

Вестник Военного инновационного технополиса «ЭРА», 2022, T. 3, № 2, стр. 169-174
ИССЛЕДОВАНИЕ ЧАСТИЦ ПОЛИ(D,L-ЛАКТИД-СО-ГЛИКОЛИДА) С ПОЛИЭЛЕКТРОЛИТНОЙ ОБОЛОЧКОЙ МЕТОДОМ ДИНАМИЧЕСКОГО РАССЕЯНИЯ СВЕТА
М. А. Ванцян 1, *, А. В. Бусленко 1, В. В. Саруханова 1, Т. В. Букреева 1, 2
1 Национальный исследовательский центр “Курчатовский институт”
Москва, Россия
2 Институт кристаллографии им. А.В. Шубникова ФНИЦ “Кристаллография и фотоника” РАН
Москва, Россия
* E-mail: Vantsyan_MA@nrcki.ru
Поступила в редакцию 17.05.2022
После доработки 17.05.2022
Принята к публикации 27.06.2022
Аннотация
Методом динамического рассеяния света изучены субмикронные частицы поли(D,L-лактид-со-гликолида) с оболочками, нанесенными послойной адсорбцией синтетических и природных полиэлектролитов. Исследовано влияние нанесения полиэлектролитных слоев на средний размер частиц и их ζ-потенциал. Сделаны выводы об оптимальном составе комплексных частиц с точки зрения их размеров для использования в живых системах и агрегативной устойчивости.
ВВЕДЕНИЕ
Субмикронные частицы сополимеров молочной и гликолевой кислот – поли(D,L-лактид-со-гликолидов) (PLGA) – являются высокоперспективными носителями лекарственных веществ благодаря биосовместимости и биоразлагаемости этого полимерного материала [1, 2]. Однако потенциал их биомедицинского применения ограничен из-за ряда недостатков, к которым, в частности, относятся сложность модификации поверхности частиц PLGA из-за отсутствия поверхностно-активных функциональных групп, а также характерное “взрывное” высвобождение содержимого (“burst release”), особенно при доставке гидрофильных биоактивных веществ [3]. В качестве простого, но эффективного способа покрытия поверхности частиц PLGA, решающего эти проблемы, было предложено использовать послойное нанесение противоположно заряженных полиэлектролитов (метод Layer-by-layer, LbL) [4].
Уникальное преимущество модификации LbL по сравнению с другими методами заключается в том, что покрытие поверхности происходит за счет электростатического притяжения, что делает этот подход применимым к широкому кругу заряженных плоских поверхностей и коллоидных частиц [5–7]. Проницаемость активных агентов через такие покрытия можно регулировать изменением состава и количества полиэлектролитных слоев [8, 9]. Помимо контроля высвобождения инкапсулированных веществ метод LbL позволяет модифицировать химический состав поверхности и придавать носителям дополнительные свойства. Например, LbL-оболочка может стимулировать взаимодействие капсул с клеточной мембраной [10].
Было показано, что LbL-нанесение полиэлектролитов является эффективным способом формирования оболочек на частицах PLGA [11]. Такие системы “ядро–оболочка” предложено применять для доставки противоопухолевых препаратов [12–14], лечения диабета [15], стимуляции адаптивного иммунитета [16], сосудистой терапии [17, 18], тканевой инженерии [19, 20].
Для создания LbL-покрытий на частицах P-LGA использовали широкий ряд синтетических и природных полимеров, например поли(стиролсульфонат натрия) и поли(аллиламин гидрохлорид) [21], альгинат натрия [22], гиалуроновую кислоту [17, 23, 24], хитозан [22, 23], поли-L-гистидин [16], поли-L-лизин [21, 25]. Компонентами LbL-оболочек могут быть белки и нуклеиновые кислоты, предназначенные для доставки. Связывание таких молекул в многослойной структуре оболочки приводит к снижению их диффузии через слои и защищает от преждевременной деструкции [23, 26, 27 ].
Поверхностная модификация частиц PLGA методом LbL повышает возможности реализации стратегии адресной доставки лекарственных веществ. Например, химическая модификация фолиевой кислотой наночастиц PLGA, покрытых полиакриловой кислотой и полиэтиленимином [28] или хитозаном и альгинатом [29], приводила к увеличению захвата частиц раковыми клетками печени человека HepG2. LbL-оболочку с внешним слоем из гиалуроновой кислоты использовали для специфического взаимодействия частиц PLGA с мезенхимальными стволовыми клетками костного мозга человека, содержащими поверхностные CD44-рецепторы [17].
При нанесении LbL-оболочки размеры нано- и субмикрочастиц PLGA могут существенно увеличиться за счет адсорбции полимеров в виде петель и глобул [30]. Кроме того, может происходить агрегация частиц [22]. Такие последствия могут привести к существенным ограничениям применения разрабатываемых носителей в качестве средств доставки лекарств. Для использования частиц PLGA в живых системах они должны обладать достаточной агрегативной устойчивостью и размерами не более 200–300 нм [31]. Поэтому необходимо модифицировать частицы таким образом, чтобы они имели относительно небольшой размер и высокий по модулю ζ‑потенциал, свидетельствующий об электростатической стабилизации системы.
В работе для формирования полиэлектролитной оболочки на субмикронных частицах PLGA методом LbL использовали синтетические полиэлектролиты поли(аллиламин гидрохлорид), поли(диаллилдиметиламмоний хлорид) (поликатионы), поли(стиролсульфонат натрия) (полианион), а также природные полиэлектролиты, обладающие определенной биосовместимостью и биодеградируемостью – диэтиламиноэтилдекстран (поликатион), натриевую соль сульфата декстрана, альгинат и гиалуронат натрия (полианионы). Методом динамического рассеяния света (ДРС) проведено исследование влияния нанесения полиэлектролитных слоев на средний размер частиц и их ζ-потенциал, и сделаны выводы об оптимальном составе и условиях получения системы.
ЭКСПЕРИМЕНТАЛЬНО-МЕТОДИЧЕСКАЯ ЧАСТЬ
Ядром для формирования полиэлектролитной оболочки служили субмикронные частицы из синтетического сополимера поли(D,L-лактид-со-гликолида) с мольным соотношением звеньев лактид:гликолид = 75:25, среднемассовой молекулярной массой Mw = 92 кДа, полученные по методике, описанной в [32]. Образец P01 представлял собой частицы PLGA, стабилизированные поливиниловым спиртом (PVA), массовое соотношение PLGA/PVA составляло 2:1. Частицы F01 PVA не содержали.
Для формирования оболочки использовали следующие полиэлектролиты: поли(аллиламин гидрохлорид) (PAH), Mw = 120–200 кДа, поли(диаллилдиметиламмоний хлорид) (PDADMAC), Mw = 200–350 кДа, поли(стиролсульфонат натрия) (PSS), Mw = 75 кДа, диэтиламиноэтилдекстран (DEAE), Mw = 150 кДа, натриевую соль сульфата декстрана (DS), Mw = 500 кДа, альгинат натрия (Alg), Mw = 300 кДа, гиалуронат натрия (Hyal), Mw = 260 кДа.
Формирование полиэлектролитной оболочки проводили по следующим методикам.
Адсорбция синтетических поликатионов. 0.25 мл исходной суспензии наночастиц центрифугировали при 20 000g в течение 30 мин, затем удаляли супернатант. К полученному осадку приливали определенный объем раствора PAH или PDADMAC известной концентрации таким образом, чтобы массовое соотношение частиц и полиэлектролита составляло примерно 1:1. В случае P01 концентрация и объем раствора PAH составляли 3.84 г/л (в 0.5 М растворе NaCl) и 0.25 мл соответственно, PDADMAC – 2.76 г/л (в 0.5 М растворе NaCl) и 0.35 мл.
Для F01 концентрация и объем раствора PAH составляли 3.84 г/л (в 0.5 М растворе NaCl) и 0.5 мл соответственно, PDADMAC – 2.76 г/л (в 0.5 М растворе NaCl) и 0.7 мл.
Для удаления несвязанного с наночастицами полиэлектролита суспензию наночастиц центрифугировали при 20 000g в течение 30 мин. Затем удаляли супернатант, заливали осадок дистиллированной водой, ресуспендировали ультразвуком (10 мин) и перемешивали на шейкере в течение 20–30 мин с частотой 700–800 об./мин. Данную процедуру повторяли 3 раза.
Адсорбция синтетического полианиона. К осадку частиц приливали определенный объем раствора PSS известной концентрации (0.4 мл, 2.84 г/л, в 0.5 М растворе NaCl) таким образом, чтобы массовое соотношение частиц и полиэлектролита составляло примерно 1:1. Все дальнейшие манипуляции осуществляли так же, как и в случае поликатиона.
Адсорбцию природных полиэлектролитов проводили аналогично подобной процедуре с PAH, PDADMAC и PSS. При этом DEAE, DS, Alg и Hyal растворяли в 0.2 М растворе NaCl; концентрации данных полиэлектролитов составляли 2.5–2.8 г/л.
Гидродинамический диаметр (Zav), индекс полидисперсности (PDI) и ζ-потенциал частиц определяли на приборе Malvern Zetasizer ZS (Великобритания).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Свойства исходных частиц P01 и F01 приведены в табл. 1. Можно заключить, что введение PVA в частицы приводит к некоторому увеличению их размеров и повышению ζ-потенциала по абсолютной величине, что, в целом, характерно для подобных систем [33]. Поскольку все исходные частицы имели отрицательный заряд, для формирования первого полиэлектролитного слоя применяли поликатионы.
Таблица 1.
Свойства исходных частиц
| Образец | Zav, нм | PDI | ζ-потенциал, мВ | Концентрация суспензии, мг/мл |
|---|---|---|---|---|
| P01 | 145 ± 1 | 0.07 | –26 ± 1 | 3.6 |
| F01 | 130 ± 1 | 0.054 | –22 ± 1 | 6.8 |
Сначала изучали частицы PLGA с оболочкой из синтетических полиэлектролитов. Результаты исследования методом ДРС показали, что модификация частиц P01 поликатионами РАН и PDADMAC вызывает небольшое увеличение их размеров на 9–14 нм (табл. 2). При этом индекс PDI практически не изменяется, т.е. система остается довольно монодисперсной. Адсорбция поликатионов приводит к изменению ζ-потенциала на несколько десятков милливольт, и знак данной величины меняется на противоположный, что свидетельствует об успешной адсорбции и PAH, и PDADMAC. Отметим, что величина изменения ζ-потенциала при нанесении обоих поликатионов практически одинакова, и конечное значение составляет +18…+19 мВ (табл. 2). Такое невысокое абсолютное значение, в целом, характерно для нанесения поликатионов [4, 29]. Тем не менее агрегативная устойчивость частиц не снижается, о чем свидетельствуют лишь небольшое смещение пика кривой распределения по размерам в сторону бóльших значений в результате нанесения поликатиона (рис. 1а, 1б) и его неизменность в течение нескольких суток.
Таблица 2.
Свойства частиц PLGA с оболочкой из синтетических полиэлектролитов в сравнении с исходными
| Образец | Число слоев | Zav, нм | PDI | ζ-потенциал, мВ |
|---|---|---|---|---|
| P01 | 0 | 145± 1 | 0.07 | –26 ± 1 |
| P01–PAH | 1 | 159 ± 1 | 0.09 | 18 ± 2 |
| P01–PDADMAC | 1 | 154 ± 2 | 0.07 | 19 ± 3 |
| P01–PAH–PSS | 2 | 204 ± 1 | 0.177 | –36 ± 5 |
| P01–PDADMAC–PSS | 2 | 228 ±5 | 0.196 | –31 ± 6 |
| F01 | 0 | 130 ± 1 | 0.054 | –22 ± 1 |
| F01–PAH | 1 | 599 ± 7 | 0.222 | 49 ± 3 |
| F01–PDADMAC | 1 | 377 ± 51 | 0.392 | –3 ± 3 |
| F01–PAH–PSS | 2 | 518 ± 12 | 0.268 | –29 ± 5 |
Рис. 1.
Распределение частиц по размерам для образцов P01 (а), P01–PAH (б), P01–PAH–PSS (в) (на основе данных ДРС).
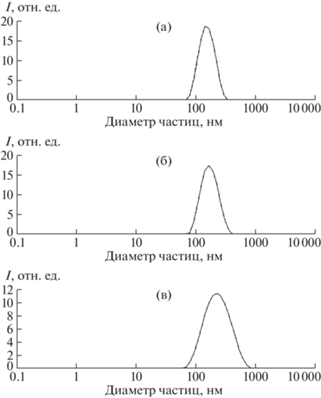
В результате последующей адсорбции полианиона PSS гидродинамический диаметр частиц увеличивается на несколько десятков нанометров (на 45 нм для РАН и 74 нм для PDADMAC), а ζ‑потенциал частиц меняет знак на противоположный, его значение составляет –36 мВ в случае РАН и –31 мВ в случае PDADMAC (табл. 2). При этом частицы становятся более полидисперсными – пик распределения по размерам уширяется (рис. 1в), индекс PDI увеличивается в 2 и 2.8 раза соответственно (табл. 2). По-видимому, несмотря на наименьшее значение Mw для PSS, его адсорбция на поверхности частиц, модифицированных поликатионами, происходит неравномерно.
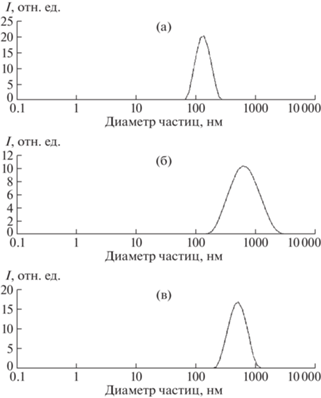
В случае образца F01 размеры модифицированных частиц в несколько раз превышают размеры исходных (рис. 2, табл. 2); вероятно, модифицированные частицы обладают невысокой агрегативной устойчивостью. Кроме того, на наличие агрегатов указывает достаточно широкий пик на кривой распределения частиц по размерам (рис. 2б). Тем не менее в случае F01–PAH наблюдается наибольшее значение ζ-потенциала, что может указывать на высокую агрегативную стабильность модифицированных частиц. Такое несоответствие вызвано, вероятно, тем, что существенный вклад в рассеяние вносят крупные частицы, в то время как высокой электрофоретической подвижностью обладают мелкие. Снижение среднего размера частиц при адсорбции PSS на F01–PAH с 599 до 518 нм можно объяснить частичным разрушением агрегатов, а также конформационными перестройками макромолекул полиэлектролитов (вследствие достаточно высокой гибкости цепей PAH и PSS), что приводит к формированию более компактной полиэлектролитной оболочки. На частичное разрушение агрегатов также указывает менее широкий (по сравнению с F01–PAH) пик на графике распределения частиц по размерам (рис. 2в). Высокая склонность к агрегации отмечается и для F01–PDADMAC, при этом наиболее крупные агрегаты оседали на дне пробирки в виде хлопьев, сгустков, поэтому измерение ζ-потенциала не показало переключения заряда поверхности частиц и дальнейшая модификация данного образца не представлялась целесообразной.
Рис. 2.
Распределение частиц по размерам для образцов F01 (а), F01–PAH (б), F01–PAH–PSS (в) (на основе данных ДРС).
Далее в работе были рассмотрены частицы P-LGA с оболочками из природных полиэлектролитов. Нанесение всех использованных полиэлектролитов вызывало изменение ζ-потенциала частиц на несколько десятков милливольт с переменой знака на противоположный, что свидетельствует об их успешной адсорбции (табл. 3). Адсорбция DEAE на частицах P01 привела к увеличению гидродинамического диаметра частиц на 82 нм (табл. 3). При этом на кривой распределения по размерам частиц P01–DEAE (рис. 3б) наблюдается существенное уширение пика по сравнению с исходным для P01 (рис. 3а), что, по-видимому, соответствует образованию агрегатов, состоящих из нескольких частиц.
Таблица 3.
Свойства наночастиц с оболочкой из природных полиэлектролитов в сравнении с исходными
| Образец | Число слоев | Zav, нм | PDI | ζ-потенциал, мВ |
|---|---|---|---|---|
| P01 | 0 | 145± 1 | 0.07 | –26 ± 1 |
| P01–DEAE | 1 | 227 ± 6 | 0.22 | 25 ± 1 |
| P01–DEAE–DS | 2 | 390 ± 5 | 0.11 | –47 ± 3 |
| P01–DEAE–Alg | 2 | 318 ± 4 | 0.27 | –21 ± 5 |
| P01–DEAE–Hyal | 2 | 395 ± 6 | 0.38 | –10 ± 2 |
| F01 | 0 | 130 ± 1 | 0.054 | –22 ± 1 |
| F01–DEAE | 1 | 431 ± 84 | 0.50 | 31 ± 2 |
Рис. 3.
Распределение частиц по размерам для образцов P01 (а), P01–DEAE (б), P01–DEAE–DS (в) (на основе данных ДРС).
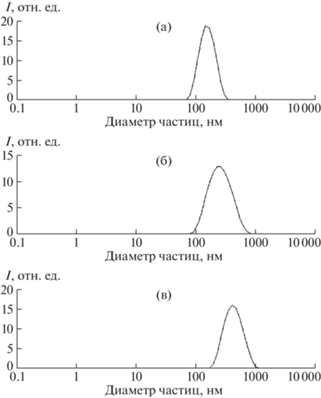
Нанесение полианионов DS, Alg и Hyal на частицы P01–DEAE увеличило их средний размер на 163, 91 и 168 нм соответственно. PDI при нанесении DS снизился в 2 раза, в случае Alg практически не изменился, а для Hyal увеличился в 1.7 раза (табл. 3). При этом изменение PDI хорошо согласуется с изменением ζ-потенциала частиц: наибольшее абсолютное значение наблюдается для DS (–47 ± 3 мВ), наименьшее – для Hyal (–10 ± 2). Более узкий пик на графике распределения частиц P01–DEAE-DS по размерам (рис. 3в) по сравнению с пиком для частиц P01–DEAE (рис. 3б) может соответствовать частичному разрушению агрегатов при увеличении размера за счет формирования рыхлой полиэлектролитной оболочки. Бóльшая склонность к агрегации частиц P01–DEAE–Alg и P01–DEAE–Hyal по сравнению с частицами, модифицированными синтетическими полиэлектролитами, обусловлена наличием в используемых полисахаридах большего (по сравнению с PAH, PDADMAC и PSS) числа функциональных групп (гидроксильных и карбоксильных), способствующих межмолекулярному водородному связыванию [34].
При взаимодействии F01 с DEAE происходит образование крупных агрегатов, которые, так же как и в случае F01–PDADMAC, оседали в виде хлопьев. Обработка ультразвуком с последующим перемешиванием не приводит к разрушению агрегатов: по данным ДРС размеры F01–DEAE в несколько раз превышают размеры исходных частиц (табл. 3). Таким образом, дальнейшее формирование слоя полианиона на частицах F01–DEAE не представляется целесообразным.
Сила полиэлектролитов (как синтетических, так и природных) [35] не оказывает существенного влияния на размеры модифицированных частиц. Так, использование как слабого PAH, так и сильных PDADMAC и PSS приводит к образованию относительно компактных оболочек (табл. 2), в то время как рыхлые структуры наблюдаются и для сильного DS, и для слабых DEAE, Alg и Hyal (табл. 3). Вероятно, строение оболочек в большей степени зависит от таких факторов, как химическая природа, гибкость цепи, наличие дополнительных функциональных групп в полимере.
ВЫВОДЫ
На поверхность субмикронных частиц PLGA за счет послойной адсорбции могут быть успешно нанесены как синтетические, так и природные полиэлектролиты. Частицы на основе PLGA, содержащие PVA, проявляют меньшую склонность к агрегации, при этом наименьшими размерами и относительно высоким по модулю ζ-потенциалом обладают частицы с LbL-оболочкой из PAH–PSS и PDADMAC–PSS. В качестве наиболее перспективной системы для медико-биологического использования можно рекомендовать частицы PLGA/PVA с оболочкой DEAE–DS, средний диаметр которых составил 390 нм при PDI 0.11.
В дальнейшем планируется получение частиц PLGA с большим числом полиэлектролитных слоев, а также включение модельных соединений в ядро и/или оболочку.
Исследование выполнено при финансовой поддержке НИЦ “Курчатовский институт”, (приказ № 2755 от 28.10.2021 г.).
В работе использовано оборудование Ресурсного центра оптической микроскопии и спектроскопии НИЦ “Курчатовский институт”.
Список литературы
Danhier F., Ansorena E., Silva J.M. et al. // J. Controlled Release. 2012. V. 161. № 2. P. 505.
Седуш Н.Г., Кадина Ю.А., Разуваева Е.В. и др. // Российские нанотехнологии. 2021. Т. 16. № 4. С. 462. https://doi.org/10.1134/S1992722321040117
Kakade S., Manickam D.S., Handa H. et al. // Int. J. Pharm. 2009. V. 365. P. 44.
Luo R., Neu B., Venkatraman S.S. // Small. 2012. V. 16. № 8. P. 2585.
Decher G. // Science. 1997. V. 277. P. 1232.
Sukhorukov G.B., Donath E., Davis S. et al. // Polym. Adv. Technol. 1998. V. 9. P. 759.
Peyratout C.S., Dahne L. // Angew. Chem. Int. Ed. 2004. V. 43. P. 3762.
Berg M.C., Zhang L., Cohen R.E. et al. // Biomacromolecules. 2006. V. 7. P. 357.
Yu X., Pishko M.V. // Biomacromolecules. 2011. V. 12. P. 3205.
Mironov E.P., Borodina T.N., Yurina D.G. et al. // Colloids Surf. B. 2019. V. 184. P. 110464.
Букреева Т.В., Марченко И.В., Тимаева О.И. // Российские нанотехнологии. 2021. Т. 16. № 4. С. 482. https://doi.org/10.1134/S1992722321040026
Domínguez-Ríos R., Sanchez-Ramirez D.R., Ruiz-Sa-ray K. et al. // Colloids Surf. B. 2019. V. 178. P. 199.
Chai F., Sun L., He X. et al. // Int. J. Nanomed. 2017. V. 12. P. 1791.
Loya-Castro M.F., Sanchez-Mejia M., Sanchez-Ramirez D.R. et al. // J. Colloid Interface Sci. 2018. V. 518. P. 122.
Wu J.Z., Williams G.R., Li H.Y. et al. // Drug Deliv. 2017. V. 24. № 1. P. 1513.
Ou W., Jiang L., Thapa R.K. et al. // Theranostics. 2018. V. 8. № 17. P. 4574.
Lee P.C., Zan B.S., Chen L.T. et al. // Int. J. Nanomedicine. 2019. V. 14. P. 1533.
Meng R., Li K., Chen Z. et al. // J. Huazhong Univ. Sci. Technol. Med. Sci. 2016. V. 36. № 1. P. 14.
Park J.S., Park K., Woo D.G. et al. // Biomacromolecules. 2008. V. 9. № 8. P. 2162.
Kong J., Wei B., Groth T. et al. // J. Biomed. Mater. Res. A. 2018. V. 106. № 10. P. 2714.
Zhao Q., Fang Zh.X., Chen M.M. et al. // Micro Nano Lett. 2018. V. 13. № 6. P. 835.
Wang F., Yuan J., Zhang Q. et al. // J. Biomater. Sci. Polym. Ed. 2018. V. 29. № 13. P. 1566.
Zhang B.J., Han Z.W., Duan K. et al. // J. Biomed. Mater. Res. A. 2018. V. 106. № 1. P. 95.
Guter M., Breunig M. // Methods Mol. Biol. 2019. V. 1943. P. 153.
Kapadia C.H., Ioele S.A., Day E.S. // J. Biomed. Mater. Res. A. 2020. V. 108. № 3. P. 601.
Go D.P., Palmer J.A., Mitchell G.M. et al. // J. Biomed. Mater. Res. A. 2015. V. 103. № 5. P. 1849.
Zuo Q., Guo R., Hona A. et al. // Biomed. Mater. 2015. V. 10. № 3. P. 35008.
Zhou J., Romero G., Rojas E. et al. // Macromol. Chem. Phys. 2010. V. 211. № 4. P. 404.
Zhou J., Romero G., Rojas E. et al. // J. Colloid Interface Sci. 2010. V. 345. № 2. P. 241.
Chai F., Sun L., He X. et al. // Int. J. Nanomed. 2017. V. 12. P. 1791.
Gaumet M., Vargas A., Gurny R. et al. // Eur. J. Pharm. Biopharm. 2008. V. 69. P. 1.
Fessi H., Puisieux F., Devissaguet J.P. et al. // Int. J. Pharm. 1989. V. 55. P. R1.
Vandervoort J., Ludwig A. // Int. J. Pharm. 2002. V. 238. P. 77.
Ануфриева Е.В., Некрасова Т.Н., Браудо Е.Е. и др. // Высокомол. соединения. А. 1985. Т. XXVII. № 11. С. 2347.
Sharma V., Sundaramurthy A. // Beilstein J. Nanotechnol. 2020. V. 11. P. 508.
Дополнительные материалы отсутствуют.
Инструменты
Вестник Военного инновационного технополиса «ЭРА»