

Вестник Военного инновационного технополиса «ЭРА», 2023, T. 4, № 1, стр. 3-19
Гипоксия и HIF-зависимый каскад в регуляции генов ранозаживления в дермальных фибробластах
К. А. Дариенко 1, *, А. А. Пантелеев 1, **, ***
1 Национальный исследовательский центр “Курчатовский институт”
Москва, Россия
* E-mail: darcristall@gmail.com
** E-mail: a.a.pantel@gmail.com
*** E-mail: Panteleev_AA@nrcki.ru
Поступила в редакцию 10.10.2023
После доработки 10.10.2023
Принята к публикации 10.10.2023
Аннотация
Разработка новых подходов к лечению кожных ран и ожогов остается актуальной задачей современной биомедицины. Одним из перспективных направлений в этой области является использование алло- и аутологичных дермальных фибробластов человека, культивируемых перед их трансплантацией в условиях гипоксии (низкого парциального давления кислорода). Фибробласты являются основными клетками, участвующими в формировании внеклеточного матрикса дермального слоя кожи, функциональность которого нарушается при повреждении. В обзоре кратко описаны различные популяции фибробластов кожи и их функции, основные этапы ранозаживления и роль фибробластов в этом процессе, а также физиологическая роль гипоксии и ее влияние на ранозаживляющие функции фибробластов. Особое внимание уделено регуляторному HIF1-зависимому молекулярному кас-каду, являющемуся ключевым элементом клеточной адаптации к гипоксии, а также механизму HIF1-опосредованной регуляции генов, принимающих участие в ранозаживлении.
ОГЛАВЛЕНИЕ
Введение
1. Структура кожных покровов
2. Гетерогенность популяций фибробластов дермы
3. Фибробласты – основные участники процесса ранозаживления
4. Фибробласты в тканевой инженерии
5. HIF-1-фактор как регулятор клеточной адаптации к условиям гипоксии
6. Гипоксия и HIF-1 в контроле функций фибробластов
Заключение
ВВЕДЕНИЕ
Кожа – это крупнейший орган человеческого тела, составляющий до 15% от общей массы тела взрослого человека и являющийся барьером между внутренней и внешней средой, обеспечивающим защиту от экзогенных механических, физических, химических и биологических воздействий [1, 2]. Функциональное значение кожи не переоценимо, особенно учитывая, что разнообразные факторы внешней среды воздействуют на организм постоянно, вызывая соответствующие адаптивные реакции. Именно в коже, подвергающейся непосредственному воздействию этих факторов, происходит первичный иммунный ответ [3]. Повреждение целостности кожного покрова и формирование раны приводят к нарушению трофики из-за повреждения сосудов, развитию воспалительной реакции и к последующей активации ранозаживляющего потенциала неповрежденных структур кожи с дальнейшим восстановлением целостности и функциональности ткани.
Известен ряд заболеваний, при которых регенеративный потенциал кожи значительно снижен. В результате возникающие раны и дефекты не только не способны к естественному восстановлению, но и трудно поддаются различным медицинским воздействиям. Это часто приводит к развитию хронических ран/язв, некрозу ткани и необходимости хирургического вмешательства. Например, одним из самых распространенных осложнений у пациентов с диабетом 2 типа является диабетическая язва стопы. Этот синдром (синдром диабетической стопы – СДС) развивается примерно у 5–10% пациентов-диабетиков чаще всего при длительном стаже заболевания [4, 5]. СДС также может проявляться при диабете 1 типа. У 2–16% пациентов с СДС осложнения заболевания приводят к ампутации стопы [4, 6]. Согласно [7] от 50 до 70% всех ампутаций нижних конечностей приходится на долю больных сахарным диабетом.
Трофической язвой (длительно незаживающей раной) называют дефект тканей со значительно сниженной способностью к заживлению из-за нарушения трофики и иннервации кожи и подлежащих тканей [8]. Патогенез трофических язв сводится к формированию так называемой “фибриновой манжетки” вокруг капилляров кожи, что вызывает аноксию тканей (полное отсутствие кислородного питания) и, соответственно, развитие и прогрессирование трофических патологий. Традиционными способами лечения подобных заболеваний являются средства местного воздействия на поверхность раны [9], а также средства системной фармакотерапии, направленной в основном на коррекцию свертывающей способности крови, достижение сосудорасширяющего действия и на улучшение доставки О2 в раневое поле для устранения местной гипоксии (дефицита O2) [10].
Ранозаживление является сложным, комплексным процессом. В зависимости от его длительности рана (язва) приобретает острый или хронический характер [11]. С начала 1960-х гг. в связи с резким ростом количества случаев диабета постоянно растет интерес к разработке новых подходов к лечению хронических диабетических язв [12–14]. В частности, используются принципы биоинженерии и клеточной терапии для создания клеточных и тканевых трансплантатов, которые, временно замещая утерянные ткани, восстанавливают нормальную функцию поврежденных структур и поддерживают их архитектонику. В настоящее время одним из распространенных подходов является использование для трансплантации тканеинженерных эквивалентов кожи на основе пористых трехмерных структур из материалов искусственного и природного происхождения (скаффолдов/матриксов), используемых в качестве клеточных носителей [15, 16]. Обязательное условие успешного использования подобных матриксов – их способность обеспечивать механическую и структурную поддержку искусственной ткани, а также клеточную адгезию и пролиферацию. В итоге это стимулирует процесс регенерации.
Одним из способов повышения эффективности этого подхода является использование клеток с повышенным синтезом факторов ранозаживления. В данном обзоре рассматривается возможность использования гипоксии и активируемых ею молекулярных каскадов для повышения ранозаживляющего потенциала дермальных фибробластов.
1. СТРУКТУРА КОЖНЫХ ПОКРОВОВ
С гистологической точки зрения кожа состоит из трех слоев: эпидермиса, дермы и гиподермы [17].
Морфофункциональное формирование кожи млекопитающих в процессе онтогенеза начинается после гаструляции (одной из самых ранних стадий эмбрионального развития) на 6–9-й день развития эмбриона мыши (стадии E6.5–Е9.5) из эктодермального зародышего листка. До стадии Е12.5 мышиный эпителий состоит из одного или двух слоев кератиноцитов, а подлежащая дерма выглядит гомогенной по составу. Мультипотентные р63-позитивные кератиноциты формируют базальный слой эмбрионального эпидермиса, дающий начало всем вышележащим стратифицирующимся слоям и всем придаткам кожи, таким как волосяные фолликулы и разнообразные железы. К E17.5 эпидермис полностью сформирован и приобретает свои барьерные свойства. Мезодермальный зародышевый листок эмбриона дает начало формированию соединительной ткани верхней (папиллярной, сосочковой) части дермы с более высокой плотностью фибробластов и нижней ее части (ретикулярной, сетчатой) с низкой плотностью клеток, но высокой плотностью ECM (extracellular matrix, внеклеточного матрикса). Сразу после рождения мыши (P2) в коже формируется гиподерма (подкожная жировая клетчатка), состоящая из преадипоцитов и дифференцированных адипоцитов [18, 19].
Сформированный эпидермис млекопитающих представляет собой многослойный эпителий, образованный клетками, расположенными восходящими слоями от базальной мембраны к поверхности кожи [20]. Самый глубокий слой – базальный (stratum basale) – отделен от дермы базальной мембраной и прикреплен к ней полудесмосомами. Кератиноциты этого слоя могут иметь разную форму, от кубической до столбчатой. Они являются митотически активными, и их дочерние клетки формируют все вышележащие слои эпидермиса [21]. Кератиноциты супрабазальных слоев дифференцируются в процессе движения к поверхности кожи, формируя сначала шиповатый слой (stratum spinosum), а затем гранулярный слой (stratum granulosum). Гранулярный слой характеризуется активным накоплением в специфических гранулах различных компонентов (белки и липиды), необходимых для процесса корнификации кератиноцитов и формирования рогового слоя эпидермиса. Над гранулярным слоем может располагаться блестящий слой (stratum lucidum), имеющий вид тонкого прозрачного слоя, состоящего из элеидина. Однако он наблюдается только в толстой коже ступней и ладоней. Верхний ороговевший слой клеток эпидермиса (stratum corneum) более водонепроницаем, чем нижележащие слои, препятствует как потере влаги, так и проникновению через кожу патогенов и других посторонних веществ, обеспечивая таким образом барьерные свойства эпидермиса [22].
