

Успехи физиологических наук, 2021, T. 52, № 2, стр. 13-20
Оксидативный стресс, старение и короткие пептиды
Б. И. Кузник a, b, Н. С. Линькова c, d, e, *, О. М. Ивко c
a Кафедра нормальной физиологии, ФГБОУ ВО “Читинская государственная медицинская академия” МЗ РФ
672000 Чита, Россия
b Инновационная клиника “Академия здоровья”
672038 Чита, Россия
c Отдел биогеронтологии АНО НИЦ “Санкт-Петербургский институт биорегуляции и геронтологии”
197110 Санкт-Петербург, Россия
d Кафедра терапии, гериатрии и антивозрастной медицины академии постдипломного образования ФГБУ “Федеральный научно-клинический центр специализированных видов медицинской помощи
и медицинских технологий ФМБА”
199034 Москва, Россия
e Лаборатория “Проблем старения” ФГАОУ ВО “Белгородский государственный
национальный исследовательский университет”
308009 Белгород, Россия
* E-mail: miayy@yandex.ru
Поступила в редакцию 25.12.2020
После доработки 30.12.2020
Принята к публикации 10.01.2021
Аннотация
В обзоре оксидативный стресс рассматривается как один из механизмов нарушения функций клеток, органов и тканей. При старении снижается активность ферментов антиоксидантной системы. Активные формы кислорода, образующиеся при оксидативном стрессе, повреждают ДНК, РНК, белки и липиды, приводя к апоптозу клеток. Нейродегенеративные заболевания могут развиваться в условиях оксидативного стресса и митохондриальной дисфункции. В терапии нейродегенеративной патологии перспективным направлением является использование антиоксидантов, например, мелатонина или коротких пептидов AEDG, KE. Пептид AEDG стимулирует синтез эндогенного мелатонина при старении организма, пептиды AEDG и KE обладают антиоксидантными и геропротекторными свойствами, нормализуя длину теломер и предотвращая апоптоз клеток. Изучение оксидативного стресса на клеточном, органном и тканевом уровне имеет важное значение для геронтологии и поиска новых подходов к терапии нейродегенеративных заболеваний.
Свободнорадикальная теория старения постулирует неспособность защитных механизмов организма реагировать на повреждения, вызванные активными формами кислорода (АФК) [44]. Оксидативный стресс (ОС) – дисбаланс между прооксидантами и антиоксидантами, приводящий к нарушению окислительно-восстановительной сигнализации и повреждению функционально активных молекул в клетке [12, 45]. ОС нарастает с возрастом и влияет на нормальное функционирование тканей и органов. ОС участвует в патогенезе ассоциированных с возрастом заболеваний: нейропатологии, сахарного диабета, атеросклероза, сердечно-сосудистой патологии, нарушениях функции почек и дыхательной системы, дисфункции скелетных мышц, артритов, деменции, ожирения, остеопороза и метаболического синдрома, злокачественных новообразований [32].
Клетки генерируют АФК как побочные продукты химических реакций процесса клеточного дыхания или в ответ на стрессорное воздействие [23]. АФК включают в себя супероксидный анион-радикал, оксида азота, перекись водорода, гидроксильный радикал, пероксинитрит и другие соединения [30]. АФК продуцируются в митохондриях и пероксисомах с участием семейства NADH-оксидаз, моноаминоксидазы и других ферментов.
Антиоксидантная система включает в себя низкомолекулярные вещества, конкурирующие с АФК за клеточные структуры: витамины, глутатион, липофильные антиоксиданты, мочевую кислоту. Антиоксиданты в большинстве случаев нейтрализуют АФК в результате прямой химической реакции, приводящей к образованию большого количества менее реактивных или инертных продуктов. В ответ на появление АФК экспрессируется супероксиддисмутаза (SOD), превращающая супероксид кислорода в перекись водорода. Перекись водорода под действием каталазы или глутатионпероксидазы преобразуется в воду и кислород. Дисульфид глутатиона, продуцируемый глутатионпероксидазой во время восстановления пероксидов, затем снова восстанавливается глутатионредуктазой [43]. Другие антиоксидантные белки способны связывать редокс-активные переходные металлы, чтобы поддерживать их в неактивной форме. Железо и медь могут взаимодействовать с перекисью водорода с образованием высокоактивного гидроксильного радикала [51].
Другой механизм защиты от ОС включает активацию ядерного фактора Nrf2. Nrf2 связывается с Kelch-подобным ECH-белком 1 (KEAP1), который действует в качестве субстратного адаптера, обеспечивающего убиквитинирование и деградацию Nrf2 под действием убиквитинлигазы E3 Cullin-3 [25]. В присутствии окислителей, KEAP1 высвобождает Nrf2 с последующей его транслокацией в ядро, где он активирует транскрипцию цитопротекторных генов через промоторные последовательности, содержащие консервативные элементы антиоксидантного ответа [17]. Это увеличивает уровень антиоксидантных ферментов и белков, таких как глутатион-S-трансфераза, NAD, хиноноксидоредуктаза-1, SOD, глутатионпероксидаза, гемоксигеназа-1, глутамат-цистеинлигаза, тиоредоксин и каталаза, а также способствует митохондриальному биогенезу, обеспечивая замену поврежденных органелл [37]. Некоторые ферменты способны восстанавливать поврежденные АФК белки, однако часто это невозможно. В большинстве случаев поврежденные окислителем белки удаляются посредством протеолиза, опосредованного либо убиквитин-протеасомальной, либо лизосомальной системой.
ОКСИДАТИВНЫЙ СТРЕСС, СТАРЕНИЕ И АССОЦИИРОВАННЫЕ С ВОЗРАСТОМ ЗАБОЛЕВАНИЯ
Синтез АФК и активных форм азота (АФА) является частью нормального аэробного клеточного метаболизма. Активация синтеза АФК и АФА происходит при стрессе и развитии ассоциированных с возрастом заболеваний. В то же время АФК, являясь побочными продуктами химических реакций в организме, в низких концентрациях могут выполнять регуляторные функции [8, 20]. При ОС интенсивность синтеза АФК и АФА превосходит антиоксидантную активность защитных систем клетки. Поврежденные молекулы, не восстановленные с помощью репаративной системы клетки и не подвергнутые утилизации, приводят к апоптозу или репликативному старению клеток. Эти же процессы лежат и в основе старения [53].
Головной мозг чувствителен к окислительному дисбалансу из-за его высокой потребности в энергии, усиленного потребления кислорода и большого количества легко окисляемых полиненасыщенных жирных кислот [52]. В головном мозге по сравнению с другими органами содержится значительно меньше антиоксидантов. Это затрудняет нейтрализацию АФК, которые накапливаются в гиппокампе, стриатуме и гипоталамусе при старении, что способствует апоптозу нейронов и снижению когнитивных функций [35].
Эндотелий сосудов головного мозга также является мишенью ОС. Накопление АФК в стенке сосуда вызывает эндотелиальную дисфункцию и участвует в патогенезе цереброваскулярных заболеваний. Сосудистая сеть головного мозга становится более восприимчивой к воспалительным процессам, что способствует усилению образования свободных радикалов [40].
Установлено, что продукция АФК в фибробластах кожи генерируется за счет модуляции метаболизма фосфатидилинозитол-3,4,5-трифосфата. При репликативном старении фибробластов повышенная продукция АФК может быть заблокирована путем ингибирования 3 сигнальных путей: PI3K, протеинкиназы C и NADPH-оксидазы. Снижение уровня PTEN (рhosphatase and tensin homolog) способствует уменьшению количества АФК в фибробластах кожи человека [36].
Одной из основных мишеней ОС являются нуклеотиды. Под влиянием АФК могут возникать разрывы и модификации оснований ДНК, что приводит к изменению активности генов и нарушению метаболизма клетки. ОС способствует укорочению длины теломер, репликативному старению клеток и уменьшению продолжительности жизни. При этом снижается активность теломеразы, которая не только регулирует длину теломер, но и защищает митохондрии от ОС [16].
На длину теломер влияют следующие факторы: генетическая предрасположенность, пол (женщины имеют более длинные теломеры по сравнению с мужчинами, возможно, причина в антиоксидантной активности эстрогенов), этническая принадлежность (у европейцев теломеры длиннее, чем у представителей негроидной расы), стресс (стресс и депрессия повышают уровень окислительного стресса, что, в свою очередь, уменьшает длину теломер и активность теломеразы), физическая активность (умеренная физическая активность увеличивает длину теломер), ожирение (связано с хроническим воспалением и увеличением АФК, что ведет к уменьшению длины теломер), курение и алкоголь (уменьшают длину теломер) [49]. 8-оксогуанин, индуцированный окислительным стрессом, изменяет способность TRF1 (Telomere Repeat Factor 1 – гомодимер, связывающийся с двухцепочечной TTAGGG-областью теломер и обладающий способностью ингибировать удлинение теломер теломеразой) и TRF2 (Telomere Repeat Factor 2 – гомодимер, которые связывается с той же областью, что и TRF1, но препятствует распознаванию двуспирального разрыва участка ДНК как повреждения, нуждающегося в репарации) связывать теломерные последовательности. Снижение связывания TRF1 и TRF2 отвечает за арест вилки репликации и укорочение или дисфункцию теломер, которые, в свою очередь, вызывают старение и нестабильность хромосом (рис. 1) [16].
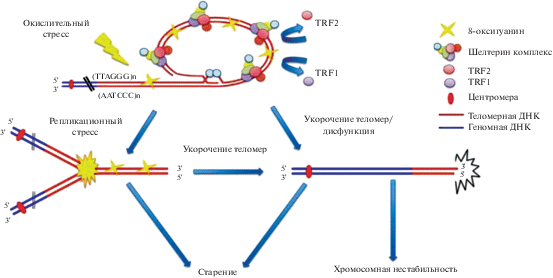
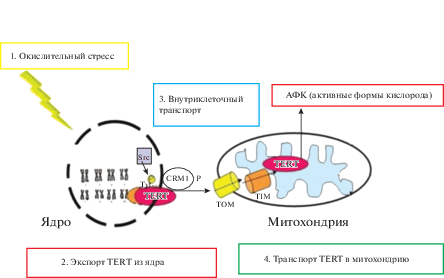
Во время окислительного стресса SRC-киназа фосфорилирует каталитическую субъединицу TERT теломеразы. Далее происходит экспорт TERT из ядра с помощью белка CRM1. TERT транспортируется в митохондрии с помощью белков TIM и TOM. В митохондриях TERT связывается с комплексом генов митохондриальной ДНК, который индуцирует потребление кислорода митохондриями (рис. 2) [42].
Однако многие продукты окисления белков или липидов могут быть полезны для выживания клеток. Такие соединения обычно образуются при умеренной интенсивности ОС. Этот тип стресса является частью адаптивного ответа (рис. 3) [13].

ОКСИДАТИВНЫЙ СТРЕСС И НЕЙРОДЕГЕНЕРАТИВНЫЕ ЗАБОЛЕВАНИЯ
Функции гиппокампа могут нарушаться при снижении антиоксидантной активности ферментов и облучении низкими дозами радиации, сопровождаемыми дефицитом внеклеточной SOD [24]. При этом снижаются функции обучения и памяти, уменьшается нейрогенез, происходит снижение численности шипиков дендритов нейронов гиппокампа [54]. Старение также связано с увеличением производства прооксидантов и накоплением конечных продуктов окисления. Нарушения функций гиппокампа при дефиците SOD, воздействии ионизирующего излучения и естественном старении имеют общие механизмы. Уровни тканевого окисления белков, липидов, ДНК и РНК повышаются с возрастом, что объясняется увеличением производства АФК и снижением активности антиоксидантной системы [40, 41].
ОС играет существенную роль в патогенезе болезней Альцгеймера (БА), Паркинсона (БП), Хантингтона (БХ), бокового амиотрофического склероза, атаксии Фридрейха. При всех указанных состояниях развивается митохондриальная дисфункция, сопровождаемая нарушением энергетического обмена и последующей гибелью нервных клеток [23, 47].
Повышенная продукция АФК, связанная с митохондриальной дисфункцией и сниженной антиоксидантной активностью защитной системы организма, снижает синаптическую активность, что приводит к когнитивной дисфункции. ОС и митохондриальная дисфункция образуют патофизиологиеский круг, который играет важную роль в патогенезе БА. Кроме того, АФК повреждают ядерную и митохондриальную ДНК, липиды, белки, регулирующие гомеостаз кальция и другие функции нейронов. Аномальный клеточный метаболизм, в свою очередь, может влиять на выработку и накопление токсичного β-амилоида и гиперфосфорилированного τ-протеина [47].
ОС и образование свободных радикалов наряду с эксайтотоксичностью приводят к апоптозу нейронов гиппокампа [38]. В то же время эксайтотоксичность и образование свободных радикалов, а также активация рецептора N-метил-D-аспартата (NMDAR) и нарушение синаптических функций, вызванные ОС, непосредственно связаны с патогенезом БА. Более того, механизм действия окислительно-восстановительного стресса на синапсы ассоциируется с измененной экспрессией рецептора NMDAR. Предполагается, что ОС, опосредованный через рецепторы NMDAR и их взаимодействие с другими молекулами, может быть движущей силой, приводящей к гиперфосфорилированию τ-протеина и дисфункции синапсов [27]. Увеличение концентрации кальция в цитоплазме, наблюдаемое при БА, запускает внутриклеточные каскады, приводящие к повышению уровня АФК, усилению ОС и ухудшению когнитивных функций [19]. Накопление токсичного β-амилоидного пептида в нейронах гиппокампа активирует связанные со стрессом киназы PKC, PKA, CaMKII, способствующие развитию ОС и апоптоза. Понимание роли ОС в нарушении метаболизма нейронов гиппокампа может иметь решающее значение для разработки терапевтических стратегий, направленных на предупреждение развития БА [26]. В патогенезе дегенеративных заболеваний важную роль играют астроциты. Нарушения функций астроцитов приводят к развитию ОС, воспалительным реакциям, глутаматной токсичности [35].
Укорочение теломер, ОС, повреждение ДНК, связанный со старением секреторный фенотип клеток и митохондриальная дисфункция способствуют более быстрому старению нейронов [18]. В других исследованиях выявлена взаимосвязь между снижением длины теломер в лейкоцитах крови и повышенным риском развития БА [21, 34].
Предполагается, что ОС является этиопатогенетическим фактором не только при БА, но и при других дегенеративных заболеваниях, что обусловлено нарушением регуляции антиоксидантного ответа, приводящего к дисфункции митохондрий [23].
Основной патологической особенностью БП является потеря дофаминергических нейронов и накопление телец Леви, содержащих α-синуклеин. При БП ингибируется активность митохондриальных комплексов I и IV в дофаминергических нейронах черной субстанции [50]. При этом α-синуклеин может быть импортирован в митохондрии и связываться со внутренней мембраной митохондрий дофаминергических нейронов. Гиперэкспрессия α-синуклеина обостряет митохондриальную дисфункцию, ОС и нейропатологию, вызванную ингибированием комплекса I, тогда как дефицит α-синуклеина ослабляет эти эффекты [39]. В то же время мономерный α-синуклеин способен взаимодействовать с АТФ-синтазой, что приводит к увеличению продукции АТФ. Кроме того, агрегированный α-синуклеин индуцирует изменение проницаемости мембран митохондрий, что приводит к их набуханию и апоптозу клеток [33].
При БХ отмечается многократное увеличение количества триплетов CAG в гене белка хантингтина (HTT), что приводит к повторениям полиглутамина в белке и снижению активности дыхательных комплексов II и III митохондрий [10]. В то же время ОС и воспаление являются другими распространенными патогенетическими факторами БХ [11]. В модели БХ у животных с ингибированием митохондриального комплекса II сверхэкспрессия гена NFE2L2 оказывает нейропротекторный эффект. Котрансфекция NFE2L2 с мутантным HTT в первичных нейронах стриатума снижает время метаболизма мутантного HTT и улучшает жизнеспособность клеток [48]. Окислительный стресс при БА и других нейродегенеративных заболеваниях является неотъемлемой частью патологического процесса и, следовательно, антиоксиданты могут быть полезны для терапии и профилактики указанных заболеваний [15, 23, 46]. Однако положительный результат действия антиоксидантов при нейродегенеративных заболеваниях может быть достигнут лишь в случае, если терапевтический агент способен преодолевать гематоэнцефалический барьер и усиливать механизмы эндогенной антиоксидантной защиты [14]. Кроме того, для лечения и профилактики БА могут быть использованы соединения, способные модулировать выработку АФК, например, мелатонин [22], или стимулирующие его эндогенный синтез пептиды эпифиза [3, 5].
ПЕПТИДЫ AEDG, KE, ГЕРОПРОТЕКЦИЯ И ОКСИДАТИВНЫЙ СТРЕСС
Короткие пептиды AEDG и KE, обладающие антиокидантными свойствами, способствуют увеличению длины теломер благодаря активации теломеразы. Пептид AEDG (Ala-Glu-Asp-Gly, Эпиталон) – регулятор функций эпифиза, сетчатки глаза и нейроиммуноэндокринной системы, обнаружен в составе полипептидного комплекса эпифиза [5]. Пептид AEDG нормализует ночной пик секреции гормонов мелатонина и кортизола у животных и лиц пожилого возраста, обладает иммунопроекторным, онкостатическим действием, повышает среднюю и максимальную продолжительности жизни животных в эксперименте [1, 9]. Добавление пептида AEDG в культуру эмбриональных фибробластов человека индуцирует экспрессию гена теломеразы и активацию собственно фермента теломеразы, что приводит к увеличению длины теломер в 2.4 раза [28, 29].
Пептид KE (Lys-Glu, Вилон) – представитель пептидных тимомиметиков, который был обнаружен в составе полипептидного комплекса, лекарственного препарата Тималина. Пептид КЕ стимулирует врожденный и адаптивный иммунитет, оказывает активирующее действие на макрофаги, лимфоциты, тимоциты и нейтрофилы [2, 4, 9]. Пептид KE при введении в организм трансгенных мышей подавляет экспрессию онкогена HER-2/neu в 2 раза, что сопровождается уменьшением диаметра опухоли [4, 9]. Кроме того, пептид KE способствует увеличению доли транскрибируемого эухроматина и снижению количества гетерохроматина в лимфоцитах крови лиц старческого возраста [4].
Установлено, что пептид иммунной системы KE и пептид эпифиза AEDG регулируют длину теломер в ФГА-стимулированных лимфоцитах крови. Этот результат коррелирует с продлением жизни у животных, получавших инъекции указанных пептидов. В большинстве случаев изменения длины теломер наблюдалось у людей среднего возраста: 8 случаев у мужчин среднего возраста против 4 случаев у молодых мужчин. Кроме того, изменение длины теломер чаще наблюдалось после применения пептида AEDG (7 случаев статистически значимых изменений) по сравнению с пептидом KE (5 случаев). Увеличение длины теломер после применения изучаемых пептидов наблюдалось в 2 раза чаще, чем ее уменьшение: 8 случаев против 4. Максимальное увеличение длины теломер (на 156%) было зарегистрировано в ФГА-стимулированных лимфоцитах человека среднего возраста после применения пептида AEDG. Таким образом, выявлена тенденция к “нормализации” длины теломер в ФГА-стимулированных лимфоцитах после применения пептидов. Пептиды AEDG и KE увеличивают длину теломер лимфоцитов по сравнению со средним значением, если оно было ниже среднего значения, и уменьшают этот параметр, если он изначально превышал среднее значение [5, 6].
Пептид AEDG нормализует значения антиоксидантной активности при возрастной патологии. После лечения пептидом AEDG происходит увеличение антирадикальной активности против АФК по сравнению с уровнем его до лечения [4]. Также наблюдается увеличение данной активности по сравнению с нормальной исследуемой группой без патологии. Кроме того, пептиды KE и AEDG снижают апоптоз и количество АФК в культуре нейронов крысы. При добавлении пероксида водорода в культуру нейронов крысы происходит увеличение количества АФК в 8 раз. При добавлении пептида KE в культуру нейронов с пероксидом водорода происходит статистически значимое уменьшение АФК в 0.8 раз, а при добавлении пептида AEDG – в 0.7 раз по сравнению с АФК в культуре нейронов с пероксидом водорода [31].
Таким образом, пептиды KE и AEDG, обладая антиоксидантным действием, снижают уровень ОС в клетке. Это способствует предотвращению следующего каскада реакций: трансформации гуанина в 8-оксигуанин, дестабилизации комплекса теломер с белками TRF1, TRF2, укорочению теломер, хромосомной нестабильности, клеточному старению, апоптозу и онкогенезу. Кроме того, снижение уровня окислительного стресса под действием пептидов разрывает круг положительной обратной связи: окислительный стресс-экспорт TERT из ядра в митохондрию–связывание TERT с комплексом митохондриальной ДНК– окислительный стресс (рис. 4). Эта гипотеза подтверждается тем, что пептиды способствуют нормализации длины теломер и активации TERT, преодолению лимита клеточных делений.

ЗАКЛЮЧЕНИЕ
ОС является одним из механизмов нарушения функций клеток, органов и тканей при старении. При действии сильного ОС, последствия которого не могут быть устранены белками антиоксидантной системы, наблюдается повреждение структуры ДНК, РНК, белков и липидов, приводящее к апоптозу клеток. Наиболее подверженным ОС являются нейроны головного мозга. Таким образом, ассоциированные с возрастом заболевания (БА, БХ, БП и др.) могут развиваться в условиях ОС. В терапии нейродегенеративной патологии перспективным направлением является использование антиоксидантов, например, мелатонина или коротких пептидов AEDG, KE. Пептид AEDG стимулирует синтез эндогенного мелатонина при старении организма, пептиды AEDG и KE обладают антиоксидантными и геропротекторными свойствами, нормализуя длину теломер и предотвращая апоптоз клеток. Понимание молекулярных механизмов ОС имеет важное значение для поддержания физиологических функций организма при старении и нейродегенеративной патологии.
Список литературы
Коркушко О.В., Хавинсон В.Х., Шатило В.Б. Пинеальная железа: пути коррекции при старении. СПб.: Наука. 2006.
Севостьянова Н.Н., Линькова Н.С., Полякова В.О. и др. Иммуномодулирующее действие вилона и его аналога в культурах клеток тимуса человека и животных // Клеточные технологии в биологии и медицине. 2012. № 4. С. 220–223.
Трофимова С.В., Линькова Н.С., Клименко А.А. и др. Пинеамин повышает синтез мелатонина в эпифизе у лиц пожилого возраста // Успехи геронтологии. 2017. Т. 30. № 3. С. 422–426.
Хавинсон В.Х., Кузник Б.И., Рыжак Г.А. Пептидные геропротекторы – эпигенетические регуляторы физиологических функций организма. СПб.: Изд-во РГПУ им. А.И. Герцена, 2014.
Хавинсон В.Х., Копылов А.Т., Васьковский Б.В. и др. Идентификация пептида AEDG в полипептидном комплексе эпифиза // Бюллетень экспериментальной биологии и медицины. 2017. Т. 164. № 7. С. 52–55.
Хавинсон В.Х., Пендина А.А., Ефимова О.А. и др. Влияние пептида AEDG на длину теломер и митотический индекс ФГА-стимулированных лимфоцитов крови человека // Клеточные технологии в биологии и медицине. 2019. № 3. С. 175–178.
Хавинсон В.Х., Линькова Н.С., Пендина А.А. и др. Изучение влияния пептида КЕ на длину теломер хромосом ФГА-стимулированных лимфоцитов человека // Медицинский академический журнал. 2019. Специальный выпуск. С. 166–168.
Ames B.N., Shigenaga M.K. Oxidants are a major contributor to aging // Ann. N.Y. Acad. Sci. 1992. V. 663. P. 85–96.
Anisimov V.N., Khavinson V.Kh. Peptide bioregulation of aging: results and prospects // Biogerontology. 2010. № 11. P. 139–149. https://doi.org/10.1007/s10522-009-9249-8
Bates G.P., Dorsey R., Gusella J.F. et al. Huntington disease // Nature Reviews Disease Primers. 2015. V. 1. https://doi.org/10.1038/nrdp.2015
Browne S.E., Beal M.F. Oxidative damage in Huntington’s disease pathogenesis // Antioxidants & Redox Signaling. 2006. V.8. № 11–12. P. 2061–2073.
Cabello-Verrugio C., Simon F., Trollet C., Santibañez J.F. Oxidative Stress in Disease and Aging: Mechanisms and Therapies // Oxid. Med. Cell Longev. 2017. 4310469. https://doi.org/10.1155/2017/4310469
Cai Z., Yan L.J. Protein oxidative modifications: beneficial roles in disease and health // J. Biochem. Pharmacol. Res. 2013. V. 1. P. 15–26.
Carvalho A.N., Firuzi O., Gama M.J. et al. Oxidative Stress and Antioxidants in Neurological Diseases: Is There Still Hope? // Curr. Drug Targets. 2017. V. 18. № 6. P. 705–718. https://doi.org/10.2174/1389450117666160401120514
Chen Z., Zhong C. Oxidative stress in Alzheimer’s disease // Neurosci. Bull. 2014. V. 30. № 2. P. 271–281. https://doi.org/10.1007/s12264-013-1423-y
Coluzzi E., Leone S., Sgura A. Oxidative Stress Induces Telomere Dysfunction and Senescence by Replication Fork Arrest // Cells. 2019. V. 8. № 1. pii: E19. https://doi.org/10.3390/cells8010019
Denzer I., Munch G., Friedland K. Modulation of mitochondrial dysfunction in neurodegenerative diseases via activation of nuclear factor erythroid-2-related factor 2 by food-derived compounds // Pharmacol. Res. 2016. V. 1. № 3. P. 80–94.
Ding X., Liu X., Wang F. et al. Role of Senescence and Neuroprotective Effects of Telomerase in Neurodegenerative Diseases // Rejuvenation Res. 2019. https://doi.org/10.1089/rej.2018.2115
Farooqui A.A., Yi Ong W., Lu X.R. et al. Neurochemical consequences of kainate-induced toxicity in brain: involvement of arachidonic acid release and prevention of toxicity by phospholipase A(2) inhibitors // Brain Res. Rev. 2001. V. 38. P. 61–78.
Ferrer M.D., Sureda A., Mestre A. et al. The double edge of reactive oxygen species as damaging and signaling molecules in HL60 cell culture // Cell Physiol. Biochem. 2010. V. 25. P. 241–252. https://doi.org/10.1159/000276558
Guo Y., Yu H. Leukocyte Telomere Length Shortening and Alzheimer’s Disease Etiology // J. Alzheimers Dis. 2019. V. 69. № 3. P. 881–885. https://doi.org/10.3233/JAD-190134
Hossain M.F., Uddin M.S., Uddin G.M.S. et al. Melatonin in Alzheimer’s Disease: A Latent Endogenous Regulator of Neurogenesis to Mitigate Alzheimer’s Neuropathology // Mol. Neurobiol. 2019. V. 56. № 12. P. 8255–8276. https://doi.org/10.1007/s12035-019-01660-3
Huang M.L., Chiang S., Kalinowski D.S. et al. The Role of the Antioxidant Response in Mitochondrial Dysfunction in Degenerative Diseases: Cross-Talk between Antioxidant Defense, Autophagy, and Apoptosis // Oxid. Med. Cell Longev. 2019. 2019:6392763. https://doi.org/10.1155/2019/6392763
Huang T.T., Leu D., Zou Y. Oxidative stress and redox regulation on hippocampal-dependent cognitive functions // Arch. Biochem. Biophys. 2015. V. 576. P. 2–7. https://doi.org/10.1016/j.abb.2015.03.014
Joshi G., Johnson J.A. The Nrf2-ARE pathway: A valuable therapeutic target for the treatment of neurodegenerative diseases // Recent. Pat. CNS Drug Discov. 2012. V. 7. P. 218–229.
Kamat P.K., Kalani A., Rai S. et al. Mechanism of Oxidative Stress and Synapse Dysfunction in the Pathogenesis of Alzheimer’s Disease: Understanding the Therapeutics Strategies // Mol. Neurobiol. 2016. V. 53. № 1. P. 648–661.
Kamat P.K., Rai S., Swarnkar S. et al. Okadaic acid-induced tau phosphorylation in rat brain: role of NMDA receptor // Neuroscience. 2013. V. 238. P. 97–113.
Khavinson V.Kh, Bondarev I.E., Butyugov A.A., Smirnova T.D. Peptide Promotes Overcoming of the Division Limit in Human Somatic Cell // Bulletin of Experimental Biology and Medicine. 2004. V. 137. № 5. P. 613–616.
Khavinson V.Kh., Bondarev I.E., Butyugov A.A. Epithalon Peptide Induces Telomerase Activity and Telomere Elongation in Human Somatic Cells // Bulletin of Experimental Biology and Medicine. 2003. V. 135. № 6. P. 590–592.
Korovila I., Hugo M., Castro J.P. et al. Proteostasis, oxidative stress and aging // Redox. Biol. 2017. V. 13. P. 550–567. https://doi.org/10.1016/j.redox.2017.07.008
Kozina L.S., Arutjunyan A.V., Khavinson V.Kh. Antioxidant properties of geroprotective peptides of the pineal gland // Adv. Gerontol. Geriatr. Suppl.1. 2007. P. 213–216.
Liu Z., Zhou T., Ziegler A.C. et al. Oxidative stress in neurodegenerative diseases: from molecular mechanisms to clinical applications // Oxid. Med. Cell. Longev. 2017. 2525967 https://doi.org/10.1155/2017/2525967
Ludtmann M.H.R., Angelova P.R., Horrocks M.H. et al. α-Synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease // Nature Communications. 2018. V. 9. № 1. P. 2293. https://doi.org/10.1038/s41467-018-04422-2
Ma F., Lv X., Du Y. et al. Association of Leukocyte Telomere Length with Mild Cognitive Impairment and Alzheimer’s Disease: Role of Folate and Homocysteine // Dement. Geriatr. Cogn. Disord. 2019. P. 1–12. https://doi.org/10.1159/000501958
Mythri R.B., Venkateshappa C., Harish G. et al. Evaluation of markers of oxidative stress, antioxidant function and astrocytic proliferation in the striatum and frontal cortex of Parkinson’s disease brains // Neurochemical research. 2011. V. 36. P. 1452–1463. https://doi.org/10.1007/s11064-011-0471-9
Noh E.M., Kim J.M., Hong O.Y. et al. PTEN inhibits replicative senescence-induced MMP-1 expression by regulating NOX4-mediated ROS in human dermal fibroblasts // J. Cell Mol. Med. 2017. V. 21. № 11. P. 3113–3116. https://doi.org/10.1111/jcmm.13220
Patel M. Targeting oxidative stress in central nervous system disorders // Trends Pharmacol. Sci. 2016. V. 37. P. 768–778. https://doi.org/10.1016/j.tips.2016.06.007
Rai S., Kamat P.K., Nath C., Shukla R. Glial activation and post-synaptic neurotoxicity: the key events in streptozotocin (ICV) induced memory impairment in rats // Pharmacol. Biochem. Behav. 2014. V. 117. P. 104–117.
Reeve A.K., Ludtmann M.H.R., Angelova P.R. et al. Aggregated α-synuclein and complex I deficiency: exploration of their relationship in differentiated neurons // Cell Death & Disease. 2015. V. 6. e1820. https://doi.org/10.1038/cddis.2015.166
Salminen L.E., Paul R.H. Oxidative stress and genetic markers of suboptimal antioxidant defense in the aging brain: a theoretical review // Rev. Neurosci. 2014. V. 25. № 6. P. 805–819. https://doi.org/10.1515/revneuro-2014-0046
Santos R.X., Correia S.C., Zhu X. et al. Mitochondrial DNA oxidative damage and repair in aging and Alzheimer’s disease // Antioxid. Redox. Signal. 2013. V. 18. № 18. P. 2444–2457. https://doi.org/10.1089/ars.2012.5039
Saretzki G. Cellular senescence in the development and treatment of cancer // Current Pharmaceutical Design. 2010. V. 16. № 1. P. 79–100.
Sautin Y.Y., Johnson R.J. Uric acid: the oxidant-antioxidant paradox // Nucleosides Nucleotides Nucleic Acids. 2008. V. 27. P. 608–619. https://doi.org/10.1080/15257770802138558
Sies H. Oxidative stress: a concept in redox biology and medicine // Redox Biol. 2015. V. 4. P. 180–183. https://doi.org/10.1016/j.redox.2015.01.002
Sies H., Jones D. Oxidative stress. In: Fink G., editor. 2nd ed. Vol. 3. Elsevier; Amsterdam: 2007. P. 45–48.
Thapa A., Carroll N.J. Dietary Modulation of Oxidative Stress in Alzheimer’s Disease // Int. J. Mol. Sci. 2017. V. 18. № 7. pii: E1583. https://doi.org/10.3390/ijms18071583
Tönnies E., Trushina E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease // J. Alzheimers Dis. 2017. V. 57. № 4. P. 1105–1121. https://doi.org/10.3233/JAD-161088
Tsvetkov A.S., Arrasate M., Barmada S. et al. Proteostasis of polyglutamine varies among neurons and predicts neurodegeneration // Nature Chemical Biology. 2013. V. 9. № 9. P. 586–592. https://doi.org/10.1038/nchembio.1308
Turner K.J., Vasu V., Griffin D.K. Telomere Biology and Human Phenotype // Cells. 2019. V. 8. № 1. pii: E73. https://doi.org/10.3390/cells8010073
Tysnes O.-B., Storstein A. Epidemiology of Parkinson’s disease // J. Neural Transmission. 2017. V. 124. № 8. P. 901–905. https://doi.org/10.1007/s00702-017-1686-y
Valko M., Jomova K., Rhodes C.J. et al. Redox- and non-redox-metal-induced formation of free radicals and their role in human disease // Arch. Toxicol. 2016. V. 90. P. 1–37. https://doi.org/10.1007/s00204-015-1579-5
Wang X., Wang W., Li L. et al. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease // Biochim. Biophys. Acta. 2014. V. 1842. № 8. P. 1240–127. https://doi.org/10.1016/j.bbadis.2013.10.015
Yan L.J. Positive oxidative stress in aging and aging-related disease tolerance // Redox Biol. 2014. V. 2. P. 165–169. https://doi.org/10.1016/j.redox.2014.01.002
Zou Y., Zou Y., Corniola R. et al. Extracellular superoxide dismutase is important for hippocampal neurogenesis and preservation of cognitive functions after irradiation // Proc. Natl. Acad. Sci. USA. 2012. V. 109. № 52. P. 21522–21527. https://doi.org/10.1073/pnas.1216913110
Дополнительные материалы отсутствуют.
Инструменты
Успехи физиологических наук