

Успехи физиологических наук, 2021, T. 52, № 2, стр. 61-82
Факторы риска и генетические предпосылки развития воспалительных заболеваний кишечника: современное состояние проблемы
К. А. Дворникова a, *, Е. Ю. Быстрова a, О. Н. Платонова a, А. Д. Ноздрачев a
a Лаборатория интероцепции, ФГБУН Институт физиологии им. И.П. Павлова РАН
199034 Санкт-Петербург, Россия
* E-mail: 691442@gmail.com
Поступила в редакцию 20.10.2020
После доработки 01.11.2020
Принята к публикации 12.01.2021
Аннотация
За последнее время возросло количество молекулярно-биологических и генетических исследований, направленных на системное изучение этиологии и патогенеза воспалительных заболеваний кишечника, в первую очередь болезни Крона и язвенного колита. Активно изучаются факторы риска и генетические предпосылки развития воспалительных заболеваний кишечника. В частности, проводится поиск возможных ассоциаций болезни Крона и язвенного колита с экспрессией генов рецепторов врожденного иммунитета, например, Toll-подобных рецепторов (TLRs), а также с их полиморфизмом (SNP). Согласно последним данным, можно говорить о существовании определенной корреляции между наличием конкретных SNP в генах TLRs и дебютом болезни Крона и/или язвенного колита. В настоящей работе рассматриваются результаты современных исследований по изучению патогенеза, этиологии и генетики воспалительных заболеваний кишечника – болезни Крона и язвенного колита, в том числе в контексте существующих ассоциаций с TLRs и SNP в генах TLRs.
ВВЕДЕНИЕ
За последнее время возросло количество молекулярно-биологических и генетических исследований, направленных на системное изучение этиологии и патогенеза воспалительных заболеваний кишечника (inflammatory bowel disease, IBD). Активно изучаются возможные ассоциации IBD с экспрессией генов рецепторов врожденного иммунитета, в первую очередь, Toll-подобных рецепторов (TLRs), а также с их полиморфизмом (SNP) [146]. Поиск ассоциаций между заболеваниями, TLRs и SNP в генах TLRs позволяет формировать понимание путей и механизмов развития IBD, в том числе на ранних стадиях. Установлено, что наличие конкретных SNP в генах TLRs может приводить к нарушению функционирования некоторых ключевых сигнальных путей и повышает риск развития аутоиммунных, онкологических и желудочно-кишечных заболеваний, вследствие существования определенных ассоциаций между ними [1, 31]. Обнаружение и изучение SNP в рецепторных структурах желудочно-кишечного тракта является важным направлением исследований [89]. На сегодняшний день можно говорить о возрастающем интересе к изучению SNP в TLRs, ассоциированных с воспалительными заболеваниями кишечника.
В настоящей работе рассматриваются результаты современных исследований по изучению патогенеза, этиологии и генетики воспалительных заболеваний кишечника – болезни Крона и язвенного колита в контексте существующих ассоциаций с TLRs и SNP в генах TLRs. Также рассматриваются существующие методы терапии заболеваний. Приводятся сведения о возможных механизмах и роли TLRs и SNP в генах TLRs в развитии болезни Крона и язвенного колита.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ БОЛЕЗНИ КРОНА И ЯЗВЕННОГО КОЛИТА
IBD – группа хронических заболеваний желудочно-кишечного тракта, этиология которых в настоящее время остается до конца неясной. Однако с каждым годом выполняется все больше исследований, направленных на изучение природы IBD. Так, проведенный анализ позволил всесторонне изучить глобальную патофизиологию IBD, в результате чего удалось обнаружить существенный сдвиг: заболеваемость в западных странах стабилизировалась, но в активно развивающихся индустриальных странах наблюдается рост случаев воспалительных заболеваний кишечника [93]. При этом распространенность заболеваний остается высокой во всем мире. Дебют IBD обычно отмечается в подростковом возрасте, и, как правило, впоследствии приводит к развитию рецидивирующего и хронического заболевания. IBD включают два основных типа расстройств: язвенный колит (Ulcerative colitis, UC) и болезнь Крона (Crohn’s disease, CD). Между собой эти заболевания отличаются очагом поражения, патофизиологией, симптомами, течением заболевания, лечением и возможными осложнениями, но при этом этиология обоих заболеваний до сих пор до конца не изучена. Интересно, что пациенты с UC и CD имеют более высокий риск развития колоректальной карциномы по сравнению с общей популяцией [74]. UC характеризуется постоянным поражением только толстой кишки, воспаление носит поверхностный характер, что может приводить к эрозиям, язвам, диарее, появлению крови и слизи в стуле. UC характеризуется повышением уровня эйкозаноидов, которые играют патофизиологическую роль в воспалении. Обычное клиническое течение UC представлено периодами обострения и ремиссии. В фазе обострения может наблюдаться рост С-реактивного белка (C-reactive protein, CRP), скорости оседания эритроцитов (СОЭ), увеличение производства основных провоспалительных цитокинов (интерлейкин-1 (IL-1), интерлейкин-6 (IL-6), интерлейкин-8 (IL-8) и фактор некроза опухоли-α (TNF-α)), а также снижение гемоглобина (Hb). Однако, не было обнаружено, что уровни гемоглобина, СОЭ, CRP и провоспалительных цитокинов являются значимыми предикторами рецидива. Предикторами рецидива и тяжести заболевания являются нейтрофилы, плазматические клетки и эозинофилы. При этом существуют противоречия в представлениях о циркулирующих цитокинах при IBD. Однако профиль цитокинов показывает, что UC и CD иммунологически различны [68]. UC характеризуется преобладанием цитокинов, связанных с клетками Th2 (T-хелперы 2), а при CD преобладают Th1-ассоциированные цитокины. Терапия UC включает пероральное или местное применение 5-аминосалициловой кислоты (5-АСК) и кортикостероидов, но такое лечение доступно только в случае легкого и умеренного рецидивирующего хода заболевания. Поэтому очень важно запланировать оптимальную поддерживающую терапию до рецидива. Также трансплантация фекальной микробиоты (FMT) рассматривается в качестве новой перспективной терапии IBD, оказавшейся эффективной для индукции ремиссии при UC [25].
CD характеризуется кишечными повреждениями в любом сегменте желудочно-кишечного тракта (от ротовой полости до анального отверстия) и включает хроническое рецидивирующее трансмуральное воспаление, которое может привести к хронической боли в животе, диарее, обструкции или перианальным поражениям [110]. CD возникает в результате чрезмерного ответа клеток Th1 (T-хелперы 1) и Th17 (T-хелперы 17) на провоспалительные цитокины, такие как интерлейкины IL-12, IL-18 и IL-23, которые продуцируются антигенпрезентирующими клетками и макрофагами [142]. У 21–47% пациентов CD может выражаться внекишечными проявлениями (extraintestinal manifestations, EIMs). EIM может затрагивать любую систему органов, включая костно-мышечную, дерматологическую, почечную, гепатопанкреатобилиарную, легочную и зрительную системы [9]. Например, прогрессирующий анкилозирующий спондилит (АS) у пациентов с CD встречается в 25–75% случаев [45]. У 50% в течение 10 лет после постановки диагноза CD могут развиваться кишечные осложнения – стриктуры или фистулы [6]. Дебют CD отмечается у пациентов моложе 30 лет, однако заболеваемость увеличивается и у пожилых людей. При исследовании CD в западных странах не было обнаружено половых различий в заболеваемости, но, в свою очередь, результаты исследований на азиатских популяциях продемонстрировали преобладание заболеваемости CD среди мужчин [101]. В целом, наблюдается увеличение заболеваемости CD во всем мире, в частности, промышленно развитые страны (страны Европы и Северной Америки) имеют наивысшую распространенность CD, наименьший уровень CD в популяции характерен для стран Африки, Южной Америки и Азии [92].
При CD существует острая необходимость в рациональной и оптимизированной противовоспалительной терапии для предотвращения прогрессирования и последующих осложнений заболевания. Ранее лечение было направлено на смягчение только клинических симптомов, однако за последнее время были разработаны новые эффективные методы лечения, направленные на выход в клиническую ремиссию (в том числе без стероидов) и трансмуральное заживление слизистой оболочки кишечника [66]. Разработка биологических методов, направленных против специфических медиаторов воспалительного процесса, стало прорывом в лечении CD. Так, антитела против TNF (анти-TNF-α терапия) стали первым классом веществ, одобренным для лечения CD, а в дальнейшем были одобрены антитела против интегрина α4β7 (vedolizumab), IL-12 и IL-23 [35]. Однако не все пациенты с CD проявляют клинический ответ на это лечение. В свою очередь, предполагается, что антитела против IL-23 у пациентов с резистентностью к TNF, помогут создать терапевтические подходы, основанные на понимании молекулярного фенотипа CD [120]. Но на данный момент нет клинически совместимых прогностических биомаркеров для индивидуального руководства лекарственной терапией. Тем не менее, терапия CD продолжает активно развиваться. Для измерения повреждения кишечника, связанного с CD и оценки прогрессирования повреждения в течении определенного периода времени, которое может быть обратимым с помощью анти-TNF-α терапии, применяется индекс Лемана [36]. Индекс Лемана может иметь ключевое значение в лечении заболевания. Ошибка в диагностике CD может иметь серьезные последствия в связи с использованием анти-TNF-α терапии для лечения заболевания.
Важно отметить, что существует сходство между CD и кишечным туберкулезом (Intestinal tuberculosis, ITB). В настоящее время дифференциация этих заболеваний остается основной диагностической проблемой. Препараты, используемые для лечения CD, могут оказаться фатальными при ошибочно диагностируемом ITB. Хотя между заболеваниями существуют определенные клинические, эндоскопические, гистологические, серологические различия, при этом между CD и ITB единственными отличительными признаками в диагностике являются казеозный гранулематозный некроз при биопсии, положительный мазок на кислотоустойчивую культуру бацилл (AFB) и некротический лимфатический узел при визуализации поперечного сечения в ITB. Однако эти особенности ограничены низкой чувствительностью [58]. Недавно была разработана диагностическая модель прогнозирования, с помощью которой возможно повысить точность и практичность в дифференцировке CD и ITB [48].
Изучение механизмов и патофизиологии IBD является основой для понимания путей развития CD и UC, и формирования методов эффективной терапии. На сегодняшний день известно, что воспаление при IBD включает нарушение кишечной барьерной функции, нарушения в регуляции врожденных и адаптивных иммунных реакций, а также изменение состава кишечной микробиоты [110].
Кишечная барьерная функция
Обычно термином “кишечный барьер” обозначают слизистую оболочку кишечника, а также биохимические и иммунологические элементы слизистой оболочки и подслизистого слоя. Кишечная барьерная функция заключается в способности кишечного эпителия ограничивать свободный обмен водой, ионами и макромолекулами между просветом кишечника и подлежащими тканями [96]. Важно, что на сегодняшний день нет доступных терапевтических методов, позволяющих клинически восстановить эпителиальный барьер. Однако продолжаются исследования новых перспективных подходов, в частности, на основе трансмембранных белков – клаудинов. На примере клаудина-2 (Claudin-2), который способен усиливать межклеточную диффузию ионов Ca2+ в кишечнике, был предложен альтернативный подход в поддержке кишечного барьера – нацеливание на поры. В этом случае с помощью клаудина-2 возможно регулирование плотности соединения эпителия кишечника [96]. Известно, что плотные соединения пор являются важным фактором барьерной функции в эпителии кишечника. Также изучается путь параклеточной проницаемости на примере киназы легких цепей миозина (MLCK) [42, 96]. Клаудин-2 и MLCK являются потенциальными терапевтическими мишенями для модуляции проницаемости пор. Кишечный барьер, включающий кишечные эпителиальные клетки (intestinal epithelial cells, IECs), иммунные клетки врожденного иммунитета, интраэпителиальные лимфоциты (intraepithelial lymphocytes, IELs) и слизистые оболочки, является первым физическим и химическим барьером, с которым сталкиваются кишечные бактерии, патогены и пищевые антигены, и находится в постоянном гомеостазе с содержимым кишечного просвета [18, 104, 113]. Связь между кишечным барьером и дисфункцией кишечника была впервые обнаружена в исследованиях ex vivo [50]. Было установлено, что возникновение проблем в одном из компонентов кишечного барьера может привести к обратной функции кишечного барьера – повышенной проницаемости, что является признаком IBD. Например, в случае активной формы CD, наблюдается сниженная экспрессия герметизирующего белка клаудина-8 (Сlaudin-8) и повышенная экспрессия порообразующего белка клаудина-5 (Сlaudin-5) в IECs. Установлено, что нарушения секреторной активности клеток Панета, специализированных IECs и присутствующих в основании кишечных крипт в тонкой кишке, могут приводить к развитию CD [137].
IELs находятся между эпителиальными клетками кишечника и являются первыми клетками, которые вступают в контакт с люминальным антигеном, который пересекает слизистую оболочку. Большинство IELs представляют собой CD8+ T-лимфоциты, играющие решающую роль в поддержании гомеостаза кишечника [64]. Однако роль IELs в патогенезе CD недостаточно изучена. Приводятся сведения, в которых отражается провоспалительная роль IELs в CD. Так, в одном исследовании была продемонстрирована повышенная продукция IL-17A, IFNγ и TNF в IELs у пациентов с CD [109]. В частности, дисфункция IELs может способствовать развитию CD [20].
Врожденный иммунитет (Toll-подобные рецепторы)
Иммунные клетки врожденного иммунитета экспрессируют различные паттерн-распознающие рецепторы (Pattern recognition receptors, PRRs), ключевыми из них являются Toll-подобные рецепторы (Toll-like receptors, TLRs). TLRs распознают патоген-ассоциированные молекулярные паттерны (Pathogen-associated molecular patterns, PAMPs), происходящие из микроорганизмов или представляющие собой эндогенные молекулы, приводя к активации врожденного иммунного ответа [121]. TLRs играют важную роль в поддержании кишечного гомеостаза, в созревании дендритных клеток (Dendritic cells, DC), индукции пролиферации и дифференцировке Тh-клеток (Th1, Th2) [146].