Среди эпителиальных клеток эпидермиса – кератиноцитов – располагаются и неэпителиальные клетки, такие как меланоциты, клетки Лангерганса, Т-лимфоциты и клетки Меркеля [23]. Меланоциты – клетки нейрального происхождения, образующиеся из нервной трубки и нервного гребня эмбриона [24]. В коже человека меланоциты располагаются в базальном слое эпидермиса и продуцируют пигмент меланин, который защищает делящиеся кератиноциты от УФ-излучения [25, 26]. В коже мышей эпидермальные меланоциты мигрируют в волосяной фолликул в первые дни после рождения и в эпидермисе их не остается. Клетки Лангерганса (дендритные клетки, эпидермальные макрофаги) являются частью иммунной системы кожи млекопитающих [3, 27]. Они представляет собой антигенпрезентирующие клетки костномозгового происхождения, которые локализуются в базальном или шиповатом слоях эпидермиса. Клетки Лангерганса захватывают антигены, проникающие в эпидермис, осуществляют их процессинг и передают соответствующую информацию лимфоцитам, контролируя таким образом адаптивный иммунный ответ [3, 28, 29]. Еще один вид эпидермальных клеток – тактильные клетки Меркеля, имеющие нейральное происхождение. Они связываются с афферентными нервными волокнами и выполняют функцию механорецепторов. Тела тактильных клеток расположены в базальном слое эпидермиса, при этом своими отростками они соединяются с кератиноцитами базального и шиповатого слоев с помощью десмосом [30].
Дерма является самым толстым из трех слоев кожи и представляет собой опорную соединительную ткань, имеющую специфическую архитектонику и состоящую в основном из фибробластов и некоторых других типов клеток, а также ECM (коллагеновые, эластические, ретикулярные волокна и аморфное вещество). В дерме располагаются волосяные фолликулы, потовые и сальные железы, кровеносные сосуды и нервные окончания [31]. Сосочковый (верхний) слой дермы образован рыхлой волокнистой соединительной тканью с лимфатическими и кровеносными капиллярами, нервными волокнами и окончаниями. Плотность клеток в сосочковом слое (фибробласты, тучные клетки, макрофаги, лейкоциты, пигментные клетки) значительно ниже, чем в эпидермисе. Характеризуется он и сравнительно небольшим количеством ECM (коллагеновые, эластические и ретикулярные волокна). Сетчатый (нижний) слой дермы образован плотной волокнистой соединительной тканью, характеризуется пучковым расположением коллагеновых волокон, выполняющих опорную функцию [32–34]. Для сетчатого слоя характерна еще менее плотная клеточная составляющая, чем для сосочкового слоя, а большую часть его объема занимает ECM, играющий основную роль в структурной организации дермального слоя и поддержании его архитектоники. ECM является неклеточным компонентом дермы, секретируется фибробластами и представляет собой постоянно обновляющийся комплекс фибриллярных белков, гликопротеинов и протеогликанов, имеющий различный состав в зависимости от локализации ткани и окружающих ее физиологических условий [35]. Механическую прочность дермы обеспечивает Col I (коллаген I типа), находящийся преимущественно в сетчатом слое. Col VII характерен для базального слоя эпидермиса, обеспечивает его прочную связь с соединительной тканью дермального слоя кожи посредством базальной мембраны [36]. Существует мнение, что формирование коллагеновых волокон стимулируется декорином и фибромодулином [37]. Эластичность и упругость дермы обеспечивают эластиновые волокна, обладающие хорошей способностью к растяжению. Эти свойства эластиновых волокон определяются белками эластином и фибриллином, входящими в их состав [38].
В настоящее время накоплен значительный объем данных о взаимодействии клеток эпидермиса и дермы (кератиноцитов и фибробластов). Известно, что в нормальной и поврежденной коже кератиноциты секретируют факторы роста, которые стимулируют процесс выработки компонентов ECM фибробластами, и, наоборот, фибробласты продуцируют факторы пролиферации и миграции для кератиноцитов, которые важны при эпителизации ран [39]. VEGF (vascular endothelial growth factor), эндотелиальный фактор роста сосудов, продуцируемый кератиноцитами, способствует образованию в верхних слоях дермы новых сосудов (ангиогенез). Пролиферирующие кератиноциты реагируют на факторы роста, секретируемые фибробластами, такие как KGF (кератиноцитарный фактор роста), контролирующий миграцию и пролиферацию кератиноцитов, EGF (эпидермальный фактор роста), стимулирующий пролиферацию, а также FGF (фактор роста фибробластов), участвующий в развитии кожи плода и контролирующий пролиферацию и миграцию клеток. FGF2 оказывает влияние на активацию экспрессии кератинов K6, K16 и K17, участвующих в осуществлении процесса миграции кератиноцитов [18, 34]. Известно, что провоспалительные цитокины IL-1 (интерлейкин), IL-6 и TNF-α, продуцируемые кератиноцитами, стимулируют миграцию кератиноцитов и фибробластов в раневое поле и активируют секрецию фибробластами FGF, повышающего и стимулирующего синтез компонентов ECM и придающего кератиноцитам повышенную подвижность в процессе реэпителизации. Противовоспалительный цитокин TGF-β (трансформирующий фактор роста бета), продуцируемый как кератиноцитами, так и фибробластами, стимулирует образование при ранении грануляционной ткани, контролирует феномен трансформации фибробластов в миофибробласты (см. ниже) и регулирует обратный переход дифференцирующихся кератиноцитов к базально-клеточному фенотипу (де-дифференцировка) со специфичными для базальных клеток маркерами K5 и K14 [40]. Также обнаружено, что мигрирующие по краям раны кератиноциты активно экспрессируют MMP (металлопротеиназы), ферменты, разлагающие ECM, что позволяет этим клеткам мигрировать через фибриновую пробку и поверх грануляционной ткани [41].
Клетки разных слоев кожи находятся в тесной взаимосвязи между собой и функционируют как единое целое, взаимно секретируя факторы роста и взаимно стимулируя дифференцировку, пролиферацию, миграцию, синтез структурных белков ECM и в целом определяя регенераторный потенциал кожи через ауто- и паракринные механизмы. В силу этого полноценное восстановление целостности кожных покровов после ранения не может происходить при участии лишь одного типа клеток, а вовлекает в процесс ранозаживления клетки всех типов и всех слоев кожи, что приводит к качественному обновлению структуры и восстановлению целостности ткани.
2. ГЕТЕРОГЕННОСТЬ ПОПУЛЯЦИЙ ФИБРОБЛАСТОВ ДЕРМЫ
Фибробласты являются основными и самыми многочисленными клетками в составе дермы [31]. Помимо дермы фибробласты присутствуют в других тканях, со специфичными для конкретной популяции маркерами и для конкретной ткани функциями. Поскольку существуют значительные различия в строении дермы на разных участках тела млекопитающих, локальные субпопуляции дермальных фибробластов также имеют свои индивидуальные особенности. В процессе эмбрионального развития фибробласты происходят из разных анатомических зон эмбриона, включая нервный гребень, мезодерму латеральной пластинки и дерматомиотом [42].
Дермальные фибробласты происходят в основном из недифференцированных мультипотентных мезенхимальных клеток-предшественников [43]. Под влиянием различных факторов роста и цитокинов, высвобождаемых из макрофагов, тромбоцитов крови и нейтрофилов на 12.5-й день эмбрионального развития мыши (E12.5), мезенхимные клетки-предшественники трансформируются в клетки-предшественники фибробластов, из которых, в свою очередь, к стадии Е16.5 формируются две линии зрелых фибробластов: 1) папиллярные дермальные фибробласты (PF, papillary fibroblasts) и фибробласты волосяного фолликула (HFF, hair follicle fibroblasts), а также 2) фибробласты ретикулярной (RF, reticular fibroblasts) и гиподермальной (HDF, hypodermal fibroblasts) популяций [34, 44].
