2003. V. 116. P. 1519–1525. https://doi.org/10.1242/jcs.00355
Tolias K.F., Cantley L.C., Carpenter C.L. Rho family GTPases bind to phosphoinositide kinases // J. Biol. Chem. 1995. V. 270. P. 17656–17659. https://doi.org/10.1074/jbc.270.30.17656
Vigil D., Cherfils J., Rossman K.L., Der C.J. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? // Nat. Rev. Cancer 2010. V. 10. P. 842–857. https://doi.org/10.1038/nrc2960
van de Graaf S.F., Chang Q., Mensenkamp A.R. et al. Direct interaction with Rab11a targets the epithelial Ca2+ channels TRPV5 and TRPV6 to the plasma membrane // Mol. Cell. Biol. 2006. V. 26. P. 303–312. https://doi.org/10.1128/MCB.26.1.303-312.2006
van de Graaf S.F., Hoenderop J.G., Bindels R.J. Regulation of TRPV5 and TRPV6 by associated proteins // Am. J. Physiol. Renal Physiol. 2006. V. 290. F. 1295–1302. https://doi.org/10.1152/ajprenal.00443.2005
Wang G., Zhu X., Xie W. et al. Rad as a novel regulator of excitation-contraction coupling and beta-adrenergic signaling in heart // Circ. Res. 2010. V. 106. P. 317–327. https://doi.org/10.1161/CIRCRESAHA.109.208272
Ward Y., Spinelli B., Quon M.J. et al. Phosphorylation of critical serine residues in gem separates cytoskeletal reorganization from down-regulation of calcium channel activity // Mol. Cell Biol. 2004. V. 24. P. 651–661. https://doi.org/10.1128/mcb.24.2.651-661.2004
Watabe-Uchida M., Govek E.E., Van Aelst L. Regulators of Rho GTPases in neuronal development // J. Neurosci. 2006. V. 26. P. 10633–10635. https://doi.org/10.1523/ jneurosci.4084-06.2006
Weernink P.A., Meletiadis K., Hommeltenberg S. et al. Activation of type I phosphatidylinositol 4-phosphate 5-kinase isoforms by the Rho GTPases, RhoA, Rac1, and Cdc42 // J. Biol. Chem. 2004. V. 279. P. 7840–7849. https://doi.org/10.1074/jbc.M312737200
Weernink P.A., Schulte P., Guo Y. et al. Stimulation of phosphatidylinositol-4-phosphate 5-kinase by Rho-kinase // J. Biol. Chem. 2000. V. 275. P. 10168–10174. https://doi.org/10.1074/jbc.275.14.10168
Wei L., Taffet G.E., Khoury D.S. et al. Disruption of Rho signaling results in progressive atrioventricular conduction defects while ventricular function remains preserved // FASEB J. 2004. V. 18. P. 857–859. https://doi.org/10.1096/fj.03-0664fje
Wennerberg K., Rossman K.L., Der C.J. The Ras superfamily at a glance // J. Cell Sci. 2005. V. 118. P. 843–846. https://doi.org/10.1242/jcs.01660
Wilk-Blaszczak M.A., Singer W.D., Quill T. et al. The monomeric G-proteins Rac1 and/or Cdc42 are required for the inhibition of voltage-dependent calcium current by bradykinin // J. Neurosci. 1997. V. 17. P. 4094–4100. https://doi.org/10.1523/JNEUROSCI.17-11-04094.1997
Xu X., Marx S.O., Colecraft H.M. Molecular mechanisms, and selective pharmacological rescue, of Rem-inhibited CaV1.2 channels in heart // Circ. Res. 2010. V. 107. P. 620–630. https://doi.org/10.1161/CIRCRESAHA.110.224717
Yada H., Murata M., Shimoda K. et al. Dominant negative suppression of Rad leads to QT prolongation and causes ventricular arrhythmias via modulation of L-type Ca2 channels in the heart // Circ. Res. 2007. V. 101. P. 69–77. https://doi.org/10.1161/CIRCRESAHA.106.146399
Yang S.A., Carpenter C.L., Abrams C.S. Rho and Rho-kinase mediate thrombin-induced phosphatidylinositol 4-phosphate 5-kinase trafficking in platelets // J. Biol. Chem. 2004. V. 279. P. 42331–42336. https://doi.org/10.1074/jbc.M404335200
Yatani A., Okabe K., Polakis P. et al. ras p21 and GAP inhibit coupling of muscarinic receptors to atrial K+ channels // Cell. 1990. V. 61. P. 769–776. https://doi.org/10.1016/0092-8674(90)90187-j
Yatani A., Quilliam L.A., Brown A.M., Bokoch G.M. Rap1A antagonizes the ability of Ras and Ras-Gap to inhibit muscarinic K+ channels // J. Biol. Chem. 1991. V. 266. P. 22222–22226.
Yatani A., Irie K., Otani T. et al. RhoA GTPase regulates L-type Ca2+ currents in cardiac myocytes // Am. J. Physiol. Heart Circ. Physiol. 2005. V. 288. H. 650–659. https://doi.org/10.1152/ajpheart.00268.2004
Ye X., Carew T.J. Small G protein signaling in neuronal plasticity and memory formation: the specific role of ras family proteins // Neuron. 2010. V. 68. P. 340–361. https://doi.org/10.1016/j.neuron.2010.09.013
Zamboni V., Jones R., Umbach A. et al. Rho GTPases in intellectual disability: from genetics to therapeutic opportunities // Int. J. Mol. Sci. 2018. V. 19. pii: E1821. https://doi.org/10.3390/ijms19061821
Zerial M., McBride H. Rab proteins as membrane organizers // Nat. Rev. Mol. Cell Biol. 2001. V. 2. P. 107–117. https://doi.org/10.1038/35052055
Zheng Y., Bagrodia S., Cerione R.A. Activation of phosphoinositide 3-kinase activity by Cdc42Hs binding to p85 // J. Biol. Chem. 1994. V. 269. P. 18727–18730.
Zhu J.J., Qin Y., Zhao M. et al. Ras and Rap control AMPA receptor trafficking during synaptic plasticity // Cell. 2002. V. 110. P. 443–455. https://doi.org/10.1016/s0092-8674(02)00897-8
Дополнительные материалы отсутствуют.
Инструменты
Успехи физиологических наук