

Успехи физиологических наук, 2020, T. 51, № 2, стр. 88-104
Белок клото – универсальный регулятор физиологических процессов в организме
А. А. Нестерова a, *, Е. Ю. Глинка b, **, И. Н. Тюренков c, ***, В. Н. Перфилова c, ****
a Пятигорский медико-фармацевтический институт – филиал Волгоградского государственного медицинского университета
г. Пятигорск, Россия
b Keenan Research Centre, St. Michael’s Hospital
Торонто, Канада
c Волгоградский государственный медицинский университет
г. Волгоград, Россия
* E-mail: aanesterova2013@gmail.com
** E-mail: pyat57@rogers.com
*** E-mail: fibfuv@mail.ru
**** E-mail: vnperfilova@mail.ru
Поступила в редакцию 03.09.2019
После доработки 23.12.2019
Принята к публикации 23.12.2019
Аннотация
Клото – белок, экспрессия которого ассоциируется с увеличением продолжительности жизни и положительными эффектами при различных заболеваниях, включая онкологические. Учитывая вовлеченность клото в многочисленные сигнальные цепи и его широкие регуляторные функции, изучение данного белка представляется весьма перспективным. В обзоре представлен развернутый анализ его функциональной активности и ее регуляции, рассматривается структура белка, посттрансляционная модификация как механизм регуляции его активности, лиганды и рецепторы растворимого и мембран-связанного клото, клото-зависимые сигнальные пути, а также возможности индукции экспрессии белка и перспективы использования этих регулирующих механизмов в медицине.
ИСТОРИЯ ОТКРЫТИЯ БЕЛКА КЛОТО
В науке нередко бывают случайные открытия, но они дают новое понимание многих физиологических и патологических феноменов. К таким открытиям можно отнести белок, случайно обнаруженный при исследовании трансгенных мышей, экспрессирующих натрий-водородный обменник, когда японскими исследователями непреднамеренно был поврежден локус близлежащего гена. Это привело к развитию клинических признаков, напоминающих раннее старение, которые проявлялись атрофией кожи, замедлением роста, уменьшением массы тела и множественными изменениями (поражениями) внутренних органов: инволюцией тимуса, гипогонадизмом, бесплодием, кальцификацией сосудов и мягких тканей, атеросклерозом, остеопорозом, легочной эмфиземой, нейродегенерацией. У животных отмечалось значительное сокращение продолжительности жизни, а при усиленной экспрессии данного гена, наоборот, ее увеличение, что побудило назвать его антивозрастным геном Klotho по имени греческой богини Клото (дочери Зевса), плетущей нити жизни [5, 6, 50, 52, 53].
Позже было обнаружено, что существуют три изоформы белка клото (БК), α, β и γ [42, 43, 53] (рис. 1).
Рис. 1.
Изоформы белка клото: α-клото, включает растворимые и мембран-связанную изоформы; β- и γ–мембран-связанные. Обозначения: KL1 и KL2 – эктодомены (темные прямоугольники) трансмембранный и цитоплазматический домены (белые прямоугольник).
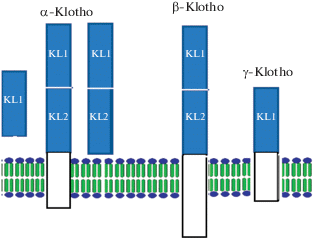
Первая из них экспрессируется в основном в почках и, в меньшей степени, в головном мозге, поджелудочной железе, вторая локализуется в печени, обе изоформы можно обнаружить в некоторых других органах, γ-клото найден в почках, бурой жировой ткани и структурах глаза [43, 60, 103].
Исследователями отмечается крайне незначительный уровень экспрессии β-изоформы этого белка. В литературе обычно α-клото именуется просто “клото” (кlotho), без упоминания изоформы, аббревиатура его гена – KL. Для обозначения β-клото, как белка, так и гена, используется полное название изоформы – “β-klotho” (KLB). Третий член семейства кодируется Lctl (Lactase-like gene), это лактазоподобный белок (Lactase-likeprotein) с альтернативным названием клото/лактаза-флоризин родственный гидролазе белок (Klotho/lactase-phlorizin hydrolase-related protein) (рис. 1).
Межвидовые различия в структуре гена, кодирующего БК, незначительны: нуклеотидные последовательности человека и мыши совпадают на 98% [95].
Учитывая вовлеченность БК в многочисленные сигнальные цепи и его широкие регуляторные функции, изучение данного белка представляется весьма перспективным.
В настоящем обзоре рассматриваются вопросы, связанные со строением белка, пост-трансляционной модификацией как механизмом регуляции его активности, характеристикой лигандов растворимого и мембран-связанного БК, клото-зависимым сигнальными путями, а также возможностями индукции экспрессии белка и перспективами использования этих регулирующих механизмов в медицине.
СТРУКТУРА БЕЛКА И ЕГО ПОСТ-ТРАНСЛЯЦИОННАЯ МОДИФИКАЦИЯ
Ген KL БК человека найден на 13 хромосоме, ген β-клото (KLB)- на 4, γ-клото – на 15. Первичные структуры соответствующих этим генам белков весьма сходны между собой. Все изоформы белка локализованы в плазматической мембране, которую пересекают один раз. Кроме того, некоторое количество БК обнаружено в цитоплазме [64, 84].
α-Клото (далее, клото или БК) человека состоит из 1012 аминокислот. Цитоплазматический домен включает 10 аминокислот, его структура не предполагает способности связываться с внутриклеточными адапторными белками. Этот домен не обладает и ферментативной (например, киназной) активностью. Вследствие этого БК, по-видимому, не является подлинным рецептором, так как не может самостоятельно передавать внешний сигнал вторичным мессенджерам. Этот вывод сделан на основании анализа первичной структуры белка по аналогии с известными белками. Известно, что БК взаимодействует с некоторыми классическими рецепторами и нерецепторными мембран-связанными молекулами. Физиологическую роль этих взаимодействий мы обсудим ниже.
Внеклеточный домен молекулы БК образуют аминокислоты с 34 по 981. Он состоит из двух частей, КЛ1 (аминокислоты 51-506) и КЛ2 (аминокислоты 515–953), которые обладают сходством между собой, однако между ними существуют некоторые функциональные различия [59].
Размер β-клото человека составляет 1044 аминокислоты. Его внеклеточный домен состоит из 996 аминокислот, цитоплазматический – из 27. Подобно БК, цитоплазматический домен β-изоформы также не взаимодействует с известными вторичными мессенджерами. KL1 и KL2 состоят из аминокислот 77–508 и 517–967 соответственно.
Внеклеточные домены обеих изоформ содержат структурные мотивы близкие к структуре β-гликозидазы, однако отсутствие остатка глутамата в гипотетическом активном центре делает эту ферментативную активность маловероятной. Рекомбинантные белки, экспрессированные с этих генов, действительно не обладают гликозидазной активностью, однако нельзя исключить возможности того, что в комплексе с какими-либо дополнительными белками и другими факторами, или вследствие пост-трансляционной модификации эта активность может проявиться.
Некоторые авторы рассматривают ферментативную активность БК в качестве одного из механизмов его действия [41, 92], хотя при этом они не обсуждают вопрос об отсутствии остатка глутамата в гипотетическом активном центре или о взаимодействии БК с какими-либо коферментами, считая гликозидазную/сиалидазную активность растворимого белка экспериментально доказанной [1, 39]. Способность БК гидролизовать стероидные глюкурониды была продемонстрирована Tohiama et al. [88].
Протеолитическая пост-трансляционная модификация ведет к появлению циркулирующих (растворимых) форм клото, состоящих из длинного (КЛ1 и КЛ2) или короткого (КЛ1) фрагментов, в дополнение к их мембран-связанным формам (рис. 2), что является важным с точки зрения функциональных различий. Протеолизу подвергаются участки полипептидной цепи между трансмембранным и внеклеточным доменами, а также между КЛ1 и КЛ2. Мембран-связанные секретазы АДАМ10 (аббревиатура: A Disintegrin And Metalloprotease 10, α-секретаза) и АДАМ17 (он же TACE, аббревиатура: Tumor necrosis factor alpha-converting enzyme) отщепляют внеклеточную часть целиком и дополнительно разрезают ее между КЛ1 и КЛ2 [17], образуя белковые фрагменты с молекулярной массой около 130 и 70 kD (рис. 2). Высвобожденный белок поступает в кровоток и доставляется к органам, которые сами его не экспрессируют, оказывая гормоноподобное воздействие [21].
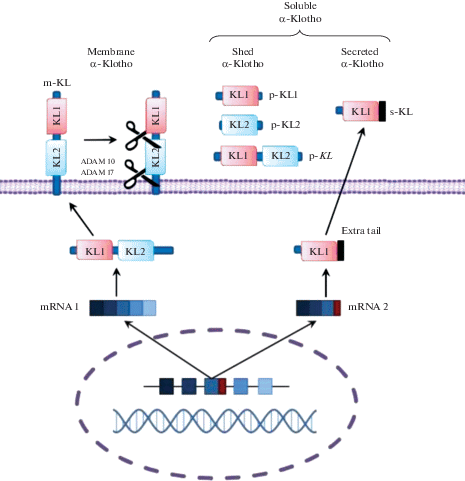
И короткая, содержащая только КЛ1, и длинная, объединяющая КЛ1 и КЛ2 формы БК обнаружены в плазме крови, спинномозговой жидкости и моче [73].
Кроме этого, постулируется существование укороченной секретируемой формы с молекулярным весом около 65 кД, содержащей КЛ1 домен и уникальный мотив из 10 аминокислот на конце. Возможно, что эта форма БК образуется в результате альтернативного сплайсинга мРНК, но ее реальное существование оспаривается некоторыми исследователями [69] (рис. 2).
Протеолитическая активность секретаз или шеддаз, “срезающих” клото с поверхности, играет важную роль в тканевом балансе между мембран-связанной и растворимой формами. Например, в клетках поджелудочной железы около 95% синтезированного клото отщепляется мембранными секретазами АДАМ10 и АДАМ17. Естественно предположить, что эффекты растворимого БК здесь могут быть более значительными, чем его мембран-связанной формы [75]. Это особенно существенно, так как эти две формы имеют функциональные различия: так, утверждается, что только мембран-связанный, но не растворимый БК взаимодействует с FGFRI (fibroblast growth factor receptor I) [50].