Врожденный иммунитет играет роль в установлении видового разнообразия микробиоты каждого человека [51]. Дефекты врожденной иммунной системы могут привести к дисбиозу кишечной микробиоты. Поэтому для поддержания гомеостаза кишечника важно взаимодействие между иммунной системой человека и кишечной комменсальной микробиотой, которое опосредуется частично через TLR. Установлено, что большинство сигнальных путей TLRs участвуют в прогрессировании IBD [76]. TLRs служат центром иммунных ответов на микроорганизмы в кишечнике, а активация передачи сигналов TLR запускает каскад сигнальных путей, которые играют важную роль в развитии IBD. Установлено, что ингибирование передачи сигналов TLR2/TLR6 снижает прогрессирование IBD [85, 126]. Сообщается о сверхэкспрессии TLR6 в кишечнике пациентов с IBD. TLR6 оказался ключевой движущей силой ответа Th1 и Th17 [85]. При дефиците TLR6 наблюдалось снижение Th1 и Th17 ответов в лимфоидной ткани, связанной с кишечником. Это может свидетельствовать о потенциальной мишени TLR6 для будущего лечения IBD. В другом исследовании было установлено, что сигнальный путь TLR1 имеет решающее значение для предотвращения хронического воспаления толстой кишки при IBD и защиты слизистой оболочки от инфекции, вызванной Yersinia enterocolitica [135]. Отсутствие или сниженная экспрессия TLR1 при острой желудочно-кишечной инфекции может привести к хронической иммунной активации и трансмутации в составе комменсальных бактерий [55]. Известно, что TLR4 важен для индукции воспалительного ответа, поскольку обеспечивает защиту от проникновения патогенных бактерий и поддерживает целостность слизистой оболочки. Однако установлена отрицательную роль сигнального пути TLR4 в IBD. В частности, сообщается об участии TLR4 в развитии IBD и разрушительном воздействии на ткани кишечного эпителия и образование язв у пациентов с UC. Также сообщается о способности стимулированных кишечных глиальных клеток выделять оксид азота через TLR4, приводя к освобождению провоспалительных цитокинов, усугубляющих воспаление кишечника [81]. Но при этом роль TLR4 при UC может быть защитной или разрушительной в зависимости от стимула. TLR2-зависимые цитопротективные ответы от резидентных клеток ткани поддерживают целостность слизистой оболочки против летальных TLR4-зависимых воспалительных реакций гемопоэтических клеток [41]. TLR5 распознает бактериальный флагеллин, и играет ряд важных защитных и вредных ролей в кишечном гомеостазе и заболеваниях, в частности IBD. Было показано, что введение флагеллина защищает от радиационно-индуцированного повреждения кишечника [125]. Установлено, что экспрессия TLR5 в IECs регулирует состав и локализацию кишечной микробиоты, а также предотвращает заболевания, связанные с воспалением кишечника. В частности, при отсутствии TLR5 наблюдается изменение кишечной микробиоты, развивается слабое воспаление и подверженность колиту [21]. Согласно имеющимся сведениям, TLR5 может быть использован в целях разработки терапевтической стратегии против IBD. Ранее было установлено, что TLR3 у пациентов с активным IBD значительно подавляется в IECs. Однако позже оказалось, что комбинированные генетические вариации TLR3 и TLR7 значительно повлияли на тяжесть язвенного колита у пациентов с IBD [157]. Агонисты TLR3 и TLR7 стимулировали секрецию интерферона-β (IFN-β) плазмоцитоидными дендритными клетками (pDCs), способствуя защитному иммунитету при воспалительных состояниях. В другом исследовании были представлены сведения об индукции интерферона I типа (IFNα и IFNβ) агонистами TLR9, TLR3 и TLR7 и дальнейшем предотвращением экспериментального колита [112]. Местное применение Имиквимода (низкомолекулярного соединения, которое действует как модификатор местного иммунного ответа и проявляет противовирусные и противоопухолевые эффекты) также уменьшало колит путем индукции экспрессии IFNα и IFNβ в слизистой оболочке толстой кишки. Однако никаких доказательств системного ответа IFNα и IFNβ не наблюдалось. Была продемонстрирована защитная функция TLR9. Оказалось, что экспрессия TLR2, TLR4, TLR8 и TLR9 увеличивается у пациентов с активным UC. В частности, уровни мРНК положительно коррелировали с тяжестью воспаления кишечника и экспрессией TLR9 [114]. Также было установлено, что активация TLR9 предотвращает развитие воспаления слизистой оболочки и способствует заживлению ран на некоторых моделях колита [151]. В свою очередь, активация сигнального пути TLR9 его агонистом DIMS0150 продемонстрировала улучшения в клинической ремиссии UC с заживлением слизистой оболочки и уменьшением симптомов. Это позволяет предположить, что применение агониста DIMS0150 может быть перспективным с точки зрения терапевтического лечения пациентов с хронической активностью UC [11].
Известно, что регенерирующие белки Reg защищают кишечник от вторжения патогенов. Так, повышенная экспрессия белков Reg у пациентов с IBD может ограничивать проникновение в слизистую оболочку кишечных микроорганизмов. TLRs обладают способностью стимулировать IECs и клетки Панета к продуцированию белков Reg3β и Reg3γ, которые специфически нацелены на грамположительные бактерии [153]. Лечение антибиотиками уменьшало общее количество бактерий. Стимуляция TLR после лечения антибиотиками увеличивала активность связывания ДНК NF-κB и снижала содержание Klebsiella pneumoniae. Врожденный иммунитет и сигнальные пути TLRs в генерации IBD имеют важное значение. TLRs и некоторые компоненты сигнального пути являются мишенями и могут применяться в терапии в качестве агонистов и антагонистов. Подавляющее большинство членов семейства TLRs вовлечены в прогрессирование IBD.
Адаптивный иммунитет
Система адаптивного иммунитета – комплекс специализированных клеток и процессов, направленных на устранение болезнетворных микроорганизмов [91]. Важно, что адаптивный и врожденный иммунитет тесно взаимосвязаны и некоторые сигнальные пути происходят под действием факторов врожденного иммунитета и наоборот. Известно, что большинство лимфоцитов активируются в кишечной лимфоидной ткани и рекрутируются в места воспаления. Сообщается о взаимосвязи адаптивного иммунитета с IBD, в частности, о стойкой иммунной активации Т-клеток при IBD [27]. Ранее сообщалось, что дебют CD может быть ассоциирован с чрезмерным ответом клеток Th1 и Th17 на провоспалительные цитокины, такие как IL-12, IL-18 и IL 23, которые продуцируются антигенпрезентирующими клетками и макрофагами [142]. При этом клетки Th1 и Th17 секретируют провоспалительные цитокины IL-17, IFNγ и TNF, которые способствуют воспалению, стимулируя выработку TNF, IL-1, IL-6, IL-8, IL-12 и IL-18 макрофагами, эндотелиальными клетками и моноцитами [102]. Активность эффекторных T-клеток регулируется регуляторными T-клетками (Tregs-клетками), подавляющим подмножеством CD4+ T-клеток, которые играют роль в поддержании иммунного гомеостаза в кишечнике и других тканях и органах. Установлено, что эффекторные Т-клетки слизистой оболочки у пациентов с IBD могут быть устойчивыми или менее чувствительными к подавлению, опосредованному Tregs-клетками. В свою очередь, продукция IL-10 Tregs-клетками может быть необходима для предотвращения воспаления кишечника [163].
Интерлейкин (IL) является эффективным цитокином, участвующим в сигнальном пути TLRs и играющим ключевую роль в патогенезе и прогрессировании IBD. Подтипы семейства IL обладают дихотомическими функциями: от поддержания нормального кишечного гомеостаза до хронического кишечного воспаления [75]. В том числе, подтипы IL имеют важные последствия, касающиеся профилактики и лечения CD и UC. В частности, IL-18 может ингибировать созревание бокаловидных клеток (enterocytus caliciformis, ECs), регулируя транскрипционные механизмы развития этих клеток. Установлено, что дисфункция ECs может способствовать основной патологии IBD [94]. Существуют сведения, указывающие на защитные функции IL в патогенезе IBD. В результате контроля пути TLR7/IL-22 происходит восстановление иммуноопосредованной устойчивости к колонизации против высокоустойчивого к антибиотикам бактериального патогена [3]. Терапевтические средства, которые нарушают передачу сигналов TLR2, могут быть неэффективны для пациентов с UC. В свою очередь, IL-11 может помочь таким пациентам, стимулируя защитные реакции цитокинов [41]. В другом исследовании было установлено, что IL-17 и IL-22, продуцируемые лимфоцитами Th17, активируют защитные пути в IECs [145]. Считается, что TLR-индуцированная активация CD3negCD127+ клеток и продуцирование Th17-связанных цитокинов могут иметь решающее значение для ранней защиты от проникновения патогенов в слизистую оболочку кишки. Однако некоторые IL могут способствовать воспалению и ускорять развитие IBD. В частности, IL-6 увеличивает выработку патогенных цитокинов кишечными внутренними лимфоцитами при хроническом кишечном воспалении. IL-6 способствует активации рецепторов, отрицательных по отношению к рецепторам естественной цитотоксичности толстой кишки, путем увеличения IL-23 и IL-1α-индуцированной продукции IL-17A и IL-22. На основе этого пути возможна разработка новых стратегий лечения IBD, направленных на внутренние лимфоциты или сигналы цитокинов [107]. IL-12 и IL-23 вырабатываются макрофагами, дендритными клетки и нейтрофилами, являются центральными факторами воспаления кишечника и основными медиаторами воспаления при IBD [110]. При этом IL-23 имеет особую важность. IL-23 передает сигналы через гетеродимерный рецептор, состоящий из IL-23R и IL-12Rβ1, и экспрессируется αβ и γδ T-клетками. Установлено, что нацеленное на IL-12 и IL-23 моноклональное анти-p40-антитело, а также несколько специфически нацеленных на IL-23 анти-p19-антител, продемонстрировали эффективность при лечении CD [115]. При этом, было показано, что IL-23 активирует патогенные CD4+ лимфоциты, продуцирующие гранулоцит-макрофагальный колониестимулирующий фактор и IFNγ, и может ингибировать Tregs-клетки кишечника [142]. Сигнальный путь IL-10 ингибирует высвобождение нескольких ключевых цитокинов и оказывает противовоспалительное действие в желудочно-кишечном тракте. Важно, что мутации в генах IL-10 и рецепторе IL-10R оказались связаны с IBD с очень ранним началом (VEO-IBD), вызывая повторные приступы диареи с примесью крови, потерю веса, задержку роста и рецидивирующие перианальные проблемы, включая абсцессы, свищи и трещины. Известно, что интегрины αLβ2, α4β1, α4β7 и αEβ7 играют ключевую роль на поверхности лейкоцитов, позволяя им связываться с молекулами клеточной адгезии на поверхности эндотелиальных клеток кишечника. Так, была обнаружена эффективность моноклональных анти-α4β7-интегриновых и анти-α4-интегриновых антител при лечении CD [34, 35].
TNF-α – провоспалительный цитокин, способствующий острым реакциям и повреждению, является основной целью биологической терапии при CD. Ранее сообщалось о анти-TNF-α терапии, которая может работать частично путем подавления IL-22BP и обеспечить эффективное лечение IBD [103]. Установлено, что макрофаги из периферической крови пациентов с CD ослабляли высвобождение TNF-α. В результате была подтверждена гипотеза о дефектной секреции провоспалительных цитокинов и врожденном иммунодефиците при CD [122].
Дисбиоз микробиоты
Микробиота кишечника человека (human gut microbiota, hGM) состоит из бактерий, вирусов, грибков, простейших и архей, которые в совокупности могут выполнять функционально различную провоспалительную или противовоспалительную роль [108]. Функциональное состояние бактериальной микробиоты при IBD тщательно изучено, за исключением некоторых видов микроорганизмов, значение и роль которых до сих пор окончательно не установлены. Дисбаланс микробиоты характеризуется сниженным биоразнообразием, уменьшением бактерий типа Firmicutes и увеличением некоторых бактерий, принадлежащих к Proteobacteria [40]. Но, в свою очередь, остается неясным, является ли дисбиоз микробиоты причиной или следствием IBD. Известно, что состав и количество микробиоты кишечника человека изменяются с возрастом. В частности, у детей микробиота кишечника значительно менее разнообразна, чем у взрослых. Микробиом, связанный со слизистой оболочкой прямой кишки, является надежным предиктором IBD. Показано, что развитие IBD тесно связано с общей повышенной распространенностью Proteobacteria и общим снижением видового разнообразия микробиоты. Так, в исследовании было показано, что у пациентов с активной формой CD наблюдается значительно меньшее видовое разнообразие кишечных бактерий по сравнению с здоровым контролем [83]. В частности, наблюдается значительное снижение Firmicutes, Enterobacteriaceae, Bacteroidales [83], а также Clostridiales – бактерий, продуцирующих бутират, что может объяснять уменьшение количества короткоцепочечных жирных кислот в образцах фекалий у пациентов с IBD [40]. Известно, что бутират усиливает барьерную функцию слизистой оболочки, индуцируя выработку муцина и антимикробных пептидов. Позже была установлена связь между IBD и снижением распространенности бутират-продуцирующего Faecalibacterium prausnitzii [130]. Ассоциированная с UC Escherichia coli, продуцирующая α-гемолизин, способна вызывать быструю потерю целостности плотного соединения в дифференцированных монослоях клеток Caco-2. Предполагается, что высокая экспрессия α-гемолизина может быть механизмом, способствующим дисфункции эпителиального барьера и патофизиологии IBD. Существуют свидетельства роли грибковой микробиоты в патогенезе IBD [130]. Так, Dectin-1 и Card9 – IBD-ассоциированные гены, участвуют в распознавании грибковой микробиоты. В частности, при отсутствии этих генов наблюдается распространение грибковой микробиоты и восприимчивость к UC, что может указывать на связь между микромицетами и патогенезом IBD. Интересно, что у пациентов с IBD и у здорового контроля в грибковой микробиоте доминируют аскомицеты и базидиомицеты (Ascomycota и Basidiomycota phyla), при этом наиболее распространены представители родов Saccharomyces, Debaryomyces, Kluyveromyces, Penicillium и Candida. Важно, что три первых рода содержатся в продуктах питания и в случае формирования правильной диеты, основанной на регулярном употреблении специфичных для этих родов продуктов питания, возможно оказать влияние на IBD [130].