Было установлено, что мультипотентная мезенхимальная популяция предшественников дермальных фибробластов экспрессирует PDGFRα (рецептор тромбоцитарного фактора роста альфа), DLK1 (дельта-подобный гомолог 1) и Lrig1 (богатый лейциновыми повторами и иммуноглобулиноподобный домен 1), являющиеся маркерами клеток всех слоев дермы. Начиная с E16.5 и до второго постнатального дня развития мыши PF, HFF, RF и HDF окончательно дифференцируются по маркерам CD26 (или DPP4, дипептидилпептидаза-4), Sca1 (антиген 1 стволовых клеток), Blimp1 (индуцирующий созревание В-лимфоцитов белок 1). При этом CD26 и Blimp1 селективно экспрессируются в фибробластах верхнего слоя дермы (PF и HFF), тогда как Sca1 – в линиях RF и HDF нижнего слоя дермы [19, 45].
Зрелые дермальные фибробласты участвуют в поддержании тканевой структуры дермы и в непрерывном обновлении ECM, поддерживая его структурный и химический гомеостаз [46]. На пролиферативную активность фибробластов оказывают влияние различные цитокины и медиаторы, в частности IL-1α, IL-lβ и IL-8 [47]. Фибробласты дермы связываются с неповрежденными коллагеновыми фибриллами через рецепторы интегринов, расположенные на клеточной мембране [48].
PF обладают более высокой ферментативной активностью по сравнению с RF, тогда как RF опосредуют процессы заживления ран и экспрессируют маркеры активации фибробластов, такие как αSMA (α-гладкомышечный актин). Популяции PF и RF имеют различные паттерны секреции белков ECM, а также факторов роста и цитокинов, необходимых для паракринных перекрестных взаимодействий с кератиноцитами [32]. Ранее считалось, что PF незначительно вовлечены в синтез ECM, однако позже было показано, что дермальные эквиваленты на основе PF обеспечивали бо́льшую жизнеспособность культивированных на них кератиноцитов, а также способствовали формированию более стратифицированного и дифференцированного эпидермиса, чем эквиваленты на основе RF. В культуре PF имеют веретенообразную морфологию, а RF – звездчатую форму [49]. Влияние RF на кератиноциты обусловлено повышенным синтезом KGF и сниженным уровнем GM-CSF (гранулоцитарно-макрофагальный колониестимулирующий фактор) по сравнению с PF. В процессе длительного культивирования и старения клеток PF приобретают ретикулярный фенотип и морфологию [50].
В свою очередь, HFF представлены двумя субпопуляциями клеток: фибробластами дермальной папиллы волосяного фолликула (DP, hair follicle dermal papilla), играющими центральную роль в развитии волос и в координации цикла волосяного фолликула, и фибробластами дермальной соединительнотканной оболочки (DS, dermal sheath), инкапсулирующими эпителиальные компоненты волосяного фолликула [42]. Фибробласты DP и DS отличаются от других дермальных фибробластов высоким уровнем экспрессии неспецифической щелочной фосфатазы, αSMA и PAR-1 (активируемого протеазой рецептора-1) [34].
Таким образом, дермальные фибробласты представляют собой гетерогенную популяцию клеток, представленную значительным числом субпопуляций, обладающих функциональными различиями. Специфичность этих популяций определяется главным образом их расположением в разных участках дермы. Функциональные различия между ними определяются экспрессией специфических компонентов ECM и различных маркеров, причем эти различия проявляются как in vivo, так и in vitro. Гетерогенность субпопуляций фибробластов имеет определяющее значение в поддержании тканевой структуры дермы, а также в репарационных процессах, имеющих место при ранозаживлении.
3. ФИБРОБЛАСТЫ – ОСНОВНЫЕ УЧАСТНИКИ ПРОЦЕССА РАНОЗАЖИВЛЕНИЯ
Фибробласты участвуют в поддержании гомеостаза в дерме, так как не только контролируют состав и структуру ECM, но и вырабатывают цитокины, факторы роста и ферменты, в том числе участвующие в синтезе и депонировании коллагена [51, 52]. Помимо создания оптимальных условий для пролиферации и жизнедеятельности других типов клеток (эндотелиальных, эпителиальных, клеток волосяных фолликулов) фибробласты координируют их функции [53].
Повреждение кожного покрова активизирует сложные адаптивные механизмы, направленные на восстановление целостности и функциональности ткани. Заживление раны представляет собой ряд последовательных стадий, которые сопровождаются такими важными процессами, как фагоцитоз, хемотаксис, апоптоз, рост и развитие сосудов, синтез белков и других компонентов ECM, пролиферация и дифференцировка клеток, а также их миграция в зону повреждения.
Процесс заживления острых ран может быть разделен на четыре основные фазы: гемостаз, воспаление (раннее и позднее), фаза клеточной пролиферации и фаза ремоделирования ткани [42]. Фибробласты принимают активное участие на всех этих этапах, играя ключевую роль в выработке и ремоделировании нового ECM, а также стягивания краев раны за счет их трансформации в миофибробласты [54, 55].
Фаза гемостаза и последующая фаза воспаления длятся от одного до трех дней, в течение которых в раневом поле резко возрастает количество клеток (инфильтрат) благодаря миграции и пролиферации фибробластов, эндотелиальных клеток, форменных элементов крови, а также кератиноцитов, осуществляющих эпителизацию раны. Сразу после нарушения целостности кожи и сосудов происходят агрегация тромбоцитов и образование фибринового сгустка (состоящего из фибрина и фибронектина), что вызывает остановку кровотечения (гемостаз) [56], которой также способствует вазоконстрикция сосудов. Тромбоциты высвобождают хемокины, участвующие в рекрутировании воспалительных клеток (нейтрофилов, и макрофагов), фибробластов и эндотелиальных клеток, помимо этого, выделяют медиаторы (такие как серотонин и гистамин), увеличивающие проницаемость клеток, а также тромбоцитарный фактор роста, который наряду с трансформирующим фактором роста стимулирует пролиферацию фибробластов, синтезирующих коллаген. Воспалительные клетки, в свою очередь, высвобождают различные медиаторы и цитокины, способствуя ангиогенезу, тромбозу и реэпителизации, обеспечивают фагоцитоз клеточного дебриса и бактерий, обеспечивая таким образом обеззараживание раны. Все указанные процессы высоко синхронизированы во времени [44, 55, 57].
Также известно, что в случае хронических ран увеличение провоспалительного клеточного инфильтрата (состоящего в основном из нейтрофилов и макрофагов) является одним из факторов задержки процесса ранозаживления. При этом повышение уровня нескольких ключевых провоспалительных цитокинов, таких как IL-1b и TNFα (фактор некроза опухоли альфа), приводит к повышению уровня MMP, нарушая таким образом миграцию клеток, продлевая воспалительную фазу и задерживая заживление раны [58].