В свою очередь, активность АДАМ10 и АДАМ17 регулируется на уровне их экспрессии, а также посредством пост-трансляционной модификации и связыванием с различными факторами, такими как мелатонин, NAD-зависимая гистон деацетилаза SIRT1, факторы транскрипции PPARγ (Peroxisome proliferator-activated receptor gamma) и PAX2 (Paired Box 2 ). Например, доказана их активация при фосфорилировании протеинкиназами PI3K (phospho-inositide 3-kinase), Akt (protein kinase B) и mTOR, индуцируемыми инсулином или инсулиноподобным фактором роста 1 (Insulin-like growth factor 1, IGF-1) [16]. АДАМ10 и АДАМ17 по-разному реагируют на внешние стимулы [101]. Детальный анализ факторов, регулирующих активность секретаз, дается в обзоре Rubinek et al., 2016 [76].
Таким образом, наличие мембран-связанной и свободной форм клото регулируется определенными факторами и не является постоянной величиной, а способность клеток реагировать на внешние стимулы, такие как FGF23, зависит как от присутствия мембран-связанного клото, так и от активности секретаз [28].
Помимо протеолитической модификации, БК и его β-изоформа могут быть подвержены также фосфорилированию, ацетилированию, гликозилированию. По-видимому, эти модификации имеют значение для функционирования белка, но их роль еще не изучена.
Концентрация БК в плазме крови человека в норме составляет в среднем 572.4 пкг/мл (с вероятностью 95% находится в интервале 541.9–604.6 пкг/мл) и снижается до 476.9 пкг/мл (с вероятностью 95% находится в интервале 416.9–545.5 пкг/мл) при сахарном диабете 2 типа, осложненном нефропатией. Уровень клото в моче в норме составляет 59.8 пкг/мг креатинина (с вероятностью 95% находится в интервале 43.6–82.0 пкг/мг креатинина) против 21.0 пкг/мг креатинина (95% доверительный интервал составляет 9.7–45.6 пкг/мг креатинина) при диабете и после 40 лет [55].
КОРРЕЛЯЦИИ МЕЖДУ ЭКСПРЕССИЕЙ КЛОТО И РАЗЛИЧНЫМИ ЗАБОЛЕВАНИЯМИ У ЧЕЛОВЕКА
БК синтезируется не во всех органах и тканях человека [95]. Его экспрессия динамически регулируется в зависимости от внешних факторов, возраста и наличия заболеваний. В силу этого, механизмы регуляции синтеза БК представляют большой интерес в медицине. Результаты клинических исследований, посвященных изучению БК, показывают, что значительное количество патологических состояний сопровождается снижением его синтеза. К ним, в первую очередь, относятся нефропатии различного происхождения и вызывающие их первичные заболевания, такие как гипертония и сахарный диабет [4, 79, 82, 105]. Авторами публикаций подчеркивается, что хронические болезни почек вызывают интоксикацию уремическими токсинами, которые в свою очередь, подавляют синтез БК [3, 55, 79, 99, 105]. Фиброзные изменения в тубулярном почечном эпителии, вызванные заболеваниями почек, сопровождаются падением как локальной экспрессии в тканях пораженной почки, так и снижением уровня этого белка в крови [78].
Подобная тенденция в снижении уровня синтеза БК наблюдалась и при таком остром воспалительном заболевании, как колит [85, 87]. Кроме этого выявлено замедление заживления ран, атрофия кожи у экспериментальных мышей с дефицитом БК, похожая на возрастную атрофию кожи человека [97]. Другое исследование выявило связанное с возрастом замедление экспрессии БК в хрусталике глаза человека, что связывают с гиперметилированием гена KL [45]. Онкологические заболевания [7, 8, 56, 80], рахит [13], окислительный стресс [20], ишемия/ реперфузия [38], гипоксия, сахарный диабет [20, 82], гиперхолистеринемия [79], нейродегенеративные процессы в центральной нервной системе и сетчатке [2, 8, 27, 96] – все эти разнообразные патологические состояния сопровождаются снижением или потерей экспрессии KL. При этом неясно, является ли ингибирование экспрессии только следствием заболевания или снижение уровня этого белка имеет отношение к патогенезу патологических состояний [54].
В феномене супрессии БК участвуют разные уровни регуляции: от влияния на факторы транскрипции до эпигенетических механизмов, включая гиперметилирование промоторного участка KL и экспрессию микроРНК, связывающуюся с 3'-UTR мРНК БК и подавляющую его трансляцию [18]. Молекулярные механизмы, ассоциированные с каждой из упомянутых патологий, сопровождающихся подавлением синтеза БК, изучены далеко не во всех случаях. Кроме того, присутствие функционально-активного белка в клетках зависит не только от его экспрессии, но и от пост-трансляционной модификации, а также деградации. Вероятно, что скорость деградации БК в лизосомах или протеасомах зависит от внешних воздействий, но этот вопрос пока остается неизученным.
В силу всего сказанного выше, механизмы, регулирующие экспрессию БК, приобретают особый интерес.
ФАКТОРЫ ТРАНСКРИПЦИИ, РЕГУЛИРУЮЩИЕ ЭКСПРЕССИЮ KL, И ИХ СВЯЗЬ С ПАТОЛОГИЧЕСКИМИ СОСТОЯНИЯМИ
Промоторный участок KL состоит из 500 нуклеотидов. Из них первые 300 связывают факторы, подавляющие экспрессию, а последующие 200 – активирующие ее. Факторы транскрипции, которые взаимодействуют с промоторным участком KL и участвуют в регуляции его экспрессии, включают активирующий протеин 1 (Activator protein 1, AP-1), глюкокортикоидные рецепторы (glucocorticoid receptor, GR), p53, рецепторы, активируемые пролифераторами пероксисом (Peroxisome proliferator-activated receptor γ1 and γ2, PPAR-γ1, PPAR-γ)2, Sp1 (specificity protein 1). Упомянутые выше факторы могут взаимодействовать друг с другом. Более детально эти вопросы рассмотрены в обзоре Xu et al., 2015 [95].
Внешние воздействия также регулируют экспрессию KL. Так, обработка клеток линии NRK-52E (normal rat kidney) эпителия почечных канальцев крыс ангиотензином 2 снижала экспрессию фактора транскрипции Sp1 in vitro и, видимо, вследствие этого, уменьшалась экспрессия KL. Необходимо отметить, что обработка ангиотензином 2 одновременно приводила к повышению экспрессии TGF-β1 (Transforming growth factor beta 1), p-p38 и p53. Таким образом, нельзя исключить, что в наблюдаемом подавлении экспрессии KL участвовали и другие факторы, например, p53 [23, 105], или TGF-β1 путем активирования фактора транскрипции SMAD3 [78]. Интересно, что гипотензивные препараты фосиноприл и валсартан предотвращали описанные эффекты ангиотензина 2 и усиливали экспрессию KL, одновременно снимая симптомы почечной недостаточности у экспериментальных животных [105]. Фиброзные изменения, являющиеся морфологическим проявлением нефропатии, также сопровождаются снижением экспрессии KL [78]. Многие исследователи полагают, что синтез мРНК, кодирующей БК, в почечном эпителии подавляется множественными факторами, в том числе, как было указано выше, TGF-β1 и зависимым от него фактором транскрипции SMAD3 [89].
Избирательный нокаут гена FGFR1 в тубулярном почечном эпителии отрицательно сказывается на экспрессии KL. Интересно, что введение рекомбинантного белка FGF23 (Fibroblast growth factor 23) также подавляет синтез БК, что указывает на наличие регуляторной связи между рецептором (FGFR1, Fibroblast growth factor receptor 1), ко-рецептором (клото) и лигандом (FGF23), которая играет важную роль в патогенезе гипертонии [33].
Другой патогенный механизм, гиперхолестеринемия, также подавляет экспрессию KL клетками эпителия почечных канальцев. Окисленные липопротеины низкой плотности (ЛПНП), поглощаясь клетками эпителия, активируют протеинкиназу ERK (extracellular-signal-regulated kinase) и ядерный фактор транскрипции “каппа-би” (nuclear factor kappa-light-chain-enhancer of activated B cells, NF-κB), который, связываясь с промоторным участком KL, может блокировать его экспрессию [89], как указывалось выше. Ингибирование образования ЛПНП восстанавливало экспрессию KL [79].
К тому же результату приводят медиаторы воспаления (Interferon gamma IFN-γ, Tumor necrosis factor alpha, TNF-α), поступающие в кровь при колите. Они активируют NF-κB и снижают концентрацию БК в крови за счет подавления его синтеза в кишечном эпителии на уровне транскрипции [87]. Помимо этого, IFN-γ активирует NO-синтазу, и образующийся оксид азота (NO) также снижает синтез БК. Аналогичным образом действует и другой провоспалительный цитокин – IL-12 (Interleukin -12) [65].
С другой стороны, FGF21, лиганд β-клото, подавляет секрецию провоспалительных цитокинов и активирует выброс противовоспалительного IL-10 макрофагами [100]. Таким образом, прослеживается отрицательная обратная связь между экспрессией клото и провоспалительными цитокинами.
По-видимому, любые процессы, активирующие NF-κB, могут подавлять синтез БК. Так, например, дефицит витамина D сопровождается избыточной активностью NF-κB и низким уровнем БК в крови [34, 40]. В свою очередь, БК, ингибируя киназы, активирующие NF-κB, предотвращает его ядерную транслокацию, и, соответственно, активацию.
В CD4 лимфоцитах пациентов с ревматоидным артритом также обнаружен низкий уровень БК. Оказалось, что показатель близок к уровню этого белка в лимфоцитах здоровых пожилых людей. Такое изменение коррелирует с подавлением синтеза ZNF334 (zinc finger protein 334), регулирующего экспрессию ряда генов, включая KL [85]. Можно заключить, что понижение экспрессии KL является общим свойством многих воспалительных процессов.