Показано, что кишечный микробиом с течением времени способен изменять состояние: от дисбиоза до полного выздоровления [44]. Эти результаты могут помочь в разработке терапии для поддержания ремиссии при IBD. Ранее было показано, что антитела против Saccharomyces cerevisiae (ASCA) оказались ассоциированы с CD. При этом антибиотики усиливают дисбактериоз, связанный с CD [40]. Установлено, что трансплантация фекальной микробиоты (FMT) может быть использована для устранения дисбиоза кишечника у пациентов с IBD [10]. Однако FMT в лечении IBD менее эффективна, чем в лечении рецидивирующей инфекции Clostridioides difficile (CDI) [100]. Был идентифицирован доминирующий видовой состав при CD, в котором выделили виды, увеличивающиеся при CD: Escherichia coli, Fusobacterium nucleatum, Haemophilus parainfluenzae (Pasteurellaceae), Veillonella parvula, Eikenella corrodens (Neisseriaceae), и Gemella moribillum; а также виды, уменьшающиеся при CD: Bacteroides vulgatus, Bacteroides caccae, Bifidobacterium bifidum, Bifidobacterium longum, Bifidobacterium adolescentis, Bifidobacterium dentum, Blautia hansenii, Ruminococcus gnavus, Clostridium nexile, Faecalibacterium prausnitzii, Ruminoccus torques, Clostridium bolteae, Eubacterium rectale, Roseburia intestinalis и Coprococcus comes [40]. Детекция видового состава микробиома кишечника является ключевым маркером для дальнейшей функциональной характеристики этих организмов и установления их роли в патогенезе IBD.
Важным открытием было установление роли аутофагии в развитии IBD. Аутофагия представляет собой процесс разложения цитоплазматических материалов внутри лизосомы и является врожденным защитным механизмом, который действует как клеточно-автономная система для уничтожения внутриклеточных патогенов [69]. Аутофагия имеет решающее значение для поддержания гомеостаза кишечника, регуляции экологии кишечника, соответствующих иммунных реакций кишечника и антимикробной защиты [69]. Стало известно, что дефектная аутофагия может приводить к нарушению функции кишечного эпителия, дисбиозу кишечника, дефекту антимикробной секреции пептидов клетками Панета, стрессовой реакции эндоплазматического ретикулума и аберрантным иммунным ответам на патогенные бактерии, а также оказывать сильное влияние на IBD, усиливая воспаление кишечника. Аутофагия необходима для внутриклеточной бактериальной обработки, ингибирования продукции провоспалительных цитокинов в IECs и иммунных клетках. Модуляция аутофагии с целью минимизации или предотвращения токсических эффектов, позволит разработать эффективную терапию для лечения IBD [69].
На течение IBD также может влиять беременность. Беременность и IBD имеют схожесть в иммунологических и микробиомных изменениях. Было обнаружено, что уровни провоспалительных цитокинов (IL-6, IL-8, IL-12, IL-17 и TNF-α) в сыворотке заметно снижаются при зачатии у беременных с IBD, а разнообразие кишечного микробиома нормализуется во время второго и третьего триместра беременности [144]. У пациентов с UC и CD проявляется специфичный для болезни микробиом до и во время ранних сроков беременности. При беременности наблюдается увеличение рода Bilophila у пациентов с CD и UC. Рост Bilophila усиливается при воспалительных и патологических состояниях, таких как воспалительные заболевания и аппендицит. F. prausnitzii был чрезмерно представлен у пациентов с UC без рецидивов. F. prausnitzii обладает противовоспалительными свойствами и может иметь клинический потенциал при IBD [129]. Обилие Faecalibacterium связано с уменьшением обилия Roseburia. Количество Oscillospira снижается, а Actinomyces увеличивается при IBD. В результате было установлено, что беременность является безопасной для пациентов с IBD и даже потенциально полезной с иммунологической и микробиологической точки зрения. В свою очередь, микробиом-направленные вмешательства во время беременности не рекомендуются во избежание осложнений с IBD. В другом исследовании, у детей, рожденных женщинами с IBD и у беременных женщин с IBD, наблюдалось более низкое бактериальное разнообразие, и измененный бактериальный состав по сравнению с контрольной группой женщин и детей [140]. В частности, у детей преобладали Gammaproteobacteria над Bifidobacteria. Также было обнаружено меньшее количество B-клеток памяти и регуляторных T-клеток в толстой кишке.
Установлено, что грудное вскармливание позволяет предотвратить CD и UC [155]. Это может объясняться улучшением развития иммунитета слизистой оболочки кишечника через взаимодействие с микробиомом. При отсутствии грудного вскармливания наблюдается колонизация Clostridium difficile и развитие иммуноопосредованных заболеваний. Отдельно отмечается связь кесарева сечения с развитием CD, но не с UC [57].
ФАКТОРЫ РИСКА ВОЗНИКНОВЕНИЯ БОЛЕЗНИ КРОНА И ЯЗВЕННОГО КОЛИТА
Существует риск возникновения и прогрессирования IBD у генетически восприимчивого человека под действием множества факторов окружающей среды [38].
Генетический фактор
Для IBD характерно семейное наследование. Среди монозиготных близнецов частота совпадений выше для CD (50%), чем для UC (15%) [110]. Проводятся генетические исследования, направленные на выявление локусов восприимчивости к CD и UC. Более 50% локусов чувствительности IBD также связаны с другими воспалительными и аутоиммунными заболеваниями [63]. В частности, в подавляющем большинстве локусы риска CD незначительно увеличивают относительный риск развития воспалений и являются общими для широкого спектра иммуномедицинских заболеваний [33]. Например, вариант PTPN22 (R620W) является сильным фактором риска для развития диабета 1 типа и ревматоидного артрита, но при этом PTPN22 (R620W) защищает от CD [148]. Локусы, содержащие гены MST1, IL-2, CARD9 и REL, ассоциированы с UC и с его осложнением – первичным склерозирующим холангитом (PSC), что, в свою очередь, позволяет выявить угрозу PSC для пациентов с UC. Для того, чтобы установить точную биологию заболеваний, важно изучение и понимание функций общих генов. Но при этом нельзя забывать, что генетические варианты CD могут иметь этническую специфичность и функциональные отличия.
Ранее было сделано основополагающее открытие кодирующей вариации в гене рецептора NOD2 (CARD15), который избирательно связан с риском развития CD [52]. Известно, что взаимодействия между NOD2 и TLRs играют важную роль в защите от бактериальной инфекции [134]. NOD2 является одним из ключевых генетических факторов, связанных с риском развития CD подвздошной кишки. При дефиците NOD2 у IECs нарушается способность уничтожать бактерии, что может приводить к нарушенным взаимодействиям между микробиотой подвздошной кишки и иммунными клетками слизистой оболочки. В свою очередь, изучение генетических мутаций в CD является главным инструментом исследования, имеющим перспективу в диагностике, прогнозировании и лечении. Выделяют три наиболее распространенных CD-ассоциированных мутации в NOD2 (Arg702Trp, Gly908Arg и Leu1007fsinsC), которые играют ключевую роль в распознавании и восприятии микробиоты. Также важную роль играют гены IL23-R и ATG16L1, увеличивающие риск CD и UC [52]. В частности, открытие варианта Thr300Ala гена ATG16L1 вместе с вариантом 3019–3020insC гена NOD2 позволило выявить новые механизмы заболевания. Оказалось, что обнаруженные мутации являются CD-специфичными, участвуют в нарушении распознавания бактерий и аутофагии, а также имеют главное значение в патогенезе CD. Ген IL23-R также имеет значение в патогенезе CD, участвуя в дифференцировке Th-17 лимфоцитов и приводя к дисрегуляции продукции цитокинов. В свою очередь, ATG16L1 и IRGM являются генами аутофагии, которые были идентифицированы как генетические факторы риска развития CD [69]. LRRK2 и MUC19 – еще 2 гена CD, связанных с аутофагией. Дисфункция LRRK2 приводит к нарушениям в переносе аутофагосом в лизосомы и увеличению апоптоза и окислительного повреждения. Важно отметить, что в японской популяции TNFSF15 оказался ключевым локусом риска CD и его эффект превышает NOD2 в других популяциях [73, 92].
На сегодняшний день полногеномный поиск ассоциаций (genome-wide association studies, GWA study, GWAS) является главным инструментом в поиске и идентификации новых генетических локусов предрасположенности к различным заболеваниям, в том числе – к желудочно-кишечным. Благодаря GWAS возможен анализ генетических факторов риска, изучение молекулярных механизмов заболеваний и выявление новых мишеней для разрабатываемых лекарственных препаратов [19]. Проведение широкомасштабных исследований GWAS на больших выборках позволяют получать данные об обнаруженных локусах в различных этнических группах и в дальнейшем объединять идентифицированные локусы, характерные для общей популяции. В свою очередь, большинство связанных с заболеванием локусов находятся в некодирующих областях генома. Остается неясным, какие гены они регулируют и в каких типах клеток это регулирование происходит. Известно, что генетическая вариация может не проявляться в зависимости от популяции.
Было проведено большое исследование GWAS, направленное на поиск и обнаружение локусов, связанных с IBD среди европейских и неевропейских групп [73]. Анализ ассоциаций позволил выявить 38 локусов чувствительности к IBD: 25 из 38 локусов перекрываются с ранее зарегистрированными для других признаков, включая иммуно-опосредованные заболевания, 13 ранее не ассоциировались с каким-либо заболеванием или признаком, 27 из 38 новых локусов ассоциированы с CD и с UC, но у семи из них выявлена неоднородность эффекта между двумя заболеваниями. У 11 локусов семь были классифицированы как специфичные для CD и четыре – для неспецифического UC. В результате были отобраны гены-кандидаты в локусах чувствительности IBD: LY75, CD28, CCL20, NFKBIZ, AHR и NFATC1, которые модулируют специфические аспекты ответа Т-клеток; OSMR, кодирующий рецептор онкостатина М, компонент рецептора цитокина – при биопсии у пациентов с активным IBD уровни онкостатина М повышаются и увеличивают барьерную функцию кишечного эпителия при воспалении кишечника; PTK2B, играющий роль в миграции моноцитов и дегрануляции нейтрофилов. Наличие общих локусов риска IBD среди европейских и неевропейских популяций позволяет предположить, что объединение данных генотипов по этническим группам в дальнейшем даст возможность обнаружить дополнительные локусы, связанные с IBD. В другом исследовании GWAS был обнаружен функциональный вариант Arg381Gln IL-23R на хромосоме 1p31, демонстрирующий пониженный риск развития CD [97]. Известно, что IL-23 действует вместе с другими цитокинами, в частности, с IL-12 и факторами транскрипции STAT3, JAK2, и является связанным с восприимчивостью к CD. Изучаются факторы транскрипции, в том числе и STAT3 – кодирующий ген, который находится внутри IBD-ассоциированного локуса. Установлено, что STAT3 активируется в IECs при IBD, но при этом специфическая для IECs делеция Stat3 может оказывать влияние на восстановление эпителия [105]. Другие факторы транскрипции – HNF4A и NKX2-3 участвуют в регенерации эпителия и контролируют пролиферацию клеток крипты, и дифференцировку IECs. UC встречается при специфической для IECs делеции Hnf4a, что указывает на возможную взаимосвязь заболевания и гена.
Изучение белков-транспортеров карнитина и органических катионов SLC22/OCTN позволило определить вариант SLC22A4/A5 как ассоциированный с CD [97]. При исследовании человеческого лейкоцитарного антигена (HLA), который кодирует белки клеточной поверхности, отвечающие за регуляцию иммунной системы, были определены аллели риска (DRB1*07) и защиты CD (DRB*0301) [149]. Эти аллели важны для определения паттерна фенотипа.
Факторы окружающей среды
Установлено, что множественные факторы окружающей среды связаны с развитием IBD [106].
Гигиена и условия жизни. Низкий уровень гигиены окружающей среды – проживание с сельскохозяйственными и домашними животными, проживание в семье и совместное использование общественных мест (туалет, ванная, кухня, спальня), наличие двух и более братьев и сестер, играет защитную роль при CD [23]. Воздействие этих факторов модифицирует риск иммуноопосредованных заболеваний, в частности IBD. Так, в случае отсутствия в детстве воздействия инфекционных антигенов, будет стимулироваться не доминирующий иммунный ответ Тh-1 (TNF-α, IFN-γ, IL-2), а Тh-2-опосредованный, который продуцирует IL-4, IL-5, IL-6, IL-13. В свою очередь, возможны другие механизмы воздействия. Например, антигенная конкуренция и влияние на Тregs-клеток. В эндемичных зонах у пациентов с IBD преобладают высокие уровни IL-10 и Tregs-клеток, но низкие уровни провоспалительных цитокинов [77]. Еще один механизм такого воздействия может быть связан с модификацией кишечного микробиома. Известно, что уменьшение видового разнообразия в микробиоме кишечника лежит в основе развития IBD [40]. Урбанизация является важным фактором риска развития IBD. Сообщается, что городская жизнь связана с развитием IBD: в большей степени CD и в меньшей – UC [131]. В частности, такая экологическая детерминанта, как загрязнение воздуха в городах, тесно связано с IBD у детей. В том числе, профессии водителя и работника на заводских производствах в городе являются фактором риска развития IBD [131].