После третьей стадии (клеточной пролиферации) следует последняя фаза ранозаживления – ремоделирование, в процессе которой происходят морфологические изменения фибробластов. Ростовые факторы и хемоаттрактанты (IGF-1, bFGF, TGF-β, PDGF, EGF) стимулируют как пролиферацию и миграцию фибробластов в область раны из прилегающих участков дермы в течение 48–72 ч с момента повреждения [15], так и феномен трансформации фибробластов в миофибробласты [35, 44, 59, 60], которые становятся самой многочисленной популяцией клеток в грануляционной ткани. Миофибробласты, как и фибробласты, являются продуцентами ECM в поврежденной ткани, но основная их роль заключается в уменьшении размера раны за счет приобретаемой ими способности к контракции [58, 61]. Отличительной чертой миофибробластного фенотипа является экспрессия αSMA, характерного для гладкомышечных и миоэпителиальных клеток эндотелия сосудов, который придает клеткам способность сокращаться [57, 62, 63]. Наиболее важными стимулами к трансформации в миофибробласты являются механическое напряжение [64] и активность TGF-β [65–68]. Провоспалительный медиатор IL-1 активирует синтез фибробластами ростовых факторов, важнейшим из которых является TGF-β, инициирующий сократительную активность, в результате чего в зоне воспаления начинает расти механическое напряжение [69]. В начальной стадии заживления, NFκB, присутствующий в фибробластах, блокирует передачу сигналов TGF-β. На этой стадии образуются протомиофибробласты, не экспрессирующие αSMA. Постепенно активность NFκB в фибробластах снижается, повышается экспрессия TGF-β-зависимых генов, а взаимодействие ET-1 и TGF-β индуцирует экспрессию αSMA и окончательную трансформацию клеток в миофибробласты [70]. Помимо αSMA миофибробласты экспрессируют N-кадгерин в отличие от кадгерин-отрицательных фибробластов. N-кадгерин – белок трансмембранной межклеточной адгезии, связанный с внутриклеточным актиновым цитоскелетом. В процессе ранозаживления N-кадгерин замещается на OB-кадгерин (кадгерин-11), который играет роль в координации сокращения миофибробластов [71]. Известно, что FGF2 ингибирует TGF-β-опосредованную трансформацию фибробластов в миофибробласты [72].
Тканевые фибробласты и миофибробласты играют ключевую роль в секреции различных ростовых факторов и структурных белков, поддерживающих функциональную целостность ECM [62, 73]. Среди них проколлаген (Col I и Col III), эластин, многодоменные адгезивные гликопротеины (фибронектин, витронектин, ламинин), гликозаминогликаны (гиалуроновая кислота, гиалуронан, хондроитинсульфат, гепарансульфат), а также протеогликаны (версикан, синдеканы, глипиканы и перлекан).
Фибробласты также секретируют многочисленные ростовые факторы и клеточные белки: тромбоспондин 1 (TSP1) и 2 (TSP2), тенасцин C, остеопонтин [74–76]. Не менее важными белками являются также синтезируемые фибробластами коллаген-пролилгидроксилазы (P4ha1 и P4ha2) и коллаген-лизилгидроксилаза (Plod2), способствующие синтезу коллагена и осуществляющие его посттрансляционные модификации в процессе ремоделирования ECM [77, 78]. PDGF, секретируемый фибробластами, помимо стимуляции пролиферации этих клеток увеличивает экспрессию ими коллагеназы, а TGF-β регулирует хемотаксис фибробластов, их трансформацию в миофибробласты, а также синтез и накопление компонентов ECM, в основном коллагена [53, 79]. Тенасцин-C и ламинин связываются с рецепторами EGF, усиливая миграцию фибробластов в область повреждения [58, 76]. EGF, в свою очередь, после выделения фибробластами в раневое поле усиливает реэпителизацию (миграцию кератиноцитов) острой раны за счет увеличения уровня экспрессии кератинов K6 и K16, также участвовавших в пролиферации клеток. Уровень b-FGF, также синтезируемого фибробластами, возрастает при остром повреждении кожного покрова. FGF способствует формированию грануляционной ткани, улучшает реэпителизацию и ремоделирование раны, контролирует морфогенез, дифференцировку, пролиферацию и миграцию клеток [80, 81]. FGF-2 увеличивает подвижность кератиноцитов, способствует делению фибробластов и эндотелиальных клеток, стимулирует синтез коллагеназ фибробластами и способствует их миграции к зоне повреждения [82, 83].
Фибробласты также принимают активное участие в контроле процесса ангиогенеза [44, 75]. Они активно синтезируют не только VEGF, но и другие важные проангиогенные факторы, среди которых основными являются FGF, TGF-β и HGF (фактор роста гепатоцитов), который стимулирует пролиферацию клеток эндотелия сосудов [74, 84]. Таким образом, фибробласты играют важную роль в росте новых кровеносных капилляров, обеспечивающих питание и поступление кислорода к растущей новой ткани. Вновь образовавшиеся сосуды дают возможность лейкоцитам попадать в область раны для предупреждения проникновения инфекции [56].
Помимо фибробластов в восстановлении нарушенной структуры кожи участвует целый ряд других клеток, включая кератиноциты, осуществляющие восстановление эпидермального слоя (эпителизацию) и формирующие барьер между внутренней и внешней средой, а также эндотелиальные клетки, осуществляющие вместе с фибробластами синтез новых компонентов ECM и белков и принимающие участие в ангиогенезе и реваскуляризации области раны [35, 85].
Фибробласты играют существенную роль и в процессе старения кожных покровов, который связан с изменением структурных и биохимических свойств кожи, а также с изменением профиля экспрессии белков, составляющих основу ECM [36, 86]. Семейство белков MMP и TIMP (тканевых ингибиторов металлопротеиназ), синтезируемых многими клетками, включая фибробласты и макрофаги, участвует в модулировании компонентов ECM соединительных тканей, в том числе дермы кожи, контролируя постоянное обновление структурных белков. ММР разрушают ECM, тогда как TIMP ингибируют деградацию матрикса [18]. Кроме того, ММР удаляют продукты денатурации ECM, образующиеся при повреждениях, что свидетельствует о важной роли ММР в процессе заживления ран [87–89]. Этим белкам отведена главная роль в деградации волокон коллагена. Уровень ММР в нормальной неповрежденной коже очень низок. Однако при повреждении кожи и инициации процесса ранозаживления увеличивается синтез MMP-1, MMP-2, MMP-3, MMP-8, MMP-9, MMP-10, MMP-12, MMP-13, MMP-19, MMP-26 и MMP-28, поддерживающих нормальную архитектонику ECM дермы [56].
Заживление значительных по размеру ран кожи взрослого человека обычно сопровождается отложением избыточного ECM, что приводит к проявлениям фиброза и образованию рубца, и в конечном счете к определенной потере функциональности поврежденных покровов. В нормальных физиологических условиях после восстановления целостности ткани миофибробласты подвергаются апоптозу. Однако в области развития фиброза за счет подавления апоптоза и, как следствие, повышения выживаемости миофибробластов повышается внутреннее натяжение в ткани, что приводит к рубцеванию. Недавние исследования показывают, что именно отсутствие воспаления способствует безрубцовому заживлению кожных ран во время внутриутробного развития [69].
В отличие от нормальной кожи рубец (шрам) имеет характерные признаки, включая ненормальную ориентацию коллагеновых волокон, чрезмерный фиброз, исчезновение эластических волокон и придатков кожи, общее нарушение структуры кожи. Нарушение в рубцовой ткани соотношения уровня белков MMP, коллагена I и III, гиалуроновой кислоты и декорина, фибронектина и ламинина, а также фибриллиновых и эластиновых волокон приводит к развитию патологических, а порой и тяжелых форм рубцевания – гипертрофических и келоидных рубцов [56]. Гипертрофические рубцы не выступают за пределы раневого поля, регрессируют в течение 6 мес после формирования и характеризуются быстрым ростом. Для них также характерно присутствие αSMA + миофибробластов и расположенных параллельно поверхности кожи коллагеновых волокон. Келоидные рубцы в отличие от гипертрофических не склонны к самопроизвольному регрессу и разрастаются за пределы площади первоначальной раны, приобретая форму доброкачественного новообразования. Они характеризуются постоянным ростом и толстыми, бессистемно ориентированными пучками коллагеновых волокон [58].
Признаком, характерным для длительно незаживающих (хронических) ран, является их пониженная способность к реэпителизации. Это связывают со сниженной скоростью пролиферации и миграции кератиноцитов, которые регулируются на молекулярном уровне многими факторами, в том числе компонентами ECM и цитокинами, вырабатываемыми фибробластами и макрофагами в раневом поле [8]. СДС у больных диабетом 2 типа характеризуются повышенным уровнем экспрессии фибробластами воспалительных маркеров и матриксных металлопротеиназ, разрушающих необходимый для поддержания целостности ткани коллаген. В результате происходят дезактивация факторов роста и уменьшение количества фибробластов, кератиноцитов и эндотелиальных сосудистых клеток, что приводит к блокировке процесса заживления раны [90]. Показано, что фибробласты, выделенные из хронических язв, возникающих на фоне хронической венозной недостаточности, обладают гораздо более низкой пролиферативной активностью по сравнению с клетками, выделенными из образцов нормально функционирующей кожи [91]. Также было показано, что фибробласты, выделенные из ткани хронической диабетической язвы, развивающейся на фоне хронической венозной недостаточности, обладали более низкой миграционной активностью и подвижностью по сравнению с фибробластами, выделенными из здоровой кожи [92].
Известно, что цитокины, аккумулируемые в большом количестве в раневой жидкости хронических трофических язв кожи, подавляют рост нормальных фибробластов и способствуют их морфологическим изменениям [93]. В хронических ранах нарушены процессы синтеза фибробластами ECM, воспаления и образования грануляционной ткани, что в целом замедляет реэпителизацию раны и ремоделирование поврежденной ткани [94]. Также было показано снижение способности фибробластов, выделенных из хронических ран, к синтезу Col I. Очевидная причина этого – увеличение синтеза и активности тканевых ингибиторов металлопротеиназ и, соответственно, уменьшение уровня экспрессии MMP-1 и MMP-2. Это приводит к накоплению коллагена в клетках и компенсаторному снижению синтеза новых волокон [95]. MMP-ингибиторы блокируют и миграцию кератиноцитов к месту повреждения и тормозят заживление in vivo [56]. Исследования, проведенные на фибробластах из хронических диабетических язв, показали значительное усиление экспрессии фибронектина (по сравнению с фибробластами нормальных тканей), а также снижение экспрессии Col I [96].
Таким образом, именно фибробласты обеспечивают кожу необходимыми структурными компонентами, составляющими основу ее архитектоники, и продуцируют основные факторы, контролирующие регенерацию не только дермального слоя кожи, но и эпидермиса, выделяя соответствующие факторы роста и стимулируя восстановление целостности кожных покровов.
4. ФИБРОБЛАСТЫ В ТКАНЕВОЙ ИНЖЕНЕРИИ
Благодаря активному развитию методов тканевой инженерии существует возможность моделировать структуру естественного ECM путем создания искусственных биосовместимых матриксов (скаффолдов) различной структуры (гели, губки, нетканые материалы и т.д.) и состава (синтетические и природные полимеры). При использовании соответствующих клеток подобные матриксы могут служить основой тканевых эквивалентов, имитирующих нативные ткани организма, для их применения в качестве трансплантатов в регенеративной медицине. Для терапии кожных ран и ожогов особый интерес представляют дермальные фибробласты, поскольку эти клетки играют ключевую роль в выработке компонентов ECM и регуляции различных аспектов ранозаживления. Использование полимерных матриксов с посеянными на них фибробластами способствует восстановлению нормальных функций поврежденных кожных покровов и поддерживает их архитектонику. Полимерный материал помимо осуществления механической и структурной поддержки искусственной ткани обеспечивает условия для нормальной жизнедеятельности клеток – их дифференцировки, пролиферации, миграции, адгезии и других важных функций [97].
Способ лечения дефектов кожи с помощью фибробластов, иммобилизованных на различных полимерных носителях, является эффективным и в то же время безопасным [98, 99]. Важное преимущество фибробластов в клеточной и тканевой трансплантологии – низкая иммуногенность этих клеток, обусловленная отсутствием экспрессии молекул главного комплекса гистосовместимости MHC II класса [34, 100]. Это позволяет использовать для трансплантации не только аутологичные, но и аллогенные фибробласты. Известно, что аутологичные фибробласты на неклеточных носителях более эффективно стимулируют ранозаживление по сравнению с аллогенными [101]. Тем не менее возможность незамедлительного применения заранее подготовленных аллогенных клеток при жизненно опасных повреждениях кожи (например, обширные ожоги) [102] делает их более предпочтительными: быстрая клеточная терапия (сразу после термического поражения или травмы) существенно снижает негативные последствия нарушения функций кожных покровов у пациента.
В 1981 г. Burke и Yannas в целях разработки нового метода лечения ожоговых ран создали искусственный двуслойный аналог кожи, его верхний слой состоял из силиконового аналога эпидермиса (впоследствии заменяемого на аутоэпидермальный трансплантат), а нижний – из пористого коллаген-хондроитин-6-сульфатного фибриллярного аналога дермы, который вскоре после трансплантации заселялся фибробластами реципиента и прорастал сосудами, обеспечивая таким образом синтез новой соединительнотканной структуры, частично восполняющей функции поврежденной кожи [103]. На основе этой разработки был запатентован продукт Integra®, широко использующийся до сих пор. После внедрения этого продукта создание биоинженерных аналогов кожи различной структуры и сложности получило широкое распространение. В частности, в 2014 г. был разработан двуслойный кожный эквивалент, состоящий из пористого хитозан-альгинатного матрикса с кератиноцитами на его поверхности (для имитации эпидермального слоя), пропитанного термически обработанным хитозан-полиэтиленгликолевым гелем с заселенными в него фибробластами (для имитации дермального слоя). При трансплантации такого эквивалента на рану происходило увеличение скорости пролиферации кератиноцитов и фибробластов хозяина, что указывало на способность носителя имитировать микросреду ткани кожи, а ускоренное затягивание краев раны – на выделение фибробластами (эквивалента и хозяина) факторов, способствующих ускорению процесса заживления [104].
Ключевая роль фибробластов в синтезе значительного количества белков, участвующих в ранозаживлении, обусловливает эффективность использования этих клеток в лечении ран, а отсутствие реакции “трансплантат-против-хозяина” – безопасность их применения.
5. HIF-1-ФАКТОР КАК РЕГУЛЯТОР КЛЕТОЧНОЙ АДАПТАЦИИ К УСЛОВИЯМ ГИПОКСИИ
Один из перспективных подходов к стимуляции ранозаживления – использование фибробластов, предварительно культивированных в условиях гипоксии, что способствует выделению ими факторов ранозаживления [105, 106].
Центральным механизмом, посредством которого гипоксия регулирует клеточные функции, является HIF1-зависимый молекулярный каскад (Hypoxia-inducible factor 1-cascade).
Транскрипционный фактор HIF1 представляет собой гетеродимер, состоящий из двух субъединиц: кислородчувствительного белка Hif1-α и ядерного транслокатора рецептора ароматических углеводородов ARNT (aryl hydrocarbon receptor nuclear translocator), обозначаемого также Hif1-β. Димеризация этих двух родственных факторов (принадлежащих к семейству PAS-белков с доменом типа спираль–петля–спираль – helix–loop–helix) ведет к трансактивации генов-мишеней путем связывания HIF1 с их специфическими промотерными участками, содержащими HRE (hypoxia-response element) – чувствительный к гипоксии элемент, представленный последовательностью 5'‑R-CGTG-3' [107, 108]. В условиях in vivo ARNT образует гетеродимеры и с другими PAS-белками, в том числе с AhR и Hif2-α, что приводит к образованию соответствующих ДНК-связывающих комплексов, активирующих специфические наборы генов [109].
Еще в 1996 г. Huang показал, что активность связывания с HIF1 с ДНК можно значительно усилить, если подвергать клетки воздействию острой гипоксии (1% O2). При этом при переводе клеток обратно в условия нормоксии (21% O2) активность связывания резко падает из-за деградации субъединицы Hif1-α [110]. В условиях нормоксии Hif1-α-белок гидроксилируется по остаткам пролина, расположенным в О2-зависимом домене (ODD, oxygen-dependent degradation domain), который состоит примерно из 200 аминокислотных остатков и является центральной частью Hif1-α [111]. Гидроксилирование осуществляется специфическими пролил-гидроксилазами PHD (prolyl hydroxylases).
PHD являются цитоплазматическими белками [77] и кодируются соответствующими Egln-генами: PHD1 (также обозначается как HPH3 – HIF-пролил-гидроксилаза 3) кодируется геном Egln2, PHD2 (HPH2) – Egln1, а PHD3 (HPH1) – геном Egln3. Они входят в семейство 2-оксоглутарат (2OG)-зависимых оксидаз, участвующих во многих физиологических процессах, в том числе в протеосомной деградации белков [112–114]. В тканях человека наибольшей физиологической значимостью из PHD обладает PHD2. Гидроксилирование Hif1-α под действием PHD2 в присутствии кислорода происходит по одному или двум остаткам пролина, Pro-564 и Pro-402 [109], в ‑Leu-X-X-Leu-Ala-Pro-последовательности [77]. В результате активируется связывание Hif1-α с белком-онкосупрессором pVHL (Von Hippel-Lindau, фон Хиппел-Линдау) [114, 115], который существует в клетке в составе комплекса VCB (pVHL-elonginC-elonginB complex), являющегося сигнальной молекулой в E2-лигаза-опосредованном каскаде. Это связывание приводит к убиквитинированию и протеосомной деградации Hif1-α [113]. Этот механизм в условиях нормоксии обеспечивает быстрый кругооборот Hif1-α в клетке (в пределах 4–5 мин). Упрощенная схема регуляции Hif1-α на молекулярном уровне представлена на рис. 1.
Рис. 1.
Схема HIF-каскада. ECM – внеклеточный матрикс, PHD – пролилгидроксилазы, VHL – фон Хиппел-Линдау белок, HRE – чувствительный к гипоксии элемент, VEGF – сосудистый эндотелиальный фактор роста, PDGF – тромбоцитарный фактор роста.
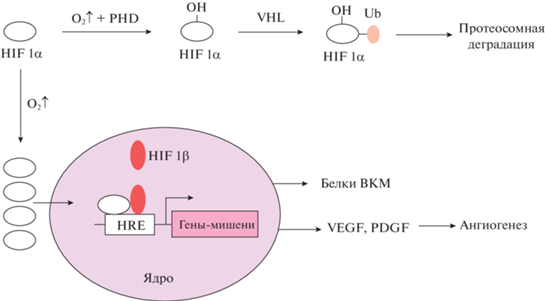
Marxsen провел анализ экспрессии mRNA различных PHD на иммортализованной линии клеток гепатомы HepG2 и первичной культуре RPTEC (эпителиальных клеток проксимальных почечных канальцев человека), культивированных в условиях нормоксии (21% O2) и острой гипоксии (1% O2). Транскрипция PHD1 оказалась конститутивной в обеих линиях клеток, в то время как уровень mRNA PHD2 и PHD3 индуцировался гипоксией, причем в культуре, выращенной в условиях 18-часовой гипоксии, индукция PHD2 и PHD3 была выше, чем в культуре, выращенной в условиях одного часа воздействия гипоксии [112]. В другом исследовании, проведенном на клетках, мутантных по гену VHL, было продемонстрировано, что подавление Hif1-α с использованием siRNA (small interfering RNA – малая интерферирующая РНК) приводит к снижению эффекта гипоксии на экспрессию PHD3. Также было показано, что непродолжительная гипоксия (4 ч) индуцирует экспрессию PHD2 независимо от наличия Hif-1α, а в 16-часовой культуре уровень PHD2 снижался на фоне siRNA-зависимой деградации Hif-1α [116].
Период полураспада Hif1-α составляет ~5–8 мин после реоксигенации (последовательном переходе из условий гипоксии в условия нормоксии) [117]. В условиях недостатка O2 происходят блокирование гидроксилирования Hif1-α и, как следствие, прекращение деградации этого белка, что обеспечивает его стабилизацию и накопление в цитоплазме, транспортировку в ядро, димеризацию с ARNT и таким образом запуск HIF1-зависимого каскада. Повышенная активность HIF1-каскада характерна для опухолевых клеток, отличающихся быстрым ростом и, соответственно, высокой потребностью в кислороде [114].
Для опухолевых клеток характерны частые мутации в гене VHL, что ведет к стабилизации в них Hif1-α и, соответственно, активации HIF1-каскада. Таким образом, подавление активности гена VHL вызывает эффект, подобный воздействию гипоксии. Известно, что аминокислотная последовательность 549–582 α-субъединицы фактора HIF1 взаимодействует с β-доменом pVHL, и мутации в этом домене, ассоциированные с опухолью, вызывают потерю взаимодействия между дефектным pVHL и нормальным HIF1-α, приводя к нарушению убиквитинирования последнего [118].
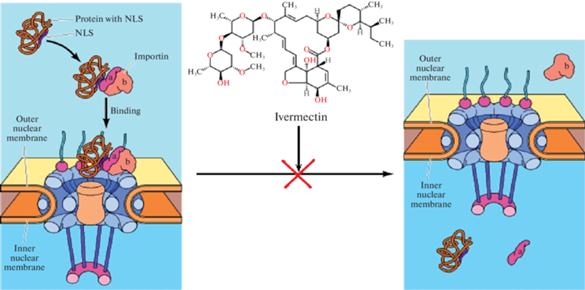
В результате вызванной гипоксией стабилизации Hif1-α этот белок транспортируется из цитоплазмы в ядро клетки посредством α/β-импортин зависимого механизма. В частности, было показано, что, ингибируя ивермектином расположенный в ядерном поровом комплексе α/β-импортин, можно предотвратить транспорт Hif1-α в ядро и тем самым снизить активность HIF1-зависимых генов (рис. 2) [119].
HIF1-зависимый каскад является важным регулятором многих процессов в клетке [120], в том числе регулирует клеточную и тканевую адаптацию к стрессовым воздействиям, в частности гипоксии. Сайты связывания с HIF1-транскрипционным фактором (HRE) обнаружены в промотерной области генов, кодирующих ферменты гликолиза, белки, контролирующие ангиогенез (рост и развитие сосудов), синтазу окиси азота, гемоксигеназу-1, тирозингидроксилазу и многих других. Это указывает на важность данного каскада для регуляции ключевых клеточных функций [121], включая апоптоз, пролиферацию и миграцию, а также эмбриональное развитие [122]. Известна и антибактериальная активность Hif1-α через индукцию ROS (reactive oxygen species) [123]. Локальная острая гипоксия приводит к повышению синтеза белков ангиогенеза (стимуляции роста и развития сосудов), усиленной подвижности и инвазивности клеток, способности выживать в условиях оксидативного стресса, а также приводит к изменению в метаболизме клеток, в том числе опосредует метаболизм глюкозы [79].
Наряду с положительным влиянием активации HIF1 на клеточные процессы известны ее многочисленные негативные эффекты, поскольку HIF1 играет роль в патогенезе различных заболеваний [124]. Так, HIF1-опосредованный метаболический сдвиг играет решающую роль в некоторых аутоиммунных заболеваниях, таких как ревматоидный артрит, воспалительные заболевания кишечника (болезнь Крона), системный и множественный склероз [125]. Среди аутоиммунных заболеваний кожи наибольшую негативную роль активация HIF1-каскада играет в патогенезе псориаза [126, 127]. Одним из патогенетических коррелятов псориаза является ненормальный ангиогенез, индуцированный VEGF-фактором – геном-мишенью HIF1 [128]. Обнаружено, что помимо активации VEGF псориатическое поражение кожи усугубляет и повышенная экспрессия других проангиогенных факторов: FLT-1 и FLK-1 (рецепторы VEGF), PAI-1 (ингибитор активатора плазминогена 1), MMP-2 и MMP-9 (матриксные металлопротеиназы) [129], также зависимых от активности HIF1.
Известно также, что сигнальный путь Hif1-α/ARNT контролирует метастазирование [122, 130]. Повышенная активность HIF1-каскада очень характерна для опухолевых клеткок [114, 131, 132]. Рост опухоли сопровождается гипоксией, что приводит к повышенной экспрессии VEGF, играющего ключевую роль в прогрессировании опухолевого роста [133–135]. Помимо VEGF в процесс роста опухоли вовлечены другие активируемые гипоксией белки: гликолитические ферменты (PGK, ALDA), переносчики глюкозы (GLUT1), белки, регулирующие метастазирование (CXCR4, E-cadherin) [136]. Опосредованный активностью HIF1-каскада избыточный рост сосудов делает опухоль устойчивой к действию многих противоопухолевых препаратов, поэтому анти-VEGF-терапия является одним из основных подходов к лечению опухоли [137, 138]. HIF1-зависимая активация IL-11 также может способствовать ангиогенезу, усугубляющему прогрессирование опухолевого перерождения клеток [139, 140].
Таким образом, транскрипционная активность HIF1 является основным механизмом, обеспечивающим адаптацию клеток к условиям острой и хронической гипоксии. Существенную роль HIF1-каскад играет и в регуляции физиологических и патологических процессов в коже, в том числе затрагивающих ее развитие и регенерацию при повреждении. Использование гипоксии в качестве стрессового фактора может быть эффективным инструментом в тканевой инженерии и клеточной трансплантологии, позволяющим получать клетки с повышенным уровнем синтеза факторов ранозаживления, активируемым в результате клеточного ответа на гипоксический стресс.
6. ГИПОКСИЯ И HIF-1 В КОНТРОЛЕ ФУНКЦИЙ ФИБРОБЛАСТОВ
Одним из значимых клинических последствий травмы кожных покровов является повреждение сосудов, что вызывает резкое снижение трофики в ткани и, как следствие, развитие местной гипоксии [8]. Интересно, что клетки, особенно фибробласты, находящиеся в поверхностной зоне раны и испытывающие дефицит кислорода, обладают повышенной пролиферативной и миграционной активностью, а также повышенным потенциалом к выработке факторов, способствующих ранозаживлению [141, 142]. Повышение фибробластами экспрессии генов, контролирующих и ускоряющих процесс ранозаживления, является прямым следствием активации в них гипоксия-зависимого каскада HIF1 [79]. Среди этих генов особую значимость играют гены, кодирующие Col I и Col III, эластин и фибронектин (Fn), а также различные MMP [35, 53, 76, 143–149].
Не менее важными с точки зрения ранозаживления мишенями HIF1-каскада в фибробластах дермы кожи являются коллаген-пролилгидроксилазы – P4HA1 и P4HA2, а также коллаген-лизилгидрокилаза PLOD2, которые контролируют посттрансляционные модификации коллагена, способствующие фолдингу белка [78, 150]. P4HA1 и P4HA2 принадлежит главная роль и в биосинтезе коллагена [77, 151]. Посттрансляционная модификация проколлагена контролируется пролилгидроксилазами P4H посредством гидроксилирования остатков пролина. Гидроксилирование обеспечивает функциональность коллагена, поскольку эти остатки важны для правильного трехмерного свертывания полипептидных цепей проколлагена в стабильные трехспиральные структуры. PLOD2 контролирует характер и степень прочности волокон коллагена, также осуществляя его посттрансляционную модицикацию [150]. PLOD2 гидроксилирует остатки Lys в Y-положении повторяющихся триплетов X-Y-Gly трехспиральной структуры молекулы коллагена [152]. После гидроксилирования формируется трехспиральная молекула проколлагена, которая транспортируется из эндоплазматического ретикулума в комплекс Гольджи и секретируется в цитоплазму. Там N- и С-концы проколлагена отщепляются группой металлопротеиназ с образованием зрелой молекулы коллагена [78]. Роль P4Hs и PLOD2 в посттрансляционной модификации молекул проколлагена показана на рис. 3.
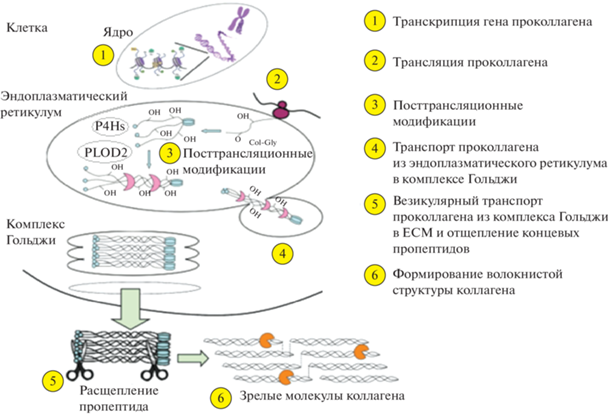
Помимо контроля экспрессии генов поцессинга коллагена комплекс HIF1-α/ARNT связывается с HRE в промоторной области генов, контролирующих ангиогенез, таких как VEGF-α [108], SDF1 (фактор 1, выделенный из стромальных клеток), IL-11, PGF (плацентарный фактор роста), PDGF-B (тромбоцитарный фактор роста B) и ANGPT (ангиопоэтин) [79]. Контроль ангиогенеза является ключевой функцией транскрипционного комплекса HIF1. В последние годы был проведен ряд исследований, в которых, блокируя различными агентами белки, направляющие Hif1-α на протеосомную деградацию, удалось не только стабилизировать уровень Hif1-α, но и активировать синтез VEGF-α и Col I [153–155].
Гипоксия контролирует не только экспрессионные, но и физиологические особенности фибробластов. Согласно [156] феномен перехода нормальных дермальных фибробластов в миофибробласты (для которых характерна высокая экспрессия αSMA, а также Col I и Col III) связан с активацией сигнального пути TGF-β1/Smad3, который, в свою очередь, активируется гипоксией. Считается, что белки Smad играют важную роль в регуляции внутриклеточных ответов на TGF-β1. Было показано, что после фосфорилирования Smad2 и Smad3, индуцированного TGF‑β1, эти белки локализуются в ядре и образуют комплекс со Smad4, который обеспечивает экспрессию профибротических генов. Блокада Smad3 и SIS3 показала значительное нарушение экспрессии индуцируемых гипоксией белков (α‑SMA, Col I и Col III). Таблица 1
Таблица 1.
Гены, экспрессируемые фибробластами дермы и играющие роль в процессе ранозаживления
| Аббревиатура | Полное название гена | Функции | Литература |
|---|---|---|---|
| Гены, кодирующие структуру белков ECM | |||
| Col I | Коллаген I | Фибриллярный коллаген, обеспечивает механическую прочность дермы, составляет 80–90% сухого веса кожи | [33, 72–74, 140, 141, 145] |
| Col VII | Коллаген VII | Обеспечивает прочную связь базального слоя эпидермиса с соединительной тканью дермального слоя кожи | [33] |
| Col III | Коллаген III | Фибриллярный коллаген, главный интерстициальный коллаген человека в эмбриональном и раннем постнатальном периоде | [72–74, 140, 141, 145] |
| ELN | Эластин | Обеспечивают эластичность и упругость дермы | [35, 72–74] |
| FN | Фибронектин | Главная функция – связывание клеток с межклеточным веществом; регулирует сборку коллагеновых фибрилл, стимулирует пролиферацию и миграцию клеток, способствует адгезии и распространению эпителиальных и мезенхимальных клеток | [72–74] |
| Гены, регулирующие HIF-каскад | |||
| PHD1, PHD2 и PHD3 | Пролил-гидроксилазы 1, 2 и 3 | Осуществляют гидроксилирование Hif1-α, активируя связывание Hif1-α с белком-онкосупрессором pVHL, что приводит к убиквитинированию и протеосомной деградации Hif1-α | [75, 109–113] |
| Гены, кодирующие белки, участвующие в процессинге белков ECM | |||
| P4ha1 и P4ha2 | Коллаген-пролилгидроксилазы 1 и 2 | Способствуют синтезу коллагена и осуществляют его посттрансляционные модификации в процессе ремоделирования ECM | [75, 76, 147–149] |
| Plod2 | Коллаген-лизилгидрокилаза | ||
| MMP | Металлопротеиназы | Ферменты, разлагающие ECM; играют главную роль в деградации волокон коллаген, удаляют продукты денатурации ECM | [15, 37, 41, 51, 84–86] |
| TIMP | Тканевые ингибиторы металлопротеиназ | Ингибируют деградацию матрикса | [15] |
| Гены, стимулирующие ангиогенез | |||
| VEGF | Эндотелиальный фактор роста сосудов | Способствует образованию в верхних слоях дермы новых сосудов | [41, 72, 73, 77, 82, 134, 135] |
| IL-11 | Интерлейкин 11 | Участвует в контроле синтеза | [77, 136, 137] |
| HGF | Фактор роста гепатоцитов | Стимулирует пролиферацию клеток эндотелия сосудов | [72, 82] |
| Гены, влияющие на активность кератиноцитов | |||
| KGF | Кератиноцитарный фактор роста | Контролирует миграцию и пролиферацию кератиноцитов | [15, 31] |
| EGF | Эпидермальный фактор роста | Стимулирует пролиферацию кератиноцитов, усиливает реэпителизацию (миграцию кератиноцитов) острой раны за счет увеличения уровня экспрессии кератинов K6 и K16 | [15, 31, 78, 79] |
| FGF | Фактор роста фибробластов | Участвует в развитии кожи плода и контролирует пролиферацию и улучшает реэпителизацию, придает кератиноцитам повышенную подвижность в процессе реэпителизации, стимулирует синтез компонентов ECM, участвует в ангиогенезе | |
| FGF2 | Фактор роста фибробластов 2 | Активирует экспрессию кератинов K6, K16 и K17, важных для миграции кератиноцитов; увеличивает подвижность кератиноцитов | [15, 31, 80, 81] |
| IL-1 и IL-6 | Интерлейкин 1 и 6 | Стимулируют миграцию кератиноцитов и фибробластов в раневое поле и активируют секрецию фибробластами FGF; участвует в ангиогенезе | [37, 41] |
| TNF-α | Фактор некро за опухоли альфа | ||
| TGF-β | Трансформирующий фактор роста бета | Контролирует феномен трансформации фибробластов в миофибробласты, стимулирует образование грануляционной ткани при ранении, участвует в ангиогенезе, регулирует обратный переход дифференцирующихся кератиноцитов к базально-клеточному фенотипу | [37, 50, 77] |
С учетом ключевой роли HIF1-зависимого регуляторного каскада в повышении экспрессии факторов ранозаживления использование фибробластов, иммобилизованных на полимерных тканеинженерных матриксах с их предварительным культивированием в условиях гипоксии, является перспективным подходом к лечению кожных ран и ожогов.
ЗАКЛЮЧЕНИЕ
Фибробласты являются важнейшим типом клеток, участвующим в заживлении кожных ран в силу их ключевой роли в синтезе компонентов внеклеточного матрикса и белков, участвующих в раневом ангиогенезе [11, 75]. Кратковременное гипоксическое воздействие на дермальные фибробласты в системе in vitro приводит к активации HIF1-каскада и, как следствие, стимуляции экспрессии генов, контролирующих синтез белков, формирующих ECM (Col I, Col III, Fn), а также их процессинг (P4HA1 и P4HA2, PLOD2, MMP). Эти механизмы играют важнейшую роль в восстановлении ECM в ходе ранозаживления. Большую важность для ранозаживления представляют и гены, участвующие в реваскуляризации раневого поля – в ангиогенезе (VEGF-α, IL-11). Таким образом, HIF1-каскад, являющийся основным регулятором физиологического ответа клетки на воздействие гипоксии, является ключевым фактором ранозаживляющего потенциала фибробластов. Из этого следует, что экспонирование фибробластов (как в клеточной взвеси, так и иммобилизованных на полимерных матриксах) гипоксии может повысить эффективность применения дермальных эквивалентов в терапии ран и ожогов. Такой подход требует подбора оптимального времени воздействия гипоксии для обеспечения максимального эффекта и одновременно исключения негативного влияния слишком продолжительного воздействия гипоксии на физиологию клеток. В целом кратковременное предварительное экспонирование фибробластов гипоксии и, соответственно, активация в них HIF1-каскада позволит не только стимулировать секрецию белков ранозаживления, но и сохранить жизнеспособность дермальных фибробластов.
Список литературы
Trompette A., Ubags N.D. Skin Barrier Immunology from early life to adulthood. Mucosal Immunology. 2023.
Быков В.Л., Юшканцева С. И. Гистология, цитология и эмбриология. Атлас. М.: ГЭОТАР–МЕДИА, 2013.
Noske K. // J. Dermatol. Sci. 2018. V. 89. № 1. P. 3.
Frykberg R.G., Zgonis T., Armstrong D.G. et al. // J. Foot Ankle Surgery. 2006. V. 45. № 5. P. 1.
Lavery L.A., Armstrong D.G., Wunderlich R.P. et al. // Diabetes Care. 2003. V. 26. № 4. P. 1069.
Armstrong D.G., Lavery L.A., Harkless L.B. // Diabetes Care. 1998. V. 21. № 5. P. 855.
Alexiadou K., Doupis J. // Diabetes Ther. 2012. V. 3. № 1. P. 1.
Винник Ю.С., Салмина А.Б., Дробушевская А.И. // Новости хирургии. 2011. Т. 19. № 3. С. 101.
Кириенкo А.И., Григорян Р.А., Богачев В.Ю., Богданец Л.И. // Сonsilium medicum. 2000. V. 2. № 4. P. 16.
Оболенский В.Н., Родоман Г.В., Никитин В.Г., Ка-рев М.А. // Трофические язвы нижних конечностей: обзор проблемы. 2011. C. 36–38.
Schultz G.S., Wysocki A. // Wound Repair Regen. 2009. V. 17. № 2. P. 153.
Winter G.D. // Nature. 1962. V. 193. P. 293.
Winter G.D. // Adv. Exp. Med. Biol. 1977. V. 94. P. 673.
Taylor J.M., Mitchell W.M., Cohen St. // J. Biol. Chem. 1972. V. 247. № 18. P. 5928.
Tottoli E.M., Dorati R., Genta I. et al. // Pharmaceutics. 2020. V. 12 (8). P. 735.
Lee S.J., Atala A. // Biomed. Mater. 2013. V. 8. № 1. P. 2.
Losquadro W.D. // Facial Plastic Surgery Clinics. 2017. V. 25. № 3. P. 283.
Hu M.S., Borrelli M.R., Hong W.X. et al. // Organogenesis. 2018. V. 14. № 1. P. 46.
Driskell R., Lichtenberger B., Hoste E. et al. // Nature. 2013. V. 504. P. 277.
Кузнецов С.Л., Горячкина В.Л., Цомартова Д.А. и др. // Российский журнал кожных и венерических болезней. 2013. № 2. С. 26.
Yousef H., Alhajj M., Sharma S. Anatomy, skin (integument), epidermis. 2017.
Alonso L., Fuchs E. // Proc. Natl. Acad. Sci. 2003. V. 100. № 1. P. 11830.
Ng K.W., Lau W.M. Skin Deep: The Basics of Human Skin Structure and Drug Penetration. Percutaneous Penetration Enhancers Chemical Methods in Penetration Enhancement. 2015. P. 3.
Delevoye C. // J. Investig. Dermatol. 2014. V. 134. № 4. P. 877.
Gasque P., Jaffar-Bandjee M.C. // J. Infect. 2015. V. 71. № 4. P. 413.
Chen T., Zhao B., Liu Y. // J. Dermatol. Sci. 2018. V. 90. № 3. P. 253.
Суханов А.Ф., Мяделец О.Д. // Архив анатомии, гистологии и эмбриологии. 1988. Т. 94. № 4. С. 79.
Van der Aar A.M.G., Picavet D.I., Muller F.J. et al. // J. Investig. Dermatol. 2013. V. 133. № 5. P. 1240.
Clayton K., Vallejo A.F., Davies et al. // Front. Immunol. 2017. V. 8. P. 1676.
Roesch-Dietlen F., Devezé-Bocardi R., Ruiz-Juárez I. et al. // Revista Médica del Instituto Mexicano del Seguro Social