Кроме того, процессы старения характеризуются снижением общего уровня циркулирующего и локального (в отдельных органах) БК, на что указывают многочисленные публикации [25, 51, 54, 77].
ЭПИГЕНЕТИЧЕСКИЙ КОНТРОЛЬ ЭКСПРЕССИИ KL
Помимо описанной выше генетической регуляции существенную роль играет также эпигенетический контроль синтеза БК. Считается, что этот вид регуляции отвечает за молчание ряда генов во многих органах. Низкий уровень БК или полное отсутствие его экспрессии обнаружены в раковых клетках. В ряде случаев эти клетки используют эпигенетический контроль, активируя посредством разных механизмов метилирование CpG островков в промоторном участке KL [56]. Известно также, что и старение сопровождается усилением метилирования этих же участков ДНК [19].
К настоящему времени выявлено, что микроРНК могут подавлять экспрессию генов, ускоряя деградацию или дестабилизируя мРНК, а также ингибируя трансляцию. Существуют данные, доказывающие, что микроРНК способны индуцировать модификацию гистонов и метилирование ДНК [35]. Всего описано девять моделей в соответствие с которыми микроРНК могут регулировать синтез белка [71].
Анализ 3'-нетранслируемого участка мРНК БК (3'-UTR) мышей выявил сайты с высоким сродством к связыванию микроРНК-339, микроРНК-556 и участки с меньшим сродством к взаимодействию с микроРНК-10 и микроРНК-199. Две первых из них непосредственно снижали синтез БК [68]. Другие молекулы, такие как микроРНК-126-5p, также способны ингибировать экспрессию KL [81]. В связи с этим встает вопрос о путях регуляции экспрессии самой микроРНК. В единственной найденной нами работе, исследующей эту проблему применительно к БК, показано, что высокая концентрация глюкозы активирует экспрессию микроРНК-199b-5p, способствуя ацетилированию гистона H3 и усилению его связывания с промоторным участком названной микроРНК, что коррелирует со снижением уровня синтезированного БК [47]. Диабетическая нефропатия также ассоциируется с повышением уровня этой микроРНК и со снижением концентрации БК в крови. Интересно, что препарат, снимающий симптомы нефропатии у мышей, атрасентан, одновременно нормализовал концентрации БК, микроРНК-199b-5p и вызывал деацетилирование гистона H3. Существуют данные, доказывающие, что действие микроРНК-199b-5p связано с NFκB [94].
К настоящему времени регуляция экспрессии самих микроРНК изучена недостаточно, хотя некоторые исследования в этом направлении ведутся. Например, показано, что экспрессия микроРНК-10 зависит от гистон-метилтрансферазы DOT1L [14] и подавляется TGFβ1 [44], а экспрессия микроРНК-126 активируется куркумином [58].
Воздействие микроРНК на экспрессию гена БК может быть противоположно влиянию других факторов, и исход возможен неоднозначный. Поскольку около 40% генов микроРНК содержится внутри других генов, их экспрессия может регулироваться теми же факторами, что и экспрессия самих этих генов. Так, например, экспрессия микроРНК-199, обнаруженная в интроне гена динамина, регулируется теми же механизмами, которые определяют экспрессию DNM1 [9].
Кроме того, появились данные, что экспрессия микроРНК-199 активируется и другими путями, например, окисью азота [10], что создает дополнительные возможности для регуляции и сложности в анализе ожидаемых эффектов. Очевидно, требуются дополнительные исследования для осуществления целенаправленного влияния на экспрессию микроРНК с тем, чтобы таким образом регулировать экспрессию интересующих нас генов.
Необходимо отметить, что присутствие функционально-активного БК в клетке зависит не только от уровня экспрессии его гена и воздействия на него различных факторов и ко-факторов, но и от скорости его деградации. В литературе данных по механизмам утилизации БК и их регулированию мы не обнаружили.
Исходя из вышеизложенного, изучение функционирования БК и его регуляции представляет интерес для клиницистов и фармакологов.
ФАРМАКОЛОГИЧЕСКИЕ СРЕДСТВА, ВЛИЯЮЩИЕ НА ЭКСПРЕССИЮ KL И СИНТЕЗ КЛОТО
В настоящее время не существует лекарственных средств, разработанных специально с этой целью, но показано, что некоторые из них, уже известные, усиливают экспрессию KL или ограничивают снижение синтеза БК при патологических состояниях. К таким средствам относятся агонисты PPARγ [61, 102], тестостерон [37], ресвератрол [36], N-ацетилцистеин [74].
Так, агонисты PPARγ, такие как росиглитазон, усиливали экспрессию БК в клетках меланомы человека, что использовалось в качестве вспомогательной терапии у пожилых пациентов [11].
Другой пример такого воздействия – природное фенольное соединение ресвератрол, который активировал комплекс факторов транскрипции ATF3 (AMP-dependent transcription factor )/c-Jun в клетках почечного эпителия крыс линии NRK-52E и тем самым усиливал экспрессию БК [36].
Нельзя не отметить работы, в которых доказывается, что тестостерон индуцирует экспрессию KL посредством стимуляции андрогенового рецептора [37, 96]. В связи с этими данными, существующая практика лечения рака простаты анти-андрогенными препаратами выглядит спорной, так как, возможно, активность опухоли и ее метастазирование вызывается снижением экспрессии KL и синтеза БК, известного своей способностью подавлять про-онкогенные метаболические пути [70].
Помимо упоминавшихся уже атрасентана [47], гипотензивных средств фосиноприла и валсартана [105], появились данные о том, что деметилирующие агенты снижают репрессию KL [46].
В результате целенаправленного поиска выявлено несколько низкомолекулярных соединений с малой токсичностью, способных усиливать экспрессию KL в культивируемых клетках: Compound G, N-[2-(1-cyclohexen-1-yl)ethyl]-6,7,8,9-tetrahydropyrido[1,2-e]purin-4-amine; Compound H, N-(2-chlorophenyl)-1H-indole-3-carboxamide; Compound I, 2-(1-propyl)amino-11-chlorothiazolo[5,4-a]acridine. Они были выбраны из 500 соединений в результате целевого скрининга [49]. Молекулярные механизмы их действия авторами не обсуждаются, но в рамках нашего обзора вызывают интерес результаты исследования одного из этих соединений, изученного другой группой авторов – “Compound H”, N-(2-chlorophenyl)-1H-indole-3-caboxamide. Установлено, что оно активировало деметилазу и способствовало деметилированию мотивов CpG в промоторном участке KL. В результате, факторы транскрипции Pax4 и Kid3 начинали связываться с KL, регулируя его экспрессию [46].
Среди активаторов экспрессии KL авторы отмечают статины (правастатин), которые стимулировали синтез мРНК клото в почках мыши [98], а также иммуносупрессор рапамицин, который влиял на синтез БК [95], возможно за счет его взаимодействия с протеин-киназой mTOR.
Отдельного внимания заслуживают взаимоотношения между витамином D и экспрессией KL. Витамин D связывается с VDR (Vitamin D-responsive element), локализованными в пределах промоторного участка KL, и активирует его экспрессию [95]. Помимо высокоаффинного кальцитриола, низкоаффинные пищевые лиганды VDR, включая куркумин, полиненасыщенные жирные кислоты и антоцианидины, инициируют, а ресвератрол и SIRT1 потенцируют передачу сигналов на VDR [34]. В свою очередь, БК ингибирует образование активной формы витамина D3 и подавляет его эффекты, такие как поглощение кальция клетками [34].
Из веществ растительного происхождения, селагинеллин, компонент, извлеченный из комнатного растения Saussurea pulvinata (Hooket Grev.) индуцировал экспрессию KL в клетках линии PC12 (феохромоцитома) [90].
Кроме того, повысить уровень циркулирующего растворимого БК возможно путем введения рекомбинантного белка или использования методов генной терапии. Оба эти метода опробованы в экспериментах на животных, но их применение для лечения людей имеет свои ограничения, как юридические, так и финансовые.
Таким образом, приведенные выше данные указывают на некоторые из возможных направлений поисков путей усиления экспрессии БК.
КЛОТО КАК ТЕРАПЕВТИЧЕСКИЙ АГЕНТ
Использование рекомбинантного БК в экспериментах с клеточными культурами или in vivo, а также его стимулированная экспрессия позволили выявить способность клото предотвращать апоптоз [62, 83], фиброзные изменения [32, 57, 86], окислительный стресс [32], пролиферацию раковых клеток [8, 56, 59, 80], старение и атрофию кожи [97], нейродегенерацию и когнитивный дефицит [2], изменения костной системы [55] и др. (табл. 1).
Таблица 1.