Вредные привычки. Среди вредных привычек курение является модифицируемым фактором риска развития CD. Давно известно, что курение (в том числе пассивное курение) вызывает увеличение риска развития CD в 2 раза [67]. Но при этом длительность воздействия курения или пассивного вдыхания дыма зависит от степени влияния на развитие IBD: в частности, курение в дородовой период и пассивное курение в детском возрасте не вызвали ассоциации с CD или UC [67]. В результате курения, в раннем начале CD может наблюдаться большое количество рецидивов, повышенная необходимость в хирургических вмешательствах и иммуносупрессии. Установлено, что никотин изменяет тонус гладкой мускулатуры, влияет на эндотелиальную функцию посредством производства оксида азота, влияет на целостность слизистой оболочки кишечника и вызывает окислительный стресс. Важно, что курение уменьшает разнообразие кишечного микробиома [119]. В частности, наблюдается увеличение Proteobacteria и Bacteroidetes phyla, Clostridium, Bacteroides и Prevotella, и уменьшение Actinobacteria и Firmicutes phyla, Bifidobacteria и Lactococcus. Курение повышает относительную распространенность Bacteroidetes при CD. Разнообразие кишечного микробиома увеличивается после прекращения курения, что выражается в увеличении ключевых представителей типа Firmicutes (Clostridium coccoides, Eubacterium rectale и Clostridium leptum) и Actinobacteria (HGC и Bifidobacteria), а также уменьшении Bacteroidetes (Prevotella spp. и Bacteroides spp.) и Proteobacteria (β- и γ-подгруппы Proteobacteria) [119].
Было установлено, что употребление алкоголя может способствовать развитию IBD. Известно, что употребление алкоголя является разрушительным для барьерной физической и иммунологической функции кишечника, создаваемой IECs и кишечной лимфоидной тканью [132]. Согласно результатам исследований, алкоголь может вызывать симптомы и/или рецидив IBD, однако влияние на патогенез CD и UC в большей степени выражено у взрослых. У пациентов с IBD и у больных алкоголизмом выражен кишечный бактериальный дисбиоз [59]. В свою очередь, дисбиоз позволяет патогенам колонизироваться и размножаться, что повышает риск инфекций. Отдельно сообщается о влиянии на симптомы IBD не самого алкоголя, а отдельных ингредиентов. Так, высокое потребление сульфита – добавки, используемой в алкогольных напитках, было связано с высоким риском рецидивов и повышенной активностью IBD, а высокое содержание сахара в алкоголе оказалось ассоциировано с распространенностью болей в животе [59].
Употребление наркотиков является одним из самых существенных факторов, осложняющих IBD. Известно, что значительная часть пациентов с IBD, употребляющих наркотики, подходит под критерии лекарственной зависимости. При этом употребление наркотиков среди пациентов с IBD связано с активностью заболевания, ухудшением качества жизни, а также психологической дисфункцией [16]. Согласно статистике, 5–13% пациентов с IBD хронически принимают наркотики в амбулаторных условиях [78]. В том числе, пациенты с IBD обращаются за наркотиками в качестве альтернативы нормальной терапии [60]. При этом наблюдаются симптоматические улучшения после использования марихуаны и каннабиса у пациентов с UC, но не с CD, при котором развивается худший прогноз заболевания [60].
Диета. Установлено, что долгосрочная диета влияет на структуру и активность микроорганизмов в кишечнике. В микробиоме кишечника происходят кратковременные изменения под влиянием макроэлементов, поступающих из пищи. В свою очередь, известно, что диета играет важную роль в патогенезе IBD [136]. В частности, при IBD необходимо соблюдение диеты с достаточным количеством железа, кальция, цинка, фолиевой кислоты, витаминов D и B12 [136]. За последнее время был проведен ряд исследований по изучению эффективности диетических стратегий при IBD: от изучения отдельных пищевых групп до конкретных питательных веществ. Причинно-следственные связи между диетой и IBD не всегда очевидны [61]. Так, эксклюзивное энтеральное питание (EEN), предполагающее использование полноценной жидкой диеты, назначаемой вместо твердых веществ и жидкостей (кроме воды) сроком до 8 нед., имеет преимущества для индукции ремиссии CD (в том числе при сложной CD) и уменьшения воспаления кишечника [4]. EEN модулирует бактериальную флору в просвете кишечника, снижая IBD, а компоненты EEN оказывают прямое противовоспалительное действие на IECs, что приводит к подавлению провоспалительных цитокинов слизистой оболочки. EEN играет роль в разрешении орофациального гранулематоза (Orofacial granulomatosis, OFG) у пациентов с CD в течение 2 дней после начала элементарной диеты [90]. EEN вызывает клиническую и биохимическую ремиссию CD у 85% пациентов и рекомендуется в качестве первоочередной терапии активной CD для детей и подростков. В другом исследовании у 3 из 14 детей с CD наблюдалось полное трансмуральное заживление [43]. EEN имеет отличный профиль безопасности, который значительно выше, чем у кортикостероидов. В свою очередь, ранее был продемонстрирован эквивалентный ответ EEN на кортикостероиды у детей с активной CD. Обычно при возвращении на нормальную диету, проводится дополнительное EEN для поддержания ремиссии. В подавляющем большинстве случаев необходима поддерживающая медикаментозная терапия детям до или после завершения EEN. Также существует частичное энтеральное питание (Partial enteral nutrition, PEN) с 50% калорий в рационе со свободной диетой, однако согласно результатам исследований, PEN является неэффективным для индукции ремиссии CD. Предполагается, что механизм зависит от исключения свободной диеты [127].
Исключающая диета CD (CDED) – новая полноценная диета в сочетании с PEN, была разработана для уменьшения негативного воздействия компонентов питания на микробиом, кишечный барьер и иммунитет [70, 71]. CDED в сочетании с PEN переносится лучше, чем EEN [71]. CDED заключается в высоком потреблении фруктов и овощей, и низком потреблении красного и обработанного мяса. В результате CDED наблюдается снижение Haemophilus, Veillonella, Anaerostipes, Prevotella, Roseburia и увеличение Oscillibacter. CDED продемонстрировала устойчивую ремиссию CD и снизила воспаление у детей и взрослых с CD и вторичной потерей ответа на анти-TNF-α терапию [127, 128]. Таким образом, CDED является перспективным диетическим вариантом питания для уменьшения воспаления при CD.
Диета на основе животного или растительного происхождения изменяет структуру микробных сообществ и подавляет межиндивидуальные различия в экспрессии микробных генов [26]. Было показано, что питание продуктами на основе животных увеличивало количество толерантных к желчи микроорганизмов (Alistipes, Bilophila, Bacteroides), в частности Bilophila wadsworthia, поддерживающую связь между диетическим жиром, желчными кислотами и нарастанием микроорганизмов, способных вызывать воспалительные заболевания кишечника [28], а также снижало Firmicutes, которые метаболизируют полисахариды пищевых растений (Roseburia, Eubacterium rectale и Ruminococcus bromii). Долгосрочное потребление пищевых волокон, полученных из фруктов, связано с низким риском развития CD [8]. Предполагается, что в основе этого защитного эффекта может лежать метаболизм клетчатки в короткоцепочечные жирные кислоты (SCFA), которые ингибируют транскрипцию провоспалительных медиаторов. В том числе, клетчатка способствует поддержанию целостности эпителиального барьера и уменьшает транслокацию E. coli. В свою очередь, высокое потребление полиненасыщенных жирных кислот омега-6 PUFA и низкое потребление омега-3 PUFA, оказалось ассоциировано с повышенным риском развития CD [38]. В результате диета на основе продуктов животного происхождения продемонстрировала большое влияние на микробиоту кишечника, чем растительная диета. В зависимости от диеты, наблюдается разница в ферментации углеводов и белков [88].
Средиземноморская диета (MedDiet) считается одной из самых здоровых в мире моделей питания благодаря сочетанию продуктов, богатых антиоксидантами и противовоспалительными веществами [39]. Было установлено, что MedDiet связана с низким риском поздней CD и играет важную роль в патогенезе IBD [62]. Также эта диета пересекается с CDED по рациону. В свою очередь, сообщается о положительных эффектах модифицированной углеводной диеты (SCD) и низкоферментируемых олигосахаридов, дисахаридов, моносахаридов и полиолов (FODMAP) на IBD. SCD и FODMAP являются хорошей дополнительной терапией в периоды обострения CD [24]. Однако еще неизвестны механизмы, лежащие в основе эффективности SCD у детей с CD.
Физическая активность. Достоверно установлено, что физическая активность защищает от последующего CD, но не от UC [150]. К тому же, физическая активность может восстанавливать дефектную аутофагию при CD и снижать уровни провоспалительных процессов [47, 95]. Отмечается связь индекса массы тела (ИМТ) с IBD [30]. Так, ИМТ оказался ниже у пациентов с CD, чем у здорового контроля и пациентов с UC. Важно, что медикаментозная терапия не позволит улучшить ИМТ пациентов с IBD.
Витамины и антибиотики. Витамин D, образующийся в результате эндогенного производства под воздействием солнечного света и поглощенный из рациона, метаболизируется в печени до 25-гидроксивитамина D (25(ОН)D). 25(OH)D является основной циркулирующей формой витамина D, а также используется для определения статуса витамина D в клинической практике [87]. Витамин D может влиять на врожденный иммунитет, а также играть иммунологическую роль при CD и UC. Проводятся исследования влияния витамина D на риск развития CD и UC. Так, ранее был продемонстрирован положительный эффект введения витамина D, который уменьшал воспаление при UC, подавляя экспрессию провоспалительных генов, включая TNF. В свою очередь, дефицит витамина D или нокаут его рецептора повышал риск развития UC. При этом среди пациентов с IBD дефицит витамина D встречается чаще, чем у здорового контроля [87]. Дефициту может способствовать несколько факторов: недостаточное воздействие солнечного света, связанное с образом жизни или постоянными симптомами IBD, ограничивающего физическую активность, недостаточное потребление пищи из-за симптомов IBD, нарушение всасывания и превращения витамина D в его активные продукты, увеличение катаболизма и усиление выведения. В другом исследовании было установлено, что высокие прогнозируемые уровни 25(OH)D в плазме у женщин значительно снижают риск развития CD и несущественно снижают риск развития UC [5]. Позже было продемонстрировано, что низкий уровень 25(OH)D связан с повышенным риском госпитализации и хирургического вмешательства при CD и UC, а нормализация уровня 25(OH)D снижала риск проведения хирургического вмешательства, связанного с CD [7]. На данном этапе исследований можно предположить возможную связь между статусом витамина D и активностью CD и UC. Было показано, что витамин D и куркумин могут вызывать аутофагию [32, 123]. В свою очередь, дефицит витамина D является критическим фактором в патологии IBD, но лечение IECs витамином D может усиливать активацию аутофагии.
Сообщается о негативном воздействии антибиотиков в детском возрасте, что может повышать риск развития CD [46, 106]. Нестабильность микробиоты кишечника в раннем детстве может повышать восприимчивость к IBD [124]. Антибиотики показаны в случае перианальных осложнений, таких как абсцессы, однако нет доказательств эффективности антибиотиков в уменьшении воспаления при CD [110]. При этом существуют исследования на азиатских популяциях, в которых сообщается о защитной связи между воздействием антибиотиков и развитием IBD [40]. Кроме того, установлено, что оральные контрацептивы, аспирин и нестероидные противовоспалительные препараты (NSAIDs) также могут повышать риск CD [84, 98].
Вирусные инфекции. Вследствие нарушения иммуноактивации и чрезмерного ответа цитокинов у пациентов с IBD, вирусные инфекции могут осложнять течение CD и UC. Сообщается, что первичная цитомегаловирусная инфекция (CMV) может вызывать тяжелые заболевания у пациентов с IBD, в особенности получающих иммуносупрессию [111]. Вирус Эпштейна-Барра (EBV), который связан с несколькими злокачественными новообразованиями, также оказался ассоциирован с IBD из-за присутствия вирусных агентов в ткани толстой кишки [12]. Показано, что вирусная репликация EBV и пролиферирующие В-клетки были увеличены у пациентов с IBD. Также сообщается о способности обострять IBD у парвовируса В19 (семейство Parvoviridae) и норовирусов (семейство Calviviridae). Парвовирус B19 локализуется в слизистой оболочке кишечника и вызывает тяжелые осложнения IBD. В свою очередь, норовирусы, которые являются основной причиной небактериального острого гастроэнтерита, также приводят к обострению IBD [53]. Следует отметить, что у пациентов с IBD наблюдается повышенная экспрессия входных молекул ACE2 и TMPRSS2 в кишечнике, с которыми связывается коронавирус SARS-CoV-2. Это может свидетельствовать о более тяжелом протекании инфекции у пациентов с IBD и возможном усугублении течения IBD [17]. Кроме того, можно ожидать, что любые вирусные инфекции, в той или иной степени поражающие ткани кишки, могут являться пусковым механизмом для дебюта IBD у индивидуумов, имеющих генетическую предрасположенность и/или риск развития воспалительного заболевания кишечника, ассоциированный с другими факторами.
В последние десятилетия появились данные, свидетельствующие о том, что одним из факторов риска развития IBD является наличие генетических изменений (в частности, однонуклеотидного полиморфизма (Single Nucleotide Polymorphism, SNP)) в генах, ассоциированных с CD и UC. В связи с этим представляет интерес выяснение возможной роли SNP в патогенезе IBD.
ОБЩИЕ ПРЕДСТАВЛЕНИЯ О СТРУКТУРЕ SNP
SNP возникает в результате точечных мутаций и представляет замену одного нуклеотида на другой [118]. Также существует однонуклеотидная вариация (Single Nucleotide Variant, SNV), представляющая собой короткие (≤50 п.н.) делеции, вставки и другие небольшие генетические изменения. SNV встречается значительно реже SNP. SNP является наиболее частым геномным вариантом, происходящим в ДНК человека с частотой 1 на 1000 оснований и встречающимся более чем у 1% населения в целом [143].