Эффекты клото и задействованные сигнальные пути при некоторых патологических состояниях
| Способ введения: генетическая модификация/растворимый белок | Экспериментальное животное | In vivo/in vitro | Патологическое состояние/заболевание | Сигнальные пути, участвующие в реализации эффекта | Полученный эффект | Ссылка |
|---|---|---|---|---|---|---|
| Растворимый белок | Мыши | In vivo | Апоптоз в кардиомиоцитах, вызванный стрессом | Ингибирование JNK, p38 | Подавление апоптоза и стресса эндоплазматического ретикулума | [83] |
| Растворимый белок | Мыши | In vivo | Экспериментальная гипертония, гипертрофия сердца при дефиците клото | FGFR1, ингибирование кальциевого канала TRPC6 | Нормализация артериального давления (АД), предотвращение гипертрофии | [33] |
| Растворимый белок | Мыши, клетки H9c2 и неонатальные кардиомиоциты | In vitro и in vivo | Повреждение сердечной мышцы, вызванное гипергликемией | Подавление окислительного стресса, митохондриальной дисфункции, фиброза, и ингибирование воспаления индуцированного ROS и активацией NF-κB | Предотвращение повреждения сердечной мышцы | [32] |
| Растворимый белок | Мыши db/db (модель диабета 2-го типа) | In vivo | Гипергликемия, повышенное систолическое давление, гипертрофия и фиброз почек | Подавление экспрессии HIF, TGF-β1, TNF-α и фибронектина, фосфорилирования Akt и mTOR в почках, усиление экспрессии супероксид дисмутазы и клото | Нормализация артериального давления, предотвращение фиброза почек | [86] |
| Растворимый белок | Мыши db/db (модель диабета 2-го типа) | In vivo | Гипергликемия | Усиление секреции инсулина, частичная нормализация уровня сахара | [30] | |
| Растворимый белок | Мыши | In vivo | Сахарный диабет, индуцированный введением стрептозотоцина | Предотвращение апоптоза в β-клетках пожелудочной железы | [62] | |
| Растворимый белок | Мыши db/db | In vivo | Сахарный диабет | Дефицит клото сопровождается активацией NF-κB | Дефицит клото сопровождается воспалительными процессами в почках | [104] |
| Генетическая модификация/трансфекция | Мезангиальные клетки человека | In vitro | Гипергликемия | Ингибирует TGFβ1/SMAD3 сигнальный путь, Egr-1 | Подавляет фиброзные процессы | [57] |
| Генетическая модификация | Крысы | In vivo | Сахарный диабет, индуцированный стрептозотоцином | Ингибирует Rho-associated coiled-coil kinase | Предотвращает гипертрофию почек, фиброз почек | [57] |
| Растворимый белок/КЛ1 домен | Мыши | In vivo, in vitro |
Рак поджелудочной железы | Модуляция IGF-I и bFGF сигнальных путей | тормозит пролиферацию раковых клеток in vitro | [8] |
| Растворимый белок/ КЛ1 домен | Иммуно-дефицитные мыши nude | In vivo, in vitro | Рак молочной железы человека | Ингибирует связывание IGF-1 с его рецептором | Тормозит развитие опухолей in vivo и рост раковых клеток в культуре | [59] |
| Генетическая модификация | Мыши | In vivo | Демиелинизация | Ингибирует Akt and ERK | Усиление ремиелинизации | [2] |
| Генетическая модификация | Мыши | In vivo | Когнитивные нарушения, эпилептическая активность | Увеличивает экспрессию GluN2B субъединиц NMDA-рецепторов | Улучшение пространственной памяти, уменьшение частоты эпилептических припадков | [27] |
Обнадеживающие данные об участии БК в механизмах, тормозящих процессы старения, получены несколькими исследовательскими группами. Показано, что недостаток его вызывает такие симптомы старения, как когнитивный дефицит, атрофию кожи, остеопороз [50]. Единая точка зрения относительно механизмов замедления старения БК пока не выработана. Одна из возможностей обеспечивается FGF21, который, связываясь с β-клото и FGFR, увеличивает чувствительность к инсулину и захват глюкозы тканями, тормозит липогенез и ускоряет окисление липидов, активирует АМФ-зависимую киназу, что способствует усилению митохондриального биогенеза и энергетического обмена [77].
Существует также точка зрения, что преждевременное старение при дефиците БК и FGF23 может быть результатом интоксикации витамином D и вызванной ею гиперкальциемии. Роль БК и его взаимодействие с FGF23 детально рассмотрена в обзоре Erben et al., 2018 [29]. Как один из факторов, замедляющих старение, также рассматривается ингибирование сигнального пути IGF1 (insulin like growth factor 1) и инсулина, вызванное БК. Кроме того, различия в продолжительности жизни могут быть связаны с полиморфизмом KL [12].
Высказывается также точка зрения, что БК предотвращает старение клеток за счет снятия блока с клеточного цикла на границе G1/S и подавления пути p53/p21, вызывающего апоптоз [84]. Это наблюдение противоречит данным Glauser and Schlegel, 2007 [31], которые показывают, что FOXO1, активированный под влиянием БК, вызывает остановку клеточного цикла, и это позволяет предотвратить апоптоз, так как дает возможность выиграть время, необходимое для репарации поврежденной ДНК. Можно предположить, что блокада клеточного цикла, индуцированная БК и FOXO1, продолжается какое-то ограниченное время, так как полная его остановка делает пролиферацию невозможной, а ее замедление характерно для процессов старения. Очевидно, противоречие снимается, если учесть, что многие процессы в клетках протекают относительно медленно в определенном временном промежутке. Это позволяет включить антагонистический механизм с запаздыванием, подобно тому, как вслед за импортом факторов транскрипции в ядро через какое-то время происходит их экспорт в цитоплазму. По-видимому, расхождение между процитированными выше публикациями [31, 84] может объясняться временными рамками в постановке эксперимента. В более общем виде можно предположить, что БК вносит вклад в синхронизацию клеточных процессов, но такие работы пока неизвестны.
К другим возможностям относятся FGF-независимые механизмы, которые рассмотрены ниже. Несмотря на обнадеживающие предварительные данные, полученные на экспериментальных животных, пока рано говорить об использовании БК для замедления или предотвращения старения у людей.
СИГНАЛЬНЫЕ МЕХАНИЗМЫ С УЧАСТИЕМ КЛОТО
Воздействие БК на множественные биологически важные процессы осуществляется посредством его участия в ряде сигнальных механизмов. Поскольку и растворимый, и самая значительная часть молекулы мембран-связанного БК находятся во внеклеточном пространстве, то, очевидно, что он должен действовать на клетку через рецептор, опознающий его и передающий сигнал через мембрану, если для передачи сигнала не требуется транслокации БК в цитоплазму. Если же транслокация необходима, то должна существовать специфическая транспортная молекула, переносящая его через мембрану с использованием универсальных клеточных механизмов эндоцитоза (например, клатрин- или кавеолин-зависимых) или пиноцитоза. В этом случае предполагается, что перенесенный в цитоплазму БК может связываться с какими-то адапторными молекулами и непосредственно участвовать в неких сигнальных внутриклеточных механизмах. Третий возможный вариант предполагает взаимодействие БК с лигандом или рецептором лиганда, что может вызвать модуляцию сигнала с этого рецептора. Например, БК необходим для связывания FGF23 с FGFR1. Кроме того, если БК срезает сиаловые кислоты с лиганда или рецептора, это может привести к изменению их конформации или демаскировать связывающий центр.
Значительный экспериментальный материал накоплен в пользу первого и третьего механизмов, хотя очевидно, что классические рецепторные белки тоже могут участвовать в переносе БК, если они сами подвергаются эндоцитозу.
Кроме того, выявлено, что БК может связываться с нерецепторными молекулами на поверхности клеточной мембраны и влиять на их активность [41, 92] или подвергаться трансцитозу [39].
И наконец, внутриклеточный БК также обладает самостоятельной функциональной активностью. Было показано, что он способен регулировать экспрессию генов: взаимодействует с RIG-I и ингибирует стимулируемую им экспрессию цитокинов IL-6 и IL-8, как in vitro так и in vivo, подавляя воспалительные процессы и механизмы старения/возраст-зависимые изменения [64, 84]. Ниже мы подробнее рассмотрим, с какими рецепторными белками способен связываться БК и модулировать сигналы с них. В силу разнообразия рецепторных белков возникают изменения различных внутриклеточных сигнальных путей. Присутствие рецепторов в тех или иных органах и тканях определяет направление этих модуляций.
На сегодняшний день известны белки, которые могут взаимодействовать с БК непосредственно: FGFR1 и FGF23, Wnt, IGF1R (insulin-like growth factor 1 receptor) и TGFβRII (transforming growth factor beta receptor II). Очевидно, что последствия связывания БК с этими белками определяются в значительной мере самими рецепторами и сильно разнятся между собой.
FGF21 и FGF23 принадлежат к эндокринным FGF, наряду с FGF15/19, в отличие от канонических FGF 1-20. Как известно, канонические лиганды нуждаются в гепарине для образования комплекса с рецептором, а эндокринные вместо этого используют изоформы БК. Нами уже упоминалось, что БК необходим для связывания FGF23 с его рецептором FGFR1c [50]. Предполагается, что ко-рецепторную функцию исполняет мембран-связанная форма белка. Переключение специфичности рецептора имеет физиологические последствия: канонические FGF участвуют в дифференцировке клеток в ходе эмбриогенеза, а эндокринный FGF23 регулирует обмен кальция и фосфора и гомеостаз витамина Д. Исследования показали, что COOH-конец молекулы FGF23 связывается с БК, затем они соединяются с рецептором FGFR1c и образуется тройной комплекс [5, 84]. В присутствии БК активация FGFR1 по каноническому пути ингибируется. При этом снижается активность RAS–MAPK, PI3K–Akt, PLCγ (Phosphoinositide phospholipase Cγ) и STAT сигнальных путей, зависимых от FGF1. В частности, подавляются выброс кальция в цитоплазму за счет ингибирования PLCγ и снижения PtdIns-3p (phosphatidylinositol-3-phosphate) и процессы, активируемые PI3K-Akt: пролиферация, подавление апоптоза и дифференцировки, активация CREB, p27, mTOR, ингибирование FOXO, активация секретаз АДАМ10 и АДАМ17 [28]. Как показывают исследования, именно PI3K–Akt путь стимулируется при многих видах опухолевого роста, для которых характерны активная пролиферация, потеря клеточной дифференцировки и подавление экспрессии БК [8, 59]. Замена связывания FGF1 на FGF23 усиливает фосфатурию и подавляет 1-α-гидроксилирование и активацию витамина D. Дефицит БК, напротив, сопровождается гиперфосфатемией и гиперкальциемией. Клото модулирует активность Na, K-ионных каналов и Na, Pi-ко-транспортера, предотвращает апоптоз, вызываемый витамином D, подавляя активность каспазы 3. Более детально внутриклеточные сигнальные пути, зависимые от БК, рассмотрены в обзоре Sopjani et al., 2015 [84].
Клото-зависимое предотвращение апоптоза может быть связано с подавлением активации фактора транскрипции p53, индуцирующего программируемую гибель клетки. Снижение синтеза БК индуцирует преждевременное старение человеческих клеток с одновременной активацией p53 и p21 и остановкой клеточного цикла на уровне G1/S [23], а пролиферативный эффект БК, очевидно, связан со снятием этого блока.
Тройной комплекс FGF21–β-клото–FGFR1 в адипоцитах регулирует ответ на стресс, вызванный голоданием [67]. FGF19 секретируется в кишечнике в ответ на поступление пищи и связывается с β–клото–FGFR4 в печени [72]. Очевидно, в обоих случаях мы имеем дело с взаимодействием белка с рецептором с его последующей модуляцией. Фундаментальный обзор всего обширного семейства FGF, FGFR и их взаимодействия с БК содержится в работе Ornitz, 2015 [72].
Как было показано выше, процессы, активируемые связыванием FGF23 с FGFR1, не могут происходить без участия БК. Местом действия являются кости, основной источник FGF23 и резервуар кальция, и почки, которые выводят избыточные фосфат и кальций. Регуляция их экскреции происходит в почечных канальцах, и там же синтезируется наибольшее количество БК. У мышей, страдающих дефицитом БК и/или FGF23 развивается гиперкальциемия и гиперфосфатемия [84]. β-клото и FGF21, связываясь с FGFR4, участвуют в регуляции энергетического и липидного обмена. Соответственно, β-клото находится в основном в белой жировой ткани и в печени [72].
Второй рецептор, непосредственно связывающий клото, это TGFβRII. TGFβ1 и зависимые от него факторы транскрипции SMAD2 и SMAD3 играют ключевую роль в развитии фиброза. Большое количество работ посвящено роли БК в предотвращении развития фиброза в почках и других органах. Непосредственно связываясь с TGFβRII и, предотвращая связывание TGFβ1 со своим рецепторным комплексом, циркулирующий БК полностью подавляет фосфорилирование SMAD2 и последующую экспрессию профиброзных генов с образованием соответствующих белков, таких как αSMA (α-smooth muscle actin), виментин, коллаген 1 типа [26, 70]. Показано, что в мезангиальных клетках БК ингибирует фактор транскрипции Egr-1 путем подавления TGF-β1-зависимого фактора транскрипции SMAD3 и, вследствие этого, снижается экспрессия коллагена 1 и фибронектина. Таким образом, БК замедляет развитие фиброза в почках [57].
Предполагается также, что БК подавляет синтез фибронектина и виментина посредством альтернативного механизма – за счет ингибирования экспрессии протеин-киназы ROCKI (Rho-associated protein kinase) [24].
Существует точка зрения, что предотвращение фиброза и рака играет ключевую роль в замедлении старения под влиянием БК, и она не связана с общепризнанным механизмом, контролирующим старение- влиянием на длину теломеров [84].
В развитии фиброза принимают участие также Wnt и IGF1 и, соответственно, другие механизмы, независимые от SMAD2 и SMAD3. Существенно, что БК и TGFβ1 взаимно подавляют экспрессию друг друга, а торможение синтеза TGFβ1 также вносит вклад в предотвращение фиброза [84].
Третий из упомянутых выше рецепторов, взаимодействующих с БК, это IGF1R [91]. Прямое связывание клото с этим рецептором доказано их ко-преципитацией [91]. Между клото и IGF1 наблюдается антагонизм: IGF1 активирует, а клото ингибирует пролиферацию раковых клеток человека линий MCF-7 и MDA-MB-231 и препятствует образованию ими колоний. При этом и растворимый, и мембран-связанный БК оказывали одно и то же действие [59, 91]. Связывание клото с IGF1R ингибирует его тирозин-киназную активность и предотвращает связывание IRS (insulin receptor substrate) с PI3K и Akt, подавляя их активацию, вызванную взаимодействием IGF или инсулина с этим рецептором. Соответственно, прерывается каскад реакций фосфорилирования последующих белков-субстратов активированной Akt, таких как C/EBP, FOXO1, mTor, NFκB, IKK (inhibitor of nuclear factor-κB (IκB) kinase), которые в свою очередь активируются или ингибируются вследствие фосфорилирования. Так как Akt фосфорилирует и инактивирует транскрипционный фактор C/EBP, связывающийся с CCAAT последовательностью (CCAAT/enhancer binding protein, C/EBP) и подавляющий развитие опухолей, то, вызванное БК ингибирование Akt, предотвращает инактивацию этого фактора. Соответственно, in vivo с повышением уровня синтеза БК в опухолях их размер уменьшается [84].
Одновременно, вызванное Akt фосфорилирование предотвращает ядерную транслокацию FOXO1 и индуцируемую им экспрессию Mn-супероксид дисмутазы – митохондриального фермента, необходимого для подавления окислительного стресса. FOXO1 и другие члены этого семейства активируют репаративные процессы ДНК, а также инициируют другие защитные и адаптивные механизмы. В частности, FOXO1 замедляет клеточный цикл в поврежденных клетках, позволяя осуществить клеточную репарацию. Показано, что он является важным регулятором энергетического обмена. Мутация обеих копий этого гена вызывает эмбриональную летальность вследствие нарушения васкуляризации [31].
Выше мы обсуждали антагонизм между БК и NF-κB. Как известно, этот фактор транскрипции находится в латентной форме в цитоплазме в виде комплекса со специфическим белковым ингибитором и высвобождается из него вследствие фосфорилирования. В этом процессе могут участвовать различные протеинкиназы, в том числе Akt. После диссоциации латентного комплекса NF-κB транспортируется в ядро и связывается с промоторными участками ряда генов, активируя или подавляя их экспрессию. Установлено, что экзогенный циркулирующий БК или его индуцированная экспрессия предотвращают активацию NF-κB, ингибируя фосфорилирование одного из компонентов этого комплекса – RelA. В результате RelA теряет способность связываться с промоторным участком генов IL-8 и MCP-1 (Monocyte chemoattractant protein 1). Специфическое фосфорилирование Ser536 в RelA указывает на участие Akt в происходящем процессе. Предполагается, что Akt активирует NF-κB фосфорилированием, стимуляцией p38 и рекрутированием киназ IKKa и IKKb, которые специфически фосфорилируют ингибитор NF-κB. Блокируя фосфорилирование Akt, индуцируемое инсулином, БК предотвращает активацию NF-κB [104].
Следует отметить крайне важную роль, которую играет Akt. Она активируется многими факторами роста. Обычно этот путь рассматривают как сигнальный механизм, обеспечивающий выживание, однако в некоторых случаях, вызванное Akt фосфорилирование способствует развитию воспаления, как в случае активации NF-κB, или вызывает другие нежелательные последствия, такие как ингибирование FOXO1, что было рассмотрено нами выше. Таким образом, поскольку БК блокирует сигнальный путь, активируемый инсулином и IGF1, он также может предотвращать ингибирование FOXO1, вызванное фосфорилированием при участии Akt. Стоит отметить антагонистические взаимоотношения, существующие между NFκB и FOXO1, и необходимость баланса между ними, что очень важно, так как воспалительные процессы, окислительный стресс и замедленная репарация ДНК наблюдаются как при многих патологических состояниях, так и при старении [48].
В дополнение к этому, активация Akt влияет на гомеостаз кальция. БК снижает присутствие на клеточной мембране активного ионного канала TRPC6 (transient receptor potential cation channel subfamily C, member 6), вовлеченного в развитие фиброза. Гиперактивация этого канала стимулируется IGF1-зависимой активацией PI3K/Akt сигнального пути, который БК подавляет [70].
Помимо этого, Akt активирует p53/p21-зависимый путь, способствующий процессам старения. Это было показано в экспериментах in vitro на эндотелиальных клетках пупочной вены человека (клеточная линия HUVECs – Human umbilical vein endothelial cells), где БК предотвращал передачу сигнала по этому пути [84].
Другая доказанная мишень БК – сигнальный механизм Wnt. Эта сложная система регулирует множественные процессы в эмбриогенезе и может активироваться при хроническом воспалении. Ее активация коррелирует с разнообразными онкологическими заболеваниями и фиброзными изменениями. Связывая секретируемые лиганды Wnt, БК препятствует активации этого сигнального пути, усиливает деградацию β-катенина, Wnt-зависимого эффекторного белка, и предотвращает экспрессию PAI-1 (plasminogen activator inhibitor 1), фактора для сигнальных путей TGFβ1 и Wnt, отвечающего за экспрессию профиброзных белков [69].
Одновременно при связывании с БК подавляется Wnt/β-катенин-зависимая экспрессия ангиотензина 2 и других белков ренин-ангиотензиновой системы [69]. Отметим, что в данном случае БК связывается с лигандом Wnt, а не с рецептором, в отличие от описанного выше прямого взаимодействия с FGFR и TGFβRII. При дефиците БК наблюдается повышенный уровень β-катенина и PAI-1, а также ангиотензина 2 [70].
Результатами исследований показано, что во многих типах опухолей экспрессия БК подавлена [70]. Установлено, что циркулирующий БК ингибирует пролиферацию и миграцию раковых клеток in vitro, что может быть следствием ингибирования Wnt [70]. Предотвращение миграции и метастазирования под влиянием БК может быть отнесено к ингибированию как Wnt, так и TGFβ1, поскольку они оба вызывают эпителиально-мезенхимальный переход (epithelial-to-mesenchymal transition). Кроме того, постоянная активация Wnt-сигнального пути также ведет к ускоренному старению [84].
Итак, экспериментально доказано, что БК непосредственно взаимодействует с IGF1R (и тем самым ингибирует PI3K/Akt/mTOR сигнальные пути), регулирует Wnt/β-катенин-зависимый механизм, FGF2 сигнальный путь (влияя на ERK (еxtracellular signal-regulated kinase) 1/2) и ингибирует канонический сигнальный механизм TGFβ1–SMAD2/3. Все эти сигнальные механизмы участвуют в онкогенезе, и их подавление посредством БК тормозит рост опухолей и пролиферацию раковых клеток, усиливает их апоптоз, влияет на аутофагию, замедляет миграцию и предотвращает метастазирование [70].
Еще один механизм, посредством которого осуществляется влияние БК на внутриклеточные сигнальные пути, это его связывание с α2-3-сиалил-лактозой в ганглиозидах липидных рафтов, что ведет к инактивации рафт-зависимого сигнального пути и подавлению экзоцитоза кальциевого канала TRPC6 по механизму, не зависящему от FGF23 [93], но вызывающему ингибирование PI3K/Akt [22].
Сиалидазная активность клото способствует удалению сиаловых кислот и обнажению ацетил-глюкозамина, с которым взаимодействует галектин-1. Циркулирующий БК связывает моносиалоганглиозиды и препятствует интернализации селективных потенциал-зависимых кальциевых каналов (TRPV5) и АТФ-зависимых почечных калиевых каналов (ROMK), и приводит к их накоплению на плазматической мембране [41], что способствует регуляции ионного транспорта кальция и калия. В отличие от упомянутых выше механизмов, в которых БК связывается непосредственно с рецептором или его лигандом, в этом случае белок изменяет липидное окружение рецептора или ионного канала и тем самым влияет на его функцию.
Интересные и обнадеживающие данные получены группой исследователей в отношении активности БК в эндокринной ткани поджелудочной железы: БК защищает β-клетки поджелудочной железы от апоптоза при диабете 1 типа, активируя интегрин β1-FAK (focal adhesion kinase)/Akt сигнальный путь, а также предотвращает апоптоз, вызванный стрептозотоцином [62]. Однако, их выводы относительно механизма действия БК посредством активации Akt находятся в противоречии с данными других авторов. Так, на модели диабета 2 типа, мышах db/db, установлено, что экспрессия БК в β-клетках поджелудочной железы снижает внутриклеточную концентрацию супероксида, тормозит апоптоз и стресс эндоплазматического ретикулума, усиливает экспрессию супероксид дисмутазы [63], что указывает скорее на участие FOXO1, а не на активацию интегрин-β1–FAK/Akt-зависимого пути в β-клетках. О подавлении активации Akt в разных тканях под влиянием БК свидетельствуют и данные, рассмотренные выше.
Из других эффектов БК в островках поджелудочной железы авторы отмечают усиление экспрессии Pdx-1 (фактора транскрипции, регулирующего экспрессию инсулина), PCNA (Proliferating cell nuclear antigen) (маркера пролиферации) и LC3 (light chain 3) (маркера аутофагии) [63]. Таким образом, защитная функция клото в клетках поджелудочной железы многогранна и не сводится к единственному сигнальному пути. Это, по-видимому, справедливо и для других органов и тканей.
В кардиомиоцитах БК подавляет стресс эндоплазматического ретикулума, вызванный изопротеренолом, и предотвращает апоптоз, ингибируя p38, MAP, ERK1/2 и JNK [83]. Он также регулирует активацию протеин-киназы C (Protein kinase C, PKC) и уровень кальция в цитоплазме, активирует cAMP/PKA (Protein kinase А) и зависимые от cAMP сигнальные пути, включая CREB (cAMP response element-binding protein), ACE (angiotensin 1-converting enzyme) и деградацию ангиотензина, ингибирует экспрессию Nox2 и NADPH оксидазы, накопление активных форм кислорода и окислительный стресс [84].
Было показано, что БК предотвращает апоптоз в эндотелиальных клетках, стимулируя митоген-активируемые киназы [66]. Интересно, что БК ингибирует фосфорилирование рецептора VEGF (receptor for vascular endothelial growth factor, VEGFRII) и подавляет секрецию VEGF (vascular endothelial growth factor) [84], что способствует предотвращению роста опухолей, учитывая митогенную активность и про-онкогенную роль VEGF.
Таким образом, сигнальные пути, которые непосредственно или косвенно модулируются БК, разнообразны и регулируют процессы, существенные для выживания клетки (рис. 3).
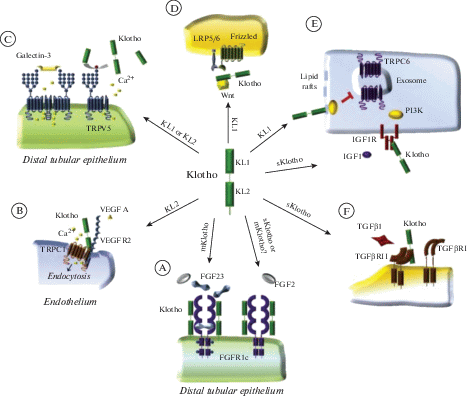
Рис. 3.
Основные сигнальные механизмы с участием белка клото [70]. (A) Мембран-связанный клото образует тройной комплекс с FGFR1c и FGF23, облегчая выведение фосфатов через эпителиальные клетки проксимальных канальцев почек. Связывание клото с FGFR1c уменьшает сродство к FGF2, ингибируя передачу сигналов от FGF2. (B) На эндотелиальных клетках домен KL2 связывается с рецептором 2 сосудистого эндотелиального фактора роста (VEGFR2) и TRPC1, образуя комплекс, который поглощается путем эндоцитоза при стимуляции с помощью VEGF. (С) В эпителии дистальных канальцев почки клото ферментативно модифицирует N-связанный гликан на TRPV5, что позволяет стабилизировать молекулы TRPV5 галектином-3 на клеточной мембране и способствует реабсорбции кальция из мочи. (D) Домен KL1 способен секвестрировать растворимые молекулы Wnt, предотвращая передачу сигналов Wnt. (E) Домен KL1 ингибирует передачу сигналов PI3K путем связывания либо IGF1 с IGFR1, либо моносиалоганглиозидов на липидных рафтах в клеточной мембране. Ингибирование передачи сигналов PI3K и Akt предотвращает экзоцитоз TRPC6 в мембранах кардиомиоцитов и подоцитов и их перегрузку кальцием. (F) Растворимый клото связывается с TGFβRII, снижает сродство к TGFβ1 и препятствует гетеродимеризации TGFβRII и TGFβRI, ингибируя, тем самым, передачу сигналов Smad2/3. Примечание: FGF – fibroblast growth factor; FGFR – fibroblast growth factor receptor; IGF1 – insulin-like growth factor 1; IGFR – insulin-like growth factor receptor; LRP5/6 – low-density lipoprotein receptor-related protein 5/6; mKlotho – membrane-bound Klotho; PI3K – phospho-inositide 3-kinase; sKlotho – soluble Klotho; TGFβ1 – transforming growth factor β1; TGFβR – transforming growth factor β receptor; TRPC1 – transient receptor potential cation channel subfamily C, member 1; TRPC6 – transient receptor potential cation channel, subfamily C, member 6; TRPV5 – transient receptor potential cation channel, subfamily V, member 5; VEGF – vascular endothelial growth factor; VEGFR2 – vascular endothelial growth factor receptor 1; Wnt – wingless-related integration site.
ЗАКЛЮЧЕНИЕ
Разнообразие клото-зависимых терапевтических эффектов соответствует многообразию внутриклеточных сигнальных путей, посредством которых эти эффекты реализуются. Как показали многочисленные исследования, дефицит БК связан с патогенезом многих заболеваний и их осложнений. Экспериментальные данные, полученные в основном на животных и с использованием клеточных культур, указывают на перспективность использования БК для лечения разнообразной патологии. В настоящее время специалистами широко обсуждаются возможности применения фармакологических подходов для повышения уровня экспрессии этого белка in vivo.
Список литературы
Бокша И. С., Прохорова Т.А., Савушкина О.К., Терешкина Е.Б. Белок клото: роль при старении организма и патологии центральной нервной системы // Биохимия. 2017. Т. 82. № 9. С. 1278–1295.
Прохорова Т.А., Бокша И.С., Савушкина О.К. и др. Белок α-клото при нейродегенеративных и психических заболеваниях // Журн. неврологии и психиатрии им. C.C. Корсакова. 2019. Т. 119. № 1. С. 80–88.
Мeльник О.О. Белок Klotho и фактор роста фибробластов Fgf23 как маркеры хронической болезни почек // Почки. 2017. Т. 6. № 3. С. 132–138.
Милованов Ю.С., Милованова Л.Ю., Саблина М.М. и др. Циркулирующая форма белка Klotho – новый ингибитор сосудистой кальцификации при хронической болезни почек // Артериальная гипертензия. 2014. Т. 20. № 6. С. 531–537.
Ильин А.В., Арбузова М.И. Фактор роста фибробластов 23 и белок Klotho в патогенезе вторичного гиперпаратиреоза // Остеопороз и остеопатии. 2013. № 3. С. 20–27.
Тимощенко О.В., Никитин Ю.П. Белок клото и атеросклероз // Атеросклероз. 2017. Т. 13. № 4. С. 38–41.
Мелехин В.В., Макеев О.Г. Ген klotho: современные представления о структуре и функциях. Возможные механизмы противоопухолевого действия // Вестник Уральской медицинской академической науки. 2018. Т. 15. № 3. С. 393–404.
Abramovitz L., Rubinek T., Ligumsky H. et al. KL1 internal repeat mediates Klotho tumor suppressor activities and inhibits bFGF and IGF-I signaling in pancreatic cancer // Clin. Cancer Res. 2011. V. 17. № 13. P. 4254–4266.
Aranda J.F., Canfrán-Duque A., Goedeke L. et al. The miR-199–dynamin regulatory axis controls receptor-mediated endocytosis // J. Cell Sci. 2015. V. 128. P. 3197–3209.
Bai Y.P., Zhang J.X., Sun Q. et al. Induction of microRNA-199 by nitric oxide in endothelial cells is required for nitrovasodilator resistance via Targeting of prostaglandin I2 synthase // Circulation. 2018. V. 138. № 4. P. 397–411.
Behera R., Kaur A., Webster M.R. et al. Inhibition of age-related therapy resistance in melanoma by rosiglitazone-mediated induction of Klotho // Clin. Cancer Res. 2017. V. 23. № 12. P. 3181–3190.
Bonafè M., Olivieri F. Genetic polymorphism in long-lived people: cues for the presence of an insulin/IGF-pathway-dependent network affecting human longevity // Mol. Cell. Endocrinol. 2009. V. 299. № 1. P. 118–123.
Borst O., Münzer P., Schmid E. et al. 1,25(OH)2vitamin D3-dependent inhibition of platelet Ca2+ signaling and thrombus formation in klotho-deficient mice // The FASEB J. 2014. V. 28. № 5. P. 2108–2119.
Bourguignon L.Y.W., Wong G., Shiina M. Up-regulation of histone methyltransferase, DOT1L, by matrix hyaluronan promotes microRNA-10 expression leading to tumor cell invasion and chemoresistance in cancer stem cells from head and neck squamous cell carcinoma // J. Biol. Chem. 2016. V. 291. № 20. P. 10571–10585.
Cararo-Lopes M.M., Mazucanti C.H.Y., Scavone C. et al. The relevance of α-KLOTHO to the central nervous system: Some key questions// Ageing Res. Rev. 2017. V. 36. P. 137–148.
Chen C.D., Podvin S., Gillespie E. et al. Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17 // PNAS. 2007. V. 104. № 50. P. 19796–19801.
Chen C.D., Tung T.Y., Liang J. et al. Identification of cleavage sites leading to the shed form of the anti-aging protein Klotho // Biochemistry. 2014. V. 53. № 34. P. 5579–5587.
Chen C.D., Zeldich E., Yuexuan L. et al. Activation of the anti-aging and cognition-enhancing gene Klotho by CRISPR-dCas9 transcriptional effector complex // J. Mol. Neurosci. 2018. V. 64. № 2. P. 175–184.
Chen K., Sun Z. Activation of DNA demethylases attenuates aging-associated arterial stiffening and hypertension // Aging Cell. 2018. V. 17. № 4. e12762.
Cheng M.F., Chen L.J., Cheng J.T. Decrease of Klotho in the kidney of streptozotocin-induced diabetic rats // J. Biomed. Biotechnol. 2010. V. 2010. P. 513853.
Cui W., Leng B., Liu W., Wang G. Suppression of apoptosis in human umbilical vein endothelial cells (HUVECs) by Klotho protein is associated with reduced endoplasmic reticulum oxidative stress and activation of the PI3K/AKT pathway // Med. Sci. Monit. 2018. V. 24. P. 8489–8499.
Dalton G., An S.W., Al-Juboori S.I. et al. Soluble klotho binds monosialoganglioside to regulate membrane microdomains and growth factor signaling // Proc. Natl. Acad. Sci. USA. 2017. V. 114. № 4. P. 752–757.
de Oliveira R.M. Klotho RNAi induces premature senescence of human cells via a p53/p21 dependent pathway // FEBS Lett. 2006. V. 580. № 24. P. 5753–5758.
Deng M., Luo Y., Li Y. et al. Klotho gene delivery ameliorates renal hypertrophy and fibrosis in streptozotocin-induced diabetic rats by suppressing the Rho-associated coiled-coil kinase signaling pathway // Mol. Med. Rep. 2015. V. 12. № 1. P. 45–54.
Dërmaku-Sopjani M., Kolgeci S., Abazi S., Sopjani M. Significance of the anti-aging protein Klotho // Mol. Membr. Biol. 2013. V. 30. № 8. P. 369–385.
Doi S., Zou Y., Togao O. et al. Klotho inhibits transforming growth factor-beta1 (TGF-beta1) signaling and suppresses renal fibrosis and cancer metastasis in mice // J. Biol. Chem. 2011. V. 286. № 10. P. 8655–8665.
Dubal D.B., Zhu L., Sanchez P.E. et al. Life extension factor Klotho prevents mortality and enhances cognition in hAPP transgenic mice // J. Neurosci. 2015. V. 35. № 6. P. 2358–2371.
Endres K., Deller T. Regulation of Alpha-Secretase ADAM10 In vitro and In vivo: Genetic, Epigenetic, and Protein-Based Mechanisms // Front. Mol. Neurosci. 2017. V. 10. Article 56.
Erben R.G. Update on FGF23 and Klotho signaling // Mol. Cell. Endocrinol. 2016. V. 432. P. 56-65.
Forsberg E.A., Olauson H., Larsson T., Catrina S.B. Effect of systemically increasing human full-length Klotho on glucose metabolism in db/db mice // Diabetes Res. Clin. Pract. 2016. V. 113. P. 208-210.
Glauser D.A., Schlegel W. The emerging role of FOXO transcription factors in pancreatic β cells // J. Endocrinol. 2007. V. 193. № 2. P. 195–207.
Guo Y., Zhuang X., Huang Z. et al. Klotho protects the heart from hyperglycemia-induced injury by inactivating ROS and NF-κB-mediated inflammation both in vitro and in vivo // Biochim. Biophys. Acta Mol. Basis Dis. 2018. V. 1864. № 1. P. 238–251.
Han X., Ross J., Kolumam G. et al. Cardiovascular effects of renal distal tubule deletion of the FGF receptor 1 gene // J. Am. Soc. Nephrol. 2018. V. 29. № 1. P. 69–80.
Haussler M.R., Whitfield G.K., Haussler C.A. et al. 1,25-Dihydroxyvitamin D and Klotho: a tale of two renal hormones coming of age // Vitam. Horm. 2016. V. 100. P. 165–230.
Hawkins P.G., Morris K.V.RNA and transcriptional modulation of gene expression // Cell Cycle. 2008. V. 7. № 5. P. 602–607.
Hsu S.C., Huang S.M., Chen A. et al. Resveratrol increases anti-aging Klotho gene expression via the activating transcription factor 3/c-Jun complex-mediated signaling pathway // Int. J. Biochem. Cell Biol. 2014. V. 53. P. 361–371.
Hsu S.C., Huang S.M., Lin S.H. et al. Testosterone increases renal anti-aging klotho gene expression via the androgen receptor-mediated pathway // Biochem. J. 2014. V. 464. № 2. P. 221-229.
Hu M.C., Shi M., Zhang J. et al. Klotho deficiency is an early biomarker of renal ischemia–reperfusion injury and its replacement is protective // Kidney Int. 2010. V. 78. № 12. P. 1240–1251.
Hu M.C., Shi M., Zhang J. et al. Renal production, uptake, and handling of circulating aKlotho // J. Am. Soc. Nephrol. 2016. V. 27. № 1. P. 79–90.
Hu M.C., Shiizaki K., Kuro-o M., Moe O.W. Fibroblast growth factor 23 and Klotho: physiology and patho-physiology of an endocrine network of mineral metabolism // Annu. Rev. Physiol. 2013. V. 75. P. 503–533.
Huang C.L. Regulation of ion channels by secreted Klotho // Adv. Exp. Med. Biol. 2012. V. 728. P. 100–106.
Ito S., Fujimori T., Hayashizaki Y., Nabeshima Y. Identification of a novel mouse membrane-bound family 1 glycosidase-like protein, which carries an atypical active site structure // Biochim. Biophys. Acta. 2002. V. 1576. № 3. P. 341–345.
Ito S., Kinoshita S., Shiraishi N. et al. Molecular cloning and expression analyses of mouse betaklotho, which encodes a novel Klotho family protein // Mech. Dev. 2000. V. 98. № 1–2. P. 115–119.
Jiajie T., Yanzhou Y., Hoi-Hung A.C. et al. Conserved miR-10 family represses proliferation and induces apoptosis in ovarian granulosa cells // Sci. Rep. 2017. V. 7. P. 41304.
Jin S.L., Zhang Y., Chen Z.H. et al. Epigenetic changes of the Klotho gene in age-related cataracts // Eur. Rev. Med. Pharmacol. Sci. 2015. V. 19. № 14. P. 2544–2553.
Jung D., Xu Y., Sun Z. Induction of anti-aging gene klotho with a small chemical compound that demethylates CpG islands // Oncotarget. 2017. V. 8. № 29. P. 46745–46755.
Kang W.L., Xu G.S. Atrasentan increased the expression of klotho by mediating miR-199b-5p and prevented renal tubular injury in diabetic nephropathy // Sci. Rep. 2016. V. 6. P. 19979.
Kim D.H., Kim J.Y., Yu B.P. et al. The activation of NF-κB through Akt-induced FOXO1 phosphorylation during aging and its modulation by calorie restriction // Biogerontology. 2008. V. 9. № 1. P. 33–47.
King G.D., Chen C.D., Huang M.M. et al. Identification of novel small molecules that elevate Klotho expression // Biochem. J. 2012. V. 441. № 1. P. 453–461.
Kuro-o M. Klotho and aging // Biochim. Biophys. Acta. 2009. V. 1790. № 10. P. 1049–1058.
Kuro-o M. Klotho and the aging process // Korean J. Intern. Med. 2011. V. 26. № 2. P. 113–122.
Kuro-o M., Matsumura Y., Aizawa H. et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing // Nature. 1997. V. 390. № 6555. P. 45–51.
Kurosu H., Yamamoto M., Clark J.D. et al. Suppression of aging in mice by the hormone klotho // Science. 2005. V. 309. № 5742. P. 1829–1833.
Lanske B., Razzaque M.S. Premature aging in klotho mutant mice: cause or consequence? // Ageing Res. Rev. 2007. V. 6. № 1. P. 73–79.
Lee E.Y., Kim S.S., Lee J.S. et al. Soluble a-Klotho as a novel biomarker in the early stage of nephropathy in patients with type 2 diabetes // PLoS One. 2014. V. 9. № 8. e102984.
Lee J., Jeong D.J., Kim J. et al. The anti-aging gene KLOTHO is a novel target for epigenetic silencing in human cervical carcinoma // Mol. Cancer. 2010. V. 9. P. 109.
Li Y., Hu F., Xue M. et al. Klotho down-regulates Egr-1 by inhibiting TGF-β1/Smad3 signaling in high glucose treated human mesangial cells // Biochem. Biophys. Res. Commun. 2017. V. 487. № 2. P. 216–222.
Li Y., Tian L., Sun D., Yin D. Curcumin ameliorates atherosclerosis through upregulation of miR-126 // J. Cell Physiol. 2019. V. 234. № 11. P. 21049–21059.
Ligumsky H., Rubinek T., Merenbakh-Lamin K. et al. Tumor suppressor activity of klotho in breast cancer is revealed by structure-function analysis // Mol. Cancer Res. 2015. V. 13. № 10. P. 1398–1407.
Lim K., Groen A., Molostvov G. et al. α-Klotho expression in human tissues // J. Clin. Endocrinol. Metab. 2015. V. 100. № 10. P. 1308–1318.
Lin W., Zhang Q., Liu L. et al. Klotho restoration via acetylation of peroxisome proliferation-activated receptor gamma reduces the progression of chronic kidney disease // Kidney Int. 2017. V. 92. № 3. P. 669–679.
Lin Y., Sun Z. Antiaging gene Klotho attenuates pancreatic β-Cell apoptosis in type 1 diabetes // Diabetes. 2015. V. 64. № 12. P. 4298–4311.
Lin Y., Sun Z. In vivo pancreatic b-cell-specific expression of antiaging gene Klotho: a novel approach for preserving β-cells in type 2 diabetes // Diabetes. 2015. V. 64. № 4. P. 1444–1458 .
Liu F., Wu S., Ren H., Gu J. Klotho suppresses RIG-I-mediated senescence-associated inflammation // Nat. Cell Biol. 2011. V. 13. № 3. P. 254–262.
Liu Y., Zhang Q. Periodontitis aggravated pancreatic b-cell dysfunction in diabetic mice through interleukin-12 regulation on Klotho // J. Diabetes Investig. 2016. V. 7. № 3. P. 303–311.
Maekawa Y., Ohishi M., Ikushima M. et al. Klotho protein diminishes endothelial apoptosis and senescence via a mitogen-activated kinase pathway // Geriatr. Gerontol. Int. 2011. V. 11. № 4. P. 510–516.
Markan K.R., Naber M.C., Ameka M.K. et al. Circulating FGF21 is liver derived and enhances glucose uptake during refeeding and overfeeding // Diabetes. 2014. V. 63. № 12. P. 4057–4063.
Mehi S.J., Maltare A., Abraham C.R., King G.D. MicroRNA-339 and microRNA-556 regulate Klotho expression in vitro // Age (Dordr). 2014. V. 36. № 1. P. 141–149.
Mencke R., Harms G., Moser J. et al. Human alternative Klotho mRNA is a nonsense-mediated mRNA decay target inefficiently spliced in renal disease // JCI Insight. 2017. V. 2. № 20. pii: 94375.
Mencke R., Olauson H., Hillebrands J.L. Effects of Klotho on fibrosis and cancer: A renal focus on mechanisms and therapeutic strategies // Adv. Drug Deliv. Rev. 2017. V. 121. P. 85–100.
Morozova N., Zinovyev A., Nonne N. et al. Kinetic signatures of microRNA modes of action // RNA. 2012. V. 18. № 9. P. 1635–1655.
Ornitz D.M., Itoh N. The Fibroblast Growth Factor signaling pathway // Wiley Interdiscip. Rev. Dev. Biol. 2015. V. 4. № 3. P. 215–266.
Pavlatou M.G., Remaley A.T., Gold P.W. Klotho: a humeral mediator in CSF and plasma that influences longevity and susceptibility to multiple complex disorders, including depression // Transl. Psychiatry. 2016. V. 6. e876.
Piao S.G., Kang S.H., Lim S.W. et al. Influence of N-acetylcysteine on Klotho expression and its signaling pathway in experimental model of chronic cyclosporine nephropathy in mice // Transplantation. 2013. V. 96. № 2. P. 146–153.
Pru'homme G.J., Glinka Y., Kurt M. et al. The anti-aging protein Klotho is induced by GABA therapy and exerts protective and stimulatory effects on pancreatic beta cells // Biochem. Biophys. Res. Commun. 2017. V. 493. № 4. P. 1542–1547.
Rubinek T., Shahmoon S., Shabtay-Orbach A. et al. Klotho response to treatment with growth hormone and the role of IGF-I as a mediator // Metabolism. 2016. V. 65. № 11. P. 1597–1604.
Salminen A., Kauppinen A., Kaarniranta K. FGF21 activates AMPK signaling: impact on metabolic regulation and the aging process // J. Mol. Med. (Berl). 2017. V. 95. № 2. P. 123–131.
Sanchez-Niño M.D., Sanz A.B., Ortiz A. Klotho to treat kidney fibrosis // J. Am. Soc. Nephrol. 2013. V. 24. № 5. P. 687–689.
Sastre C., Rubio-Navarro A., Buendıá I. et al. Hyperlipidemia-associated renal damage decreases Klotho expression in kidneys from ApoE knockout mice // PLoS One. 2013. V. 8. № 12. e83713.
Seo M., Kim M.S., Jang A. et al. Epigenetic suppression of the anti-aging gene KLOTHO in human prostate cancer cell lines // Anim. Cells Syst. (Seoul). 2017. V. 21. № 4. P. 223–232.
Shibayama Y., Kondo T., Ohya H. et al. Upregulation of microRNA-126-5p is associated with drug resistance to cytarabine and poor prognosis in AML patients // Oncol. Rep. 2015. V. 33. № 5. P. 2176–2182.
So W.Y., Cheng Q., Chen L. et al. High glucose represses β-klotho expression and impairs fibroblast growth factor 21 action in mouse pancreatic islets: involvement of peroxisome proliferator-activated receptor γ signaling // Diabetes. 2013. V. 62. № 11. P. 3751–3759.
Song S., Gao P., Xiao H. et al. Klotho suppresses cardiomyocyte apoptosis in mice with stress-induced cardiac injury via downregulation of endoplasmic reticulum stress // PLoS One. 2013. V. 8. № 12. e82968.
Sopjani M., Rinnerthaler M., Kruja J., Dërmaku-Sopjani M. Intracellular signaling of the aging suppressor protein Klotho // Curr. Mol. Med. 2015. V. 15. № 1. P. 27–37.
Soroczyńska-Cybula M., Bryl E., Smoleńska Z., Witkowski J.M. Varying expression of four genes sharing a common regulatory sequence may differentiate rheumatoid arthritis from ageing effects on the CD4(+) lymphocyte // Immunology. 2011. V. 132. № 1. P. 78–86.
Takenaka T., Kobori H., Miyazaki T. et al. Klotho protein supplementation reduces blood pressure and renal hypertrophy in db/db mice, a model of type 2 diabetes // Acta Physiol. (Oxf). 2018. V. 225. № 2. e13190.
Thurston R.D., Larmonier C.B., Majewski P.M. et al. Downregulation of aging-related Klotho gene in experimental colitis: the role of TNF and IFN-γ // Gastroenterology. 2010. V. 138. № 4. P. 1384–1394.
Tohyama O., Imura A., Iwano A. et al. Klotho is a novel glucuronidase capable of hydrolyzing steroid glucuronides // J. Biol. Chem. 2004. V. 279. № 11. P. 9777–9784.
Turan K., Ata P. Effects of intra- and extracellular factors on anti-aging klotho gene expression // Genet. Mol. Res. 2011. V. 10. № 3. P. 2009–2023.
Wang C.J., Hu C.P., Xu K.P. et al. Protective effect of selaginellin on glutamate-induced cytotoxicity and apoptosis in differentiated PC12 cells // Naunyn. Schmiedebergs. Arch. Pharmacol. 2010. V. 381. № 1. P. 73–81.
Wolf I., Levanon-Cohen S., Bose S. et al. Klotho: a tumor suppressor and a modulator of the IGF-1 and FGF pathways in human breast cancer // Oncogene. 2008. V. 27. № 56. P. 7094–7105.
Wolf M.T., An S.W., Nie M. et al. Klotho up-regulates renal calcium channel transient receptor potential vanilloid 5 (TRPV5) by intra- and extracellular N-glycosylation-dependent mechanisms // J. Biol. Chem. 2014. V. 289. № 52. P. 35849–35857.
Wright J.D., An S.W., Xie J. et al. Soluble klotho regulates TRPC6 calcium signaling via lipid rafts, independent of the FGFR-FGF23 pathway // FASEB J. 2019. V. 33. № 8. P. 9182–9193.
Wu C., Lv C., Chen F. et al. The function of miR-199a-5p/Klotho regulating TLR4/NF-κB p65/NGAL pathways in rat mesangial cells cultured with high glucose and the mechanism // Mol. Cell. Endocrinol. 2015. V. 417. P. 84–93.
Xu Y., Sun Z. Molecular basis of klotho: from gene to function in aging // Endocr. Rev. 2015. V. 36. № 2. P. 174–193.
Xuan N.T., Hai N.V. Changes in expression of klotho affect physiological processes, diseases, and cancer // Iran. J. Basic Med. Sci. 2018. V. 21. № 1. P. 3–8.
Yamauchi M., Hirohashi Y., Torigoe T. et al. Wound healing delays in α- Klotho-deficient mice that have skin appearance similar to that in aged humans – Study of delayed wound healing mechanism // Biochem. Biophys. Res. Commun. 2016. V. 473. № 4. P. 845–852.
Yoon H.E., Lim S.W., Piao S.G. et al. Statin upregulates the expression of klotho, an anti-aging gene, in experimental cyclosporine nephropathy // Nephron Exp. Nephrol. 2012. V. 120. № 4. P. 123–133.
Young G.H., Wu V.C. KLOTHO methylation is linked to uremic toxins and chronic kidney disease // Kidney Int. 2012. V. 81. № 7. P. 611–612.
Yu Y., He J., Li S. et al. Fibroblast growth factor 21 (FGF21) inhibits macrophage-mediated inflammation by activating Nrf2 and suppressing the NF-κB signaling pathway // Int. Immunopharmacol. 2016. V. 38. P. 144–152.
Zhang H., Gao Y., Zhao F.L. et al. Hydrogen sulfide-induced processing of the amyloid precursor protein in SH-SY5Y human neuroblastoma cells involves the PI3-K/Akt signaling pathway // Cell Mol. Neurobiol. 2015. V. 35. № 2. P. 265–272.
Zhang H., Li Y., Fan Y. et al. Klotho is a target gene of PPAR-gamma // Kidney Int. 2008. V. 74. № 6. P. 732–739.
Zhang Y., Wang L., Wu Z. et al. The expressions of klotho family genes in human ocular tissues and in anterior lens capsules of age-related cataract // Current Eye Research. 2017. V. 42. № 6. P. 871–875.
Zhao Y., Banerjee S., Dey N. et al. Klotho depletion contributes to increased inflammation in kidney of the db/db mouse model of diabetes via RelA (Serine)536 phosphorylation // Diabetes. 2011. V. 60. P. 1907–1916.
Zhou Q., Lin S., Tang R. et al. Role of fosinopril and valsartan on Klotho gene expression induced by angiotensin II in rat renal tubular epithelial cells // Kidney Blood Press. Res. 2010. V. 33. №3. P. 186–192.
Дополнительные материалы отсутствуют.
Инструменты
Успехи физиологических наук