SNP могут быть в кодирующих и некодирующих участках. Известно, что в подавляющем большинстве SNP в кодирующих последовательностях не имеют функциональных последствий для генома, а в некодирующих наоборот – могут влиять на процессы деградации, вырезание нуклеотидов и т.д. SNP подразделяются на синонимичные (sSNP) и несинонимичные (nsSNP): sSNP не вносят изменения в аминокислотную последовательность, а nsSNP – вносят. Также nsSNP являются полезными маркерами для ассоциативных исследований для выявления генетических вариаций, связанных с фенотипическими признаками. Изменения в последовательности происходят в зависимости от вида несинонимичного SNP: сайленс (без изменений в последовательности), нонсенс (образуются стоп-кодоны) и миссенс (изменение в последовательности).
У SNP существует номенклатура. Каждому SNP присваивается уникальный ссылочный номер (rs, RefSNP), который является идентификатором всех SNP в базах данных, формируя полную карту генетических различий. Каталог RefSNP значительно облегчает проведение широкомасштабных генетических, медицинских, фармакогенетических и эволюционных исследований, позволяет отслеживать молекулярные последствия, находить редкие клинические фенотипы и сообщать о новых результатах исследований. Помимо ссылочного номера указываются вариации SNP – аллели, генотип и частота, отражающие нуклеотид или аминокислоту дикого типа и измененную. На сегодняшний день базы данных (OMIM, dbSNP, SNPedia, Variant Annotation Integrator, Variant Effect Predictor) содержат более полумиллиарда записей SNP.
Определять SNP позволяет анализ гаплотипов. Для детекции SNP используются гибридизационные (молекулярные маяки, SNP микрочипы, динамическая аллель-специфическая гибридизация (Dynamic allele-specific hybridization, DASH)), ферментативные (ПЦР, полиморфизм длин рестрикционных фрагментов (Restriction fragment length polymorphism, RFLP), эндонуклеаза-1 (Flap endonucleases, FEN), гель-электрофорез, удлинение праймеров, TaqMan пробы) и физические методы (масс-спектрометрия, одноцепочечный конформационный полиморфизм (Single Strand Conformation Polymorphism, SSCP), электрофорез по градиенту температур (Temperature gradient gel electrophoresis, TGGE), денатурирующая высокоэффективная жидкостная хроматография (Denaturing high performance liquid chromatography, DHPLC), генотипирование SNPlex, Сюрвейерский нуклеазный анализ (Surveyor nuclease assay)), а также методы секвенирования нового поколения (Next generation sequencing, NGS) [72, 138].
Создание новых маркеров SNP позволяет автоматизировать и повысить эффективность анализа генотипов. Разрабатываются новые методы работы с гаплотипами с целью оптимизации процесса анализа SNP. В частности, проводятся исследования, направленные на создание оптимальных методов сортировки SNP для удаления избыточных и менее информативных SNP, и сохранением набора наиболее полезных генетических гаплотипов SNP [143]. Также была разработана новая технология генотипирования SNP-seq, которая имеет высокую точность при генотипировании генома с высоким SNP и консервативными фланкирующими последовательностями [161]. В дальнейшем эта технология может быть использована для генотипирования SNP человека и животных.
SNP имеет важное практическое и научное значение. SNP выступает маркером в генетических, медицинских, фармакогенетических исследованиях, направленных на изучение этиологии, патофизиологии и патогенеза различных заболеваний, анализ фармакокинетики лекарственных препаратов, создание новых перспективных методов диагностики, эффективной терапии и лечения. Также SNP играет ключевую роль для GWAS: значительно упрощается поиск локусов предрасположенности к заболеваниям в различных этнических группах. Анализ SNP имеет важное значение для судебно-медицинских расследований [56]. Короткие тандемные повторы (short tandem repeat, STR – микросаттелит с короткими повторяющимися единицами (2–6 п.н.)) ранее широко использовались при генетической идентификации личности. STR являются высокоинформативными из-за большого количества аллелей, присутствующих даже в генетически однородных популяциях. Однако STR имеют ограничения, в частности, требуемый большой размер ампликона, высокую частоту мутаций и артефактов [65]. В результате решением этих ограничений стало применение SNP для генетической идентификации. SNP по сравнению с STR имеют низкую частоту мутаций, быстрое генотипирование, высокую распространенность в геноме и простое обнаружение с использованием высокопроизводительных технологий [99]. В области судебной медицины разработка новых и мощных технологий играет ключевую роль как в генетической идентификации, так и в скорости расследования. Создаются мощные индивидуальные идентификационные SNP (Individual Identification Single Nucleotide Polymorphism, IISNP) панели для разных групп населения в мире [152]. Эти панели позволяют проводить мультиплексный анализ для однозначной идентификации личности по образцам ДНК и РНК. В последнее время рассматривается разработка глобальной панели IISNP с большим количеством маркеров [159].
Существует ряд исследований, направленных на изучение этиологии и патогенеза воспалительных заболеваний кишечника, в частности исследований SNP в генах TLRs и поиска ассоциаций с CD. Интерес основан на отсутствии четкого понимания этиологии заболевания и возможности обнаружить ассоциации SNP с CD, а также на установлении механизмов развития патологии.
SNP В ГЕНАХ TLRS, АССОЦИИРОВАННЫЕ С БОЛЕЗНЬЮ КРОНА И ЯЗВЕННЫМ КОЛИТОМ
Известно, что SNP в генах TLRs могут возникнуть из-за различных процессов, включая дупликацию, делецию или мутацию, спровоцированных факторами окружающей среды, патогенными процессами в организме, стрессом и многими другими факторами [146]. В результате полиморфизм, образовавшийся в генах TLRs, может привести к нарушению функционирования некоторых ключевых сигнальных путей и в конечном итоге повысить риск развития аутоиммунных, онкологических и желудочно-кишечных заболеваний, вследствие существования определенных ассоциаций между ними [1, 31]. SNP в TLRs в основном нарушают способность распознавания патогена и иммунные функции этих рецепторов. Такие SNP были обнаружены во внеклеточном и внутриклеточном домене TLR [13, 133, 154]. Важно, что каждый отдельно взятый SNP может быть ассоциирован сразу с несколькими воспалительными расстройствами [31]. Такая полиморфность может объясняться схожестью этиологии и патогенеза заболеваний. Но у подавляющего большинства обнаруженных SNP в генах TLRs до сих пор не известны функции. Такие SNP могут оставаться нефункционирующими и не причинять вред здоровью человека, либо могут играть решающую роль в защите или восприимчивости к инфекционным заболеваниям. Подобные SNP используются в качестве маркера в исследованиях, направленных на изучение патофизиологии и генетики заболеваний. В дальнейшем на основе полученных сведений станет возможным создание лечебных препаратов на основе генной терапии. Однако существуют SNP, которые могут влиять на рецидив существующих заболеваний или развитие новых. На данный момент, таких SNP идентифицировано больше, чем SNP с положительным эффектом. В свою очередь, SNP с негативным воздействием на здоровье человека позволяют проще проводить дифференцировку носителей таких аллелей от общей популяции, лучше понимать этиологию заболеваний и разрабатывать новые методы лечения.
Обнаружение и изучение SNP в рецепторных структурах желудочно-кишечного тракта остается важным направлением исследований [89]. На сегодняшний день можно говорить о возрастающем интересе к изучению SNP в TLRs, ассоциированных с IBD (табл.1).
Таблица 1.
SNP в TLRs и ассоциированные воспалительные заболевания кишечника
| Рецептор | Идентификатор и генотип SNP | Заболевание | Ссылка |
|---|---|---|---|
| TLR1 | rs5743618 (I602S) | CD | [156] |
| TLR4 | rs4986790 (Asp299Gly (+896 A/G)) rs4986791 (Thr399Ile (1196 С/Т)) |
CD, UC CD, UC |
[37], [49], [80], [147], [158] [80], [158] |
| TLR5 | rs5744168 (392STOP;R392X) rs2072493 (N592S) rs5744174 (L616F) |
CD, UC UC CD |
[82] [82] [125] |
| TLR8 | rs2109134 (A/T) rs1548731 (+3121T > C) |
CD CD |
[117] [117] |
| TLR9 | rs5743836 (–1237C) | CD | [139] |
| TLR10 | rs7658893 (G/A) rs7653908 (C > G) |
CD CD |
[2] [2] |
Существует ряд выявленных аллелей, наличие которых единично, или в составе комплекса других SNP в генах TLRs, может означать большую предрасположенность и восприимчивость к CD и UC, чем у не носителей этих аллелей. Но также отдельно выделяют аллели, наличие которых может формировать защиту на уровне генов от развития этого заболевания. Так, ранее было установлено, что наличие аллелей Asp299Gly (rs4986790) и Thr399Ile (rs4986791) в TLR4 обуславливает более высокий риск развития заболеваний кишечника, включая CD [37, 158] (рис. 1).
Рис. 1.
Схематичное изображение расположения разных SNP в генах TLR1, TLR 4, TLR 5, TLR 8, TLR 9, TLR10. Желтые блоки – экзоны (не в масштабе) в генах TLRs, синяя область – интроны. Стрелки показывают расположение однонуклеотидного полиморфизма TLRs.
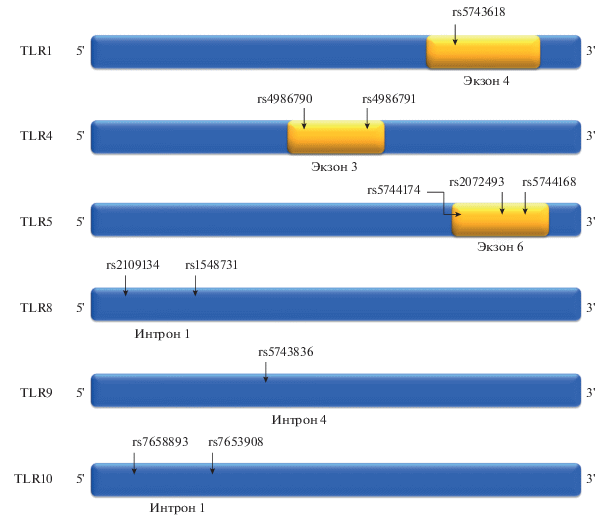
Позже также было показано, что SNP rs4986790 в TLR4 связан с повышенной восприимчивостью к CD [49]. Однако результаты, полученные в исследовании на корейской популяции, показали, что у пациентов с CD не было обнаружено SNP rs4986790 в TLR4 и, соответственно, такая генетическая вариация может не проявляться в зависимости от популяции, следовательно rs4986790 нельзя назвать полиморфизмом, ассоциированным с CD для общей популяции [158]. В свою очередь, SNP rs4986790 оказался достоверно связан с MAP-положительными пациентами с CD по сравнению с MAP-отрицательными пациентами с CD [147]. Известно, что подвид патогенных микобактерий – Mycobacterium avium subspecice paratuberculosis (MAP) участвует в патогенезе CD. В результате проведенного исследования предполагается, что CD может приводить к тому, что ряд специфических SNP предрасполагает человека к заболеванию, а последующее заражение одним или несколькими конкретными бактериями будет вызывать начало заболевания. Интересно, что между SNP rs4986790 и rs4986791 в TLR4, и SNP rs2066844 (R702W) и rs2066845 (G908R) в NOD2, обнаружена эпистатическая связь [80]. Эти SNP увеличивают риск развития CD в испанской популяции. У пациентов с SNP в NOD2 увеличивается восприимчивость к CD и заболеваемость подвздошной кишки, которая в присутствии вариантов SNP в TLR4 усиливает воспаление. Проведение генотипирования при диагностике CD может позволить выявить заболевание на ранней стадии и предупредить серьезные воспаления.
SNP rs5743618 (I602S) в гене TLR1 показал ассоциацию с CD, оказывая влияние на функцию и экспрессию рецептора на клеточной поверхности, а также на процессы его транспортировки к клеточной мембране [156] (рис. 1). Предполагается, что нефункциональный генотип 602S/S в TLR1 связан со сниженным риском Helicobacter pylori-индуцированных желудочных заболеваний через уменьшение Th-1 ответа.
Роль TLR5 в воспалении кишечника и гомеостазе остается неясной. В свою очередь, SNP rs5744168 (392STOP(R392X)) в TLR5 может защищать лиц еврейской национальности от CD (рис. 1). Это относительно распространенный полиморфизм с частотой аллеля 5%. Для rs5744168 характерен стоп-кодон, делающий TLR5 неактивным. Однако частота rs5744168 значительно ниже у пациентов с CD. SNP rs5744168 и rs2072493 (N592S) в TLR5 оказались связаны с UC в американских и индийских популяциях [82]. Уровень цитокинов оказался значительно модулирован у пациентов с разными генотипами SNP rs5744168 и rs2072493. У SNP rs5744174 (L616F) в TLR5 была обнаружена слабая связь с развитием CD у детей [125]. SNP rs5744174 оказался связан с повышенной экспрессией хемокина CCL20 в ответ на флагеллин, но при этом различий в экспрессии IL-8 или активации NF-κB не наблюдалось. В целом, большая функциональная активность rs5744174 является фактором риска развития CD у детей.
В свою очередь, SNP rs2109134 (A/T) и rs1548731 (+3121T>C) в TLR8 в блоке гаплотипа 1 (H1) оказались достоверно ассоциированы с CD у женщин [117] (рис. 1). Частота H1 составляла 72% в контроле, 59% с CD, 59% с UC и 59% в IBD, что может свидетельствовать о защитном эффекте SNP. Таким образом, TLR8 является геном восприимчивости к IBD, связанным с Х-хромосомой.
Ранее было установлено, что TLR9 может играть роль при заболеваниях с врожденными иммунными реакциями, таких как астма (рис. 1). Также сообщается о роли SNP в генах TLR9 в развитии IBD. В частности, аллель –1237C в TLR9 значительно чаще обнаруживается у пациентов с CD, что может свидетельствовать о корреляции наличия этого SNP с высокой предрасположенностью к развитию заболевания [139].
Генетическая изменчивость в TLR10 играет роль в межиндивидуальных различиях в восприимчивости к CD и клиническом исходе. Была обнаружена восприимчивость к CD при наличии SNP rs7658893 и rs7653908 в TLR10 в популяции Новой Зеландии [2] (рис. 1). При этом такой эффект TLR10 оказался независимым от NOD2, что предполагает разные пути передачи сигналов для обоих генов.
SNP в других генах
Следует отдельно отметить функциональное значение SNP rs11209026 (1142G>A;R381Q) в кодирующей последовательности рецептора интерлейкина-23 (IL-23R) [116]. Оказалось, что rs11209026 обеспечивает защиту от CD и UC, а также от аутоиммунных реакций, приводя к снижению первичных функциональных ответов CD4+ и CD8+ Т-клеток. SNP rs11209026 является одним из наиболее значительных SNP при аутоиммунных патологиях.
SNP в NOD2 (CARD15) представляет особый интерес. Известно, что SNP в NOD2 включают мутацию сдвига рамки (L1007fsinsC), которая приводит к усеченному белку NOD2 и двум аминокислотным заменам (R702W и G908R), которые тесно связаны с началом развития CD [134]. SNP в NOD2 расположены в богатом лейцином повторяющемся домене, который является микро-ассоциированной областью распознавания молекулярных паттернов и имеет молекулярную структуру, сходную с богатыми лейцином повторяющимися доменами TLRs [134]. Полученные данные могут указывать на существование общих сигнальных путей разных рецепторов и генов, оказывающих влияние на развитие IBD.
Ранее сообщалось, что нарушения секреторной активности клеток Панета, специализированных IECs и присутствующих в основании кишечных крипт в тонкой кишке, могут приводить к развитию CD. Важно, что указанные нарушения могут возникать вследствие наличия SNP в генах NOD2, ATG16L1, IRGM и LRRK2 [137, 162]. SNP в ATG16L1 (Thr300Ala) в IECs показал значительную связь с CD независимо от статуса NOD2 или IL-23R [79]. При этом ингибитор NF-κB-киназы-α (IKKα) фосфорилирует ATG16L1 и предотвращает его деградацию, при отсутствии которой фермент IRE1α накапливает и передает патологический белковый ответ (UPR) [29]. Гиперактивация IRE1α, в свою очередь, может спровоцировать спонтанный илеит, определяющий ключевые особенности CD [141].
В свою очередь, SNP в генах аутофагии ATG16L1 и IRGM были идентифицированы как генетические факторы риска развития CD. В частности, SNP в гене ATG16L1 (Thr300Ala) был идентифицирован как аллель риска CD, выраженный в аутофагической активности при ксенофагии. SNP IRGM (313C>T) представляет аллель, связанный с CD, который приводит к потере связывания микроРНК-196. Кроме того, микроРНК-196 сверхэкспрессируется в воспаленном эпителии пациентов с CD и подавляет защитный вариант IRGM [14]. Кроме того, IRGM играет главную роль в формировании и активации комплекса инициации аутофагии, включая ULK1 и BECN1, и соединяется с NOD2 и ATG16L1, что имеет важное значение для антимикробных и противовоспалительных функций аутофагии [22].
ЗАКЛЮЧЕНИЕ
Проведенный анализ исследований факторов риска и генетических предпосылок развития IBD позволил выявить некоторые функциональные последствия в генах TLRs и других генах (рис. 2). В частности, была рассмотрена роль ассоциаций SNP в генах TLRs и других генах в развитии, патогенезе и генетике CD и UC. Однако существующих исследований в области SNP генов TLRs недостаточно для однозначного понимания взаимосвязи между генетической изменчивостью и IBD, так как большинство проанализированных исследований были проведены на небольших выборках, принадлежащих к одной этнической группе. Поэтому нельзя утверждать, что результаты являются надежными и применимыми для IBD в общей популяции.

Тем не менее, согласно исследованиям, можно говорить о существующей тенденции к корреляции между SNP в генах TLRs и CD, и UC. В частности, эта корреляция основана на обнаруженных локусах риска CD и UC, которые могут увеличивать или снижать относительный риск развития воспалений. Установлено, что SNP в генах TLRs может проявляться в виде измененной клеточной продукции цитокинов и хемокинов, а связь между SNP в генах TLRs и IBD может быть опосредована явлением перекрестной иммунореактивности (перекрестной специфичности) с экзогенными антигенами.
Важно, что локусы SNP могут являться общими для широкого спектра иммуномедицинских заболеваний, что может вызывать определенные проблемы в идентификации, классификации, диагностике и терапии IBD. Такая полиморфность может объясняться схожестью этиологии и патогенеза заболеваний. Но у подавляющего большинства обнаруженных SNP в генах TLRs до сих пор не известны функции.
Изучение механизмов и патофизиологии является основой для понимания путей становления и развития IBD, формирования методов эффективной терапии. На сегодняшний день нет четкого понимания этиологии и патогенеза CD и UC. Однако известно, что воспаление при CD включает нарушение кишечной барьерной функции, нарушения в регуляции врожденных и адаптивных иммунных реакций, и кишечной микробиоты, а UC характеризуется постоянным поражением только толстой кишки и воспаление носит поверхностный характер. При этом, наличие факторов риска (недостаточная физическая активность, неполноценная диета, перенесенные вирусные или бактериальные инфекции, вредные привычки, условия жизни, прием антибиотиков и витамина D) у индивидуума, имеющего генетическую предрасположенность, в том числе ассоциированную с SNP в генах TLRs, может послужить пусковым механизмом для дебюта заболевания.
Проведение дальнейших исследований позволит получить новые сведения о роли SNP в генах TLRs в этиологии и патогенезе CD и UC. В свою очередь, использование SNP в качестве маркера в эпидемиологических и генетических исследованиях позволит создать эффективные методы генной терапии IBD.
Список литературы
Дворникова К.А., Быстрова Е.Ю., Платонова О.Н., Ноздрачев А.Д. Полиморфизм генов Toll-подобных рецепторов и ассоциированные с ним заболевания // Молекулярная медицина. 2019. Т. 17. № 6. С. 5–12.
Abad C., González-Escribano M.F., Diaz-Gallo L.M. et al. Association of Toll-like receptor 10 and susceptibility to Crohn’s disease independent of NOD2 // Genes Immun. 2011. V. 12. P. 635–642.
Abt M.C., Buffie C.G., Sušac B. et al. TLR-7 activation enhances IL-22–mediated colonization resistance against vancomycin-resistant enterococcus // Science Translational Medicine. 2016. V. 8. № 327. P. 1–12.
Adamji M., Day A.S. An overview of the role of exclusive enteral nutrition for complicated Crohn’s disease // Intest Res. 2019. V. 17. № 2. P. 171–176.
Ananthakrishnan A. N., Khalili H., Higuchi L.M. et al. Higher Predicted Vitamin D Status Is Associated With Reduced Risk of Crohn’s Disease // Gastroenterology. 2012. V. 142. № 3. P. 482–489.
Ananthakrishnan A.N., Bernstein C.N., Iliopoulos D. et al. Environmental triggers in IBD: a review of progress and evidence // Nat. Rev. Gastroenterol. Hepatol. 2018. V. 15. № 1. P. 39–49.
Ananthakrishnan A.N., Cagan A., Gainer V.S. et al. Normalization of plasma 25-hydroxy vitamin D is associated with reduced risk of surgery in Crohn’s disease // Inflamm. Bowel Dis. 2013. V. 19. № 9. P. 1921–1927.
Ananthakrishnan A.N., Khalili H., Konijeti G.G. et al. A prospective study of long-term intake of dietary fiber and risk of Crohn’s disease and ulcerative colitis // Gastroenterology. 2013. V. 145. № 5. P. 970–977.
Annese V.A. Review of Extraintestinal Manifestations and Complications of Inflammatory Bowel Disease // Saudi J. Med. Med. Sci. 2019 V. 7. № 2. P. 66–73.
Antushevich H. Fecal microbiota transplantation in disease therapy // Clinica Chimica Acta. 2020. V. 503. P. 90–98.
Atreya R., Bloom S., Scaldaferri F. et al. Clinical Effects of a Topically Applied Toll-like Receptor 9 Agonist in Active Moderate-to-Severe Ulcerative Colitis // J. Crohn’s and Colitis. 2016. V. 10. № 11. P. 1294–1302.
Bedri S., Sultan A.A., Alkhalaf M. et al. Epstein-Barr virus (EBV) status in colorectal cancer: a mini review // Human Vaccines & Immunotherapeutics. 2018. V. 15. № 3. P. 603-610.
Bharti D., Kumar A., Mahla R.S. et al. The role of TLR9 polymorphism in susceptibility to pulmonary tuberculosis // Immunogenetics. 2014. V. 66. P. 675–681.
Brest P., Lapaquette P., Souidi M. et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease // Nature Genetics. 2011. V. 43. № 3. P. 242–245.
Bruce A., Black M., Bhattacharya S. Mode of delivery and risk of inflammatory bowel disease in the offspring: systematic review and meta-analysis of observational studies // Inflamm. Bowel Dis. 2014. V. 20. P. 1217–1226.
Buckley J.P., Cook S.F., Allen J.K., Kappelman M.D. Prevalence of Chronic Narcotic Use Among Children With Inflammatory Bowel Disease // Clinical Gastroenterology and Hepatology. 2015. V. 13. № 2. P. 310–315.
Burgueño J.F., Reich A., Hazime H. et al. Expression of SARS-CoV-2 Entry Molecules ACE2 and TMPRSS2 in the Gut of Patients With IBD // Inflam. Bowel Diseases. 2020. V. 26. № 6. P. 797–808.
Camilleri M. Leaky gut: mechanisms, measurement and clinical implications in humans // Gut. 2019. V. 68. P. 1516–1526.
Cano-Gamez E., Trynka G. From GWAS to Function: Using Functional Genomics to Identify the Mechanisms Underlying Complex Diseases // Frontiers in Genetics. 2020. V. 11. № 424. P. 1–21.
Catalan-Serra I., Sandvik A.K., Bruland T. et al. T cells in Crohn’s disease: a new player in the disease pathogenesis? // J. Crohns Colitis. 2017. V. 11. P. 1135–1145.
Chassaing B., Ley R.E., Gewirtz A.T. Intestinal Epithelial Cell Toll-like Receptor 5 Regulates the Intestinal Microbiota to Prevent Low-Grade Inflammation and Metabolic Syndrome in Mice // Gastroenterology. 2014. V. 147. № 6. P. 1363–1377.
Chauhan S., Mandell M.A., Deretic V. IRGM Governs the Core Autophagy Machinery to Conduct Antimicrobial Defense // Molecular Cell. 2015. V. 58. № 3. P. 507–521.
Cholapranee A., Ananthakrishnan A.N. Environmental hygiene and risk of inflammatory bowel diseases: a systematic review and meta-analysis // Inflamm. Bowel Dis. 2016. V. 22. P. 2191–2199.
Cohen S.A., Gold B.D., Oliva S. et al. Clinical and Mucosal Improvement With Specific Carbohydrate Diet in Pediatric Crohn Disease // J. Pediatric Gastroenterology and Nutrition. 2014. V. 59. № 4. P. 516–521.
Costello S.P., Soo W., Bryant R.V. et al. Systematic review with meta-analysis: faecal microbiota transplantation for the induction of remission for active ulcerative colitis // Alimentary Pharmacology & Therapeutics. 2017. V. 46. № 3. P. 213–224.
David L.A., Maurice C.F., Carmody R.N. et al. Diet rapidly and reproducibly alters the human gut microbiome // Nature. 2013. V. 505. № 7484. P. 559–563.
de Souza H.S.P., Fiocchi C. Immunopathogenesis of IBD: current state of the art // Nat. Rev. Gastroenterol. Hepatol. 2016. V. 13. P. 13–27.
Devkota S., Wang Y., Musch M. W. et al. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10 −/− mice // Nature. 2012. V. 487. № 7405. P. 104–108.
Diamanti M.A., Gupta J., Bennecke M. et al. IKKα controls ATG16L1 degradation to prevent ER stress during inflammation // J. Exp. Med. 2017. V. 214. P. 423–437.
Dong J., Chen Y., Tang Y. et al. Body mass index is associated with inflammatory bowel disease: a systematic review and meta-analysis // PLoS One. 2015. V. 10. № 12. e0144872.
Dvornikova, K.A., Bystrova, E.Y., Platonova, O.N., Churilov, L.P. Polymorphism of toll-like receptor genes and autoimmune endocrine diseases // Autoimmunity Reviews. 2020. V. 19. № 4. P. 1–14.
El-Khider F., McDonald C. Links of Autophagy Dysfunction to Inflammatory Bowel Disease Onset // Digestive Diseases. 2016. V. 34. № 1–2. P. 27–34.
Ellinghaus D., Jostins L., Spain S.L. et al. Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci // Nat. Genet. 2016. V. 48. P. 510–518.
Feagan B.G., Sandborn W.J., D’Haens G. et al. Induction therapy with the selective interleukin-23 inhibitor risankizumab in patients with moderate-to-severe Crohn’s disease: a randomised, double-blind, placebo-controlled phase 2 study // Lancet. 2017. V. 389. P. 1699–1709.
Feagan B.G., Sandborn W.J., Gasink C. et al. Ustekinumab as Induction and Maintenance Therapy for Crohn’s Disease // New England J. Medicine. 2016. V. 375. № 20. P. 1946–1960.
Fiorino G., Bonifacio C., Peyrin-Biroulet L. Danese S. Preventing collateral damage in Crohn’s disease: the Lémann index // J. Crohns Colitis. 2016. V. 10. P. 495–500.
Franchimont D., Vermeire S., Housni E.H. et al. Deficient host–bacteria interactions in inflammatory bowel disease? The toll-like receptor (TLR)-4 Asp299gly polymorphism is associated with Crohn’s disease and ulcerative colitis // Gut. 2004. V. 53. P. 987–992.
Gajendran M., Loganathan P., Catinella A.P., Hashash, J.G. A comprehensive review and update on Crohn’s disease // Disease-a-Month. 2018. V. 64. № 2. P. 20–57.
Galbete C., Schwingshackl L., Schwedhelm C. et al. Evaluating Mediterranean diet and risk of chronic disease in cohort studies: an umbrella review of meta-analyses // European Journal of Epidemiology. 2018. V. 33. № 10. P. 909–931.
Gevers D., Kugathasan S., Denson L.A. et al. The treatment-naive microbiome in new-onset Crohn’s disease // Cell Host Microbe. 2014. V. 15. № 3. P. 382–392.
Gibson D.L., Montero M., Ropeleski M.J. et al. Interleukin-11 Reduces TLR4-Induced Colitis in TLR2-Deficient Mice and Restores Intestinal STAT3 Signaling // Gastroenterology. 2010. V. 139. № 4. P. 1277–1288.
Gopalakrishnan S., Durai M., Kitchens K. et al. Larazotide acetate regulates epithelial tight junctions in vitro and in vivo // Peptides. 2012. V. 35. № 1. P. 86–94.
Grover Z., Muir R., Lewindon P. Exclusive enteral nutrition induces early clinical, mucosal and transmural remission in paediatric Crohn’s disease // Journal of Gastroenterology. 2013. V. 49. № 4. P. 638–645.
Halfvarson J., Brislawn C.J., Lamendella R. et al. Dynamics of the human gut microbiome in inflammatory bowel disease // Nature Microbiology. 2017. V. 2. № 5. P. 1–15.
Harbord M., Annese V., Vavricka S.R., Allez M. et al. The First European Evidence-based Consensus on Extra-intestinal Manifestations in Inflammatory Bowel Disease // J. Crohn’s and Colitis. 2015. V. 10. № 3. P. 239–254.
Hashash J.G., Chintamaneni P., Ramos Rivers C.M. et al. Patterns of antibiotic exposure and clinical disease activity in inflammatory bowel disease: a 4-year prospective study // Inflamm. Bowel Dis. 2015. V. 21. № 11. P. 2576–2582.
He C., Bassik M.C., Moresi V. et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis // Nature. 2012. V. 481. № 7382. P. 511–515.
He Y., Zhu Z., Chen Y. et al. Development and Validation of a Novel Diagnostic Nomogram to Differentiate Between Intestinal Tuberculosis and Crohnʼs Disease // The American Journal of Gastroenterology. 2019. V. 114. № 3. P. 490–499.
Hold G.L., Smith M.G., McColl K.E., El-Omar E.M. A functional Toll-like receptor 4 polymorphism increases the risk of H. pylori-induced pre-malignant changes in the stomach // Gastroenterology. 2003. V. 124. P. 18–25.
Hollander D. Intestinal permeability barrier in Crohn’s disease: the difficulty in shifting the paradigm // Dig Dis Sci. 2013. V. 58. № 7. P. 1827–9.
Hooper L.V., Littman D.R., Macpherson A.J. Interactions Between the Microbiota and the Immune System // Science. 2012. V. 336. № 6086. P. 1268–1273.
Huang H., Fang M., Jostins L. et al. Fine-mapping inflammatory bowel disease loci to single-variant resolution // Nature. 2017. V. 547. P. 173–178.
Hubbard V.M., Cadwell K. Viruses, Autophagy Genes, and Crohn’s Disease // Viruses. 2011. V. 3. № 7. P. 1281–1311.
Jostins, L. et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease // 2012. V. 491 P. 119–124.
Kamdar K., Khakpour S., Chen J. et al. Genetic and Metabolic Signals during Acute Enteric Bacterial Infection Alter the Microbiota and Drive Progression to Chronic Inflammatory Disease // Cell Host & Microbe. 2016. V. 19. № 1. P. 21–31.
Kayser M., de Knijff P. Improving human forensics through advances in genetics, genomics and molecular biology // Nature Reviews Genetics. 2011. V. 12. № 3. P. 179–192.
Keag O.E., Norman J.E., Stock S.J. Long-term risks and benefits associated with cesarean delivery for mother, baby, and subsequent pregnancies: systematic review and meta-analysis // PLoS Med. 2018. V. 15. № 1. e1002494.
Kedia S., Das P., Madhusudhan K. S. et al. Differentiating Crohn’s disease from intestinal tuberculosis // World Journal of Gastroenterology. 2019. V. 25. № 4. P. 418–432.
Keefer L., Taft T. A systematic review of disease-related stigmatization in patients living with inflammatory bowel disease // Clinical and Experimental Gastroenterology. 2016. V. 49. P. 49-58.
Kerlin A.M., Long M., Kappelman M. et al. Profiles of Patients Who Use Marijuana for Inflammatory Bowel Disease // Digestive Diseases and Sciences. 2018. V. 63. № 6. P. 1600–1604.
Khalili H., Chan S.S.M., Lochhead P. et al. The role of diet in the aetiopathogenesis of inflammatory bowel disease // Nat. Rev. Gastroenterol. Hepatol. 2018. V. 15. P. 525–35.
Khalili H., Håkansson N., Chan S.S. et al. Adherence to a Mediterranean diet is associated with a lower risk of later-onset Crohn’s disease: results from two large prospective cohort studies // Gut. 2020. V. 0. P. 1–8.
Khor B., Gardet A., Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease // Nature. 2011. V. 474. № 7351. P. 307–317.
Kim C.H., Shanahan F. The Gastrointestinal Immune System* // Comprehensive Toxicology. 2010. V. 10. P. 39–52.
Kim S.M., Yoo S.Y., Nam S.H. et al. Identification of Korean-specific SNP markers from whole-exome sequencing data // Int. J. Legal Med. 2016. V. 130. № 3. P. 669–77.
Klenske E., Bojarski C., Waldner M., Rath T. et al. Targeting mucosal healing in Crohn’s disease: what the clinician needs to know // Ther Adv. Gastroenterol. 2019. V. 12. P. 1–11.
Kondo K., Ohfuji S., Watanabe K. et al. The association between environmental factors and the development of Crohn’s disease with focusing on passive smoking: A multicenter case-control study in Japan // PloS One. 2019. V. 14. № 6. e0216429.
Korolkova O.Y., Myers J.N., Pellom S.T., Wang L., M’koma A.E. Characterization of Serum Cytokine Profile in Predominantly Colonic Inflammatory Bowel Disease to Delineate Ulcerative and Crohn’s Colitides // Clinical Medicine Insights: Gastroenterology. 2015. V. 8. P. 29–44.
Larabi A., Barnich N., Nguyen H.T.T. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD // Autophagy. 2019. V. 16. № 1. P. 38–51.
Levine A., Sigall Boneh R., Wine E. Evolving role of diet in the pathogenesis and treatment of inflammatory bowel diseases // Gut. 2018. V. 67. P. 1726–1738.
Levine A., Wine E. Effects of enteral nutrition on Crohn’s disease: clues to the impact of diet on disease pathogenesis // Inflamm. Bowel Dis. 2013. V. 19. P. 1322–1329.
Liao P.Y., Lee K.H. From SNPs to functional polymorphism: The insight into biotechnology applications // Biochemical Engineering J. 2010. V. 49. № 2. P. 149–158.
Liu J.Z., van Sommeren S., Huang H. et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations // Nat. Genet. 2015. V. 47 № 9. P. 979–986.
Long A.G., Lundsmith E.T., Hamilton K.E. Inflammation and Colorectal Cancer // Current Colorectal Cancer Reports. 2017. V. 13 № 4. P. 341–351.
Lopetuso L.R., Chowdhry S., Pizarro T.T. Opposing Functions of Classic and Novel IL-1 Family Members in Gut Health and Disease // Frontiers in Immunology. 2013. V. 4. № 181. P. 1-21.
Lu Y., Li X., Liu S. et al. Toll-like Receptors and Inflammatory Bowel Disease // Frontiers in Immunology. 2018. V. 9. № 72. P. 1–9.
Maizels R.M., McSorley H.J., Smyth D.J. Helminths in the hygiene hypothesis: sooner or later? // Clinical & Experimental Immunology. 2014. V. 177. № 1. P. 38–46.
Mantzouranis G., Fafliora E., Saridi M. et al. Alcohol and narcotics use in inflammatory bowel disease // Ann. Gastroenterol. 2018. V. 31. № 6. P. 649–658.
Márquez A., Núñez C., Martínez A. et al. Role of ATG16L1 Thr300Ala polymorphism in inflammatory bowel disease: A Study in the Spanish population and a meta-analysis // Inflamm. Bowel Diseases. 2009. V. 15. № 11. P. 1697–1704.
Martinez-Chamorro A., Moreno A., Gómez-García M. et al. Epistatic interaction between TLR4 and NOD2 in patients with Crohn’s Disease: relation with risk and phenotype in a Spanish cohort // Immunobiology. 2016. V. 221. № 9. P. 927–933.
McKernan D.P., Finn D.P. An apPEAling new therapeutic for ulcerative colitis? // Gut. 2013. V. 63. № 8. P. 1207–1208.
Meena N.K., Ahuja V., Meena K., Paul J. Association of TLR5 Gene Polymorphisms in Ulcerative Colitis Patients of North India and Their Role in Cytokine Homeostasis // PloS One. 2015. V. 10. № 3. e0120697.
Mirsepasi-Lauridsen H.C., Vrankx K., Engberg J. et al. Disease-Specific Enteric Microbiome Dysbiosis in Inflammatory Bowel Disease // Frontiers in Medicine. 2018. V. 5. № 304. P. 1–8.
Moninuola O.O., Milligan W., Lochhead P., Khalili, H. Systematic review with meta-analysis: association between acetaminophen and nonsteroidal anti-inflammatory drugs (NSAIDs) and risk of Crohn’s disease and ulcerative colitis exacerbation // Aliment. Pharmacol. Ther. 2018. V. 47. P. 1428–1439.
Morgan M.E., Koelink P.J., Zheng B. et al. Toll-like receptor 6 stimulation promotes T-helper 1 and 17 responses in gastrointestinal-associated lymphoid tissue and modulates murine experimental colitis // Mucosal Immunology. 2014. V. 7. № 5. P. 1266–1277.
Morgan X.C., Tickle T.L., Sokol H. et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment // Genome Biology. 2012. V. 13. № 9. P. 1–18.
Mouli V.P., Ananthakrishnan A.N. Review article: vitamin D and inflammatory bowel diseases // Aliment Pharmacol Ther. 2014. V. 39. № 2. P. 125–136.
Muegge B.D., Kuczynski J., Knights D. et al. Diet Drives Convergence in Gut Microbiome Functions Across Mammalian Phylogeny and Within Humans // Science. 2011. V. 332. № 6032. P. 970–974.
Mukherjee S., Huda S., Sinha Babu S.P. Toll-like receptor polymorphism in host immune response to infectious diseases: A review // Scandinavian Journal of Immunology. 2019. V. 90. № 1. e12771.
Mutalib M., Bezanti K., Elawad M., Kiparissi F. The role of exclusive enteral nutrition in the management of orofacial granulomatosis in children // World Journal of Pediatrics. 2016. V. 12. № 4. P. 421–424.
Natoli G., Ostuni R. Adaptation and memory in immune responses // Nature Immunology. 2019. V. 20. № 7. P. 783–792.
Ng S.C., Leung W.K., Shi H.Y. et al. Epidemiology of inflammatory bowel disease from 1981 to 2014: results from a territory-wide population-based registry in Hong Kong // Inflamm. Bowel Dis. 2016. V. 22. P. 1954–1960.
Ng S.C., Shi H.Y., Hamidi N., Underwood F.E. et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies // Lancet. 2018. V. 390. № 10114. P. 2769–2778.
Nowarski R., Jackson R., Gagliani N. et al. Epithelial IL-18 Equilibrium Controls Barrier Function in Colitis // Cell. 2015. V. 163. № 6. P. 1444–1456.
Nys K., Agostinis P., Vermeire S. Autophagy: a new target or an old strategy for the treatment of Crohn’s disease? // Nat. Rev. Gastroenterol. Hepatol. 2013. V. 10. P. 395–401.
Odenwald M.A., Turner J.R. The intestinal epithelial barrier: a therapeutic target? // Nat. Rev. Gastroenterol. Hepatol. 2017. V. 14. P. 9–21.
Okazaki T., Wang M.H., Rawsthorne P. et al. Contributions of IBD5, IL23R, ATG16L1, and NOD2 to Crohn’s disease risk in a population-based case-control study: evidence of gene-gene interactions // Inflamm Bowel Dis. 2008. V. 14. № 11. P. 1528–41.
Ortizo R., Lee S.Y., Nguyen E.T. et al. Exposure to oral contraceptives increases the risk for development of inflammatory bowel disease: a meta-analysis of case-controlled and cohort studies // Eur. J. Gastroenterol. Hepatol. 2017. V. 29. P. 1064–1070.
Pakstis A.J., Speed W.C., Fang R. et al. SNPs for a universal individual identification panel // Hum. Genet. 2010. V. 127. № 3. P. 315–24.
Paramsothy S., Paramsothy R., Rubin D.T. et al. Faecal Microbiota Transplantation for Inflammatory Bowel Disease: A Systematic Review and Meta-analysis // Journal of Crohn’s and Colitis. 2017. V. 11. № 10. P. 1180–1199.
Park S.H., Kim Y.J., Rhee K.H. et al. A 30-year trend analysis in the epidemiology of inflammatory bowel disease in the Songpa-Kangdong district of Seoul, Korea in 1986–2015 // J. Crohns Colitis. 2019. V. 13. P. 1410–1417.
Pazmandi J., Kalinichenko A., Ardy R.C., Boztug, K. Early-onset inflammatory bowel disease as a model disease to identify key regulators of immune homeostasis mechanisms // Immunol. 2019. V. 287. P. 162–185.
Pelczar P., Witkowski M., Perez L.G. et al. A pathogenic role for T cell–derived IL-22BP in inflammatory bowel disease // Science. 2016. V. 354. № 6310. P. 358–362.
Peterson L.W., Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis // Nat. Rev. Immunol. 2014. V. 14. P. 141–153.
Pickert G., Neufert C., Leppkes M. et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing // The J. Experimental Medicine. 2009. V. 206. № 7. P. 1465–1472.
Piovani D., Danese S., Peyrin-Biroulet L. et al. Environmental risk factors for inflammatory bowel diseases: an umbrella review of meta-analyses // Gastroenterology. 2019. V. 157. P. 647–659.
Powell N., Lo J.W., Biancheri P. et al. Interleukin 6 Increases Production of Cytokines by Colonic Innate Lymphoid Cells in Mice and Patients With Chronic Intestinal Inflammation // Gastroenterology. 2015. V. 149. № 2. P. 456–467.
Quraishi M.N., Shaheen W., Oo Y.H., Iqbal T.H. Immunological mechanisms underpinning faecal microbiota transplantation for the treatment of inflammatory bowel disease // Clinical & Experimental Immunology. 2019. V. 199. P. 24–38.
Regner E.H., Ohri N., Stahly A. et al. Functional intraepithelial lymphocyte changes in inflammatory bowel disease and spondyloarthritis have disease specific correlations with intestinal microbiota // Arthritis Res. Ther. 2018. V. 20. № 149. P. 1–13.
Roda G., Chien Ng S., Kotze P.G. et al. Crohn’s disease // Nat. Rev. Dis. Primers. 2020. V. 6. № 22. P. 1–19.
Rowan C., Judge C., Cannon M.D. et al. Severe Symptomatic Primary CMV Infection in Inflammatory Bowel Disease Patients with Low Population Seroprevalence // Gastroenterology Research and Practice. 2018. V. 2018. № 1029401. P. 1–5.
Sainathan S.K., Bishnupuri K.S., Aden K. et al. Toll-like receptor-7 ligand imiquimod induces type I interferon and antimicrobial peptides to ameliorate dextran sodium sulfate-induced acute colitis // Inflam. Bowel Diseases. 2012. V. 18. № 5. P. 955–967.
Salvo R.E., Alonso C.C., Pardo C.C. et al. The intestinal barrier function and its involvement in digestive disease // Rev. Esp. Enferm. Dig. 2015. V. 107. № 11. P. 686–96.
Sánchez M.F., Fonseca C.G., Villeda R.M.A. et al. Transcript levels of Toll-Like receptors 5, 8 and 9 correlate with inflammatory activity in Ulcerative Colitis // BMC Gastroenterology. 2011. V. 11. № 138. P. 1–9.
Sands B.E., Chen J., Feagan B.G. et al. Efficacy and safety of MEDI2070, an antibody against interleukin 23, in patients with moderate to severe Crohn’s disease: a phase 2a study // Gastroenterology. 2017. V. 153. P. 77–86.
Sarin R., Wu X., Abraham C. Inflammatory disease protective R381Q IL23 receptor polymorphism results in decreased primary CD4+ and CD8+ human T-cell functional responses // Proc. Natl. Acad. Sci. 2011. V. 108. P. 9560–9565.
Saruta M., Targan S.R., Mei L. et al. High-frequency Haplotypes in the X Chromosome Locus TLR8 Are Associated With Both CD and UC in Females // Inflam. Bowel Diseases. 2009. V. 15. № 3. P. 321–327.
Sato-Otsubo A., Sanada M., Ogawa S. Single-Nucleotide Polymorphism Array Karyotyping in Clinical Practice: Where, When, and How? // Seminars in Oncology. 2012. V. 39. № 1. P. 13–25.
Savin Z., Kivity S., Yonath H., Yehuda S. Smoking and the intestinal microbiome // Archives of Microbiology. 2018. V. 200. № 5. P. 677–684.
Schmitt H., Neufert C., Neurath M.F., Atreya R. Resolution of Crohn’s disease // Seminars in Immunopathology. 2019. V. 41. № 6. P. 737–746.
Seeley J.J., Ghosh S. Molecular mechanisms of innate memory and tolerance to LPS // Journal of Leukocyte Biology. 2016. V. 101. № 1. P. 107–119.
Sewell G.W., Rahman F.Z., Levine A.P. et al. Defective tumor necrosis factor release from Crohnʼs disease macrophages in response to toll-like receptor activation: Relationship to phenotype and genome-wide association susceptibility loci // Inflam. Bowel Diseases. 2012. V. 18. № 11. P. 2120–2127.
Shakeri A., Cicero A.F.G., Panahi Y. et al. Curcumin: A naturally occurring autophagy modulator // Journal of Cellular Physiology. 2018. V. 234. № 5. P. 5643–5654.
Shaw S.Y., Blanchard J.F., Bernstein C.N. Association between the use of antibiotics in the first year of life and pediatric inflammatory bowel disease // Am. J. Gastroenterol. 2010. V. 105. № 12. P. 2687–2692.
Sheridan J., Mack D.R., Amre D.K. et al. A Non-Synonymous Coding Variant (L616F) in the TLR5 Gene Is Potentially Associated with Crohn’s Disease and Influences Responses to Bacterial Flagellin // PLoS One. 2013. V. 8. № 4. e61326.
Shmuel-Galia L., Aychek T., Fink A. et al. Neutralization of pro-inflammatory monocytes by targeting TLR2 dimerization ameliorates colitis // The EMBO J. 2016. V. 35. № 6. P. 685–698.
Sigall-Boneh R., Pfeffer G.T., Segal I. et al. Partial enteral nutrition with a Crohn’s Disease exclusion diet is effective for induction of remission in children and young adults with Crohn’s disease // Inflamm. Bowel Dis. 2014. V. 20. P. 1353–1360.
Sigall-Boneh R., Sarbagili S.C., Yanai H. et al. Dietary therapy with the Crohn’s Disease exclusion diet is a successful strategy for induction of remission in children and adults failing biological therapy // J. Crohns Colitis. 2017. V. 11. P. 1205–1212.
Sitkin S., Pokrotnieks J. Clinical Potential of Anti-inflammatory Effects of Faecalibacterium prausnitzii and Butyrate in Inflammatory Bowel Disease // Inflam. Bowel Diseases. 2018. V. 25. № 4. e40–e41.
Sokol H., Leducq V., Aschard H., Pham H.P. et al. Fungal microbiota dysbiosis in IBD // Gut. 2016. V. 66. № 6. P. 1039–1048.
Song C., Yang J., Ye W. et al. Urban-rural environmental exposure during childhood and subsequent risk of inflammatory bowel disease: a meta-analysis // Expert Rev. Gastroenterol Hepatol. 2019. V. 13. P. 591–602.
Sonnenberg G.F., Artis D. Innate lymphoid cells in the initiation, regulation and resolution of inflammation // Nature Medicine. 2015. V. 21. № 7. P. 698–708.
Stappers M., Thys Y., Oosting M. et al. TLR1, TLR2, and TLR6 gene polymorphisms are associated with increased susceptibility to complicated skin and skin structure infections // J. Infect. Dis. 2014. V. 210. P. 311–318.
Strober W., Watanabe T. NOD2, an intracellular innate immune sensor involved in host defense and Crohn’s disease // Mucosal Immunology. 2011. V. 4. № 5. P. 484–495.
Sugiura Y., Kamdar K., Khakpour S. et al. TLR1-induced chemokine production is critical for mucosal immunity against Yersinia enterocolitica // Mucosal Immunology. 2013. V. 6. № 6. P. 1101–1109.
Svolos V., Hansen R., Nichols B. et al. Treatment of Active Crohn’s Disease With an Ordinary Food-based Diet That Replicates Exclusive Enteral Nutrition // Gastroenterology. 2019. V. 156. P. 1354–67.
Thachil E., Hugot J.P., Arbeille B. et al. Abnormal activation of autophagy-induced crinophagy in Paneth cells from patients with Crohn’s disease // Gastroenterology. 2012. V. 142. P. 1097–1099.
Thomson M.J. High-throughput SNP genotyping to accelerate crop improvement // Plant. Breed. Biotechnol. 2014. V. 2. P. 195–212.
Török H.P., Glas J., Tonenchi L. et al. Crohn’s disease is associated with a toll-like receptor-9 polymorphism // Gastroenterology. 2004. V. 127. P. 365–366.
Torres J., Hu J., Seki A. et al. Infants born to mothers with IBD present with altered gut microbiome that transfers abnormalities of the adaptive immune system to germ-free mice // Gut. 2019. V. 0. P. 1–10.
Tschurtschenthaler M. et al. Defective ATG16L1-mediated removal of IRE1α drives Crohn’s disease-like ileitis // J. Exp. Med. 2017. V. 214. P. 401–422.
Uhlig H.H., Powrie F. Translating immunology into therapeutic concepts for inflammatory bowel disease // Annu. Rev. Immunol. 2018. V. 36. P. 755–781.
Vadva L., Propp T. et al. A New Pedigree-Based SNP Haplotype Method for Genomic Polymorphism and Genetic Studies // Cells. 2019. V. 8. № 835. P. 1–17.
Van der Giessen J., Binyamin D., Belogolovski A. et al. Modulation of cytokine patterns and microbiome during pregnancy in IBD // Gut. 2019. V. 69. P. 473–486.
Van Maele L., Carnoy C., Cayet D. et al. TLR5 Signaling Stimulates the Innate Production of IL-17 and IL-22 by CD3negCD127+ Immune Cells in Spleen and Mucosa // The Journal of Immunology. 2010. V. 185. № 2. P. 1177–1185.
Vijay K. Toll-like receptors in immunity and inflammatory diseases: Past, present, and future // International Immunopharmacology. 2018. V. 59. P. 391–412.
Wagner J., Skinner N.A., Catto-Smith A.G. et al. TLR4, IL10RA, and NOD2 mutation in paediatric Crohn’s disease patients: an association with Mycobacterium avium subspecies paratuberculosis and TLR4 and IL10RA expression // Medical Microbiology and Immunology. 2013. V. 202. № 4. P. 267–276.
Wang K., Baldassano R., Zhang H. et al. Comparative genetic analysis of inflammatory bowel disease and type 1 diabetes implicates multiple loci with opposite effects // Human Molecular Genetics. 2010. V. 19. № 10. P. 2059–2067.
Wang M.H., Picco M.F. Crohn’s Disease // Gastroenterology Clinics of North America. 2017. V. 46. № 3. P. 449–461.
Wang Q., Xu K.Q., Qin X.R. et al. Association between physical activity and inflammatory bowel disease risk: a meta-analysis // Dig. Liver Dis. 2016. V. 48. P. 1425–1431.
Weber A., Wasiliew P., Kracht M. Interleukin-1 (IL-1) Pathway // Science Signaling. 2010. V. 3. № 105. P. 1–6.
Wei Y.L., Qin C.J., Liu H.B. et al. Validation of 58 autosomal individual identification SNPs in three Chinese populations // Croat. Med J. 2014. V. 55. № 1. P. 10–3.
Wu Y.Y., Hsu C.M., Chen P.H. et al. Toll-Like Receptor Stimulation Induces Nondefensin Protein Expression and Reverses Antibiotic-Induced Gut Defense Impairment // Infection and Immunity. 2014. V. 82. № 5. P. 1994–2005.
Xie X., Shi X., Liu M. THE roles of TLR gene polymorphisms in atherosclerosis: a systematic review and meta-analysis of 35,317 subjects // Scand. J. Immunol. 2017. V. 86. P. 50–58.
Xu L., Lochhead P., Ko Y. et al. Systematic review with meta-analysis: breastfeeding and the risk of Crohn’s disease and ulcerative colitis // Aliment Pharmacol. Ther. 2017. V. 46. P. 780–789.
Yang C.A., Scheibenbogen C., Bauer S. et al. A frequent Toll-like receptor 1 gene polymorphism affects NK- and T-cell INF-g production and is associated with Helicobacter pylori-induced gastric disease // Helicobacter. 2013. V. 18. P. 13–21.
Yang J.Y., Kim M.S., Kim E. et al. Enteric Viruses Ameliorate Gut Inflammation via Toll-like Receptor 3 and Toll-like Receptor 7-Mediated Interferon-β Production // Immunity. 2016. V. 44. № 4. P. 889–900.
Ye B.D., Yang S.K., Song K. et al. Association of Toll-Like Receptor Gene with Crohn’s Disease in Koreans // The Korean Journal of Gastroenterology. 2009. V. 54. № 6. P. 377-383.
Yousefi S., Abbassi D.T., Kraaijenbrink T. et al. A SNP panel for identification of DNA and RNA specimens // BMC Genomics. 2018. V. 19. № 90. P. 1–12.
Zeng Z., Zhu Z., Yang Y. et al. Incidence and clinical characteristics of inflammatory bowel disease in a developed region of Guangdong province, China: a prospective populationbased study // J. Gastroenterol. Hepatol. 2013. V. 28. P. 1148–1153.
Zhang J., Yang J., Zhang L. et al. A new SNP genotyping technology Target SNP-seq and its application in genetic analysis of cucumber varieties // Sci. Rep. 2020. V. 10. № 5623. P. 1–11.
Zhang Q. et al. Commensal bacteria direct selective cargo sorting to promote symbiosis // Nat. Immunol. 2015. V. 16. P. 918–926.
Zhu L. et al. IL-10 and IL-10 receptor mutations in very early onset inflammatory bowel disease // Gastroenterology Res. 2017. V. 10. P. 65–69.
Дополнительные материалы отсутствуют.
Инструменты
Успехи физиологических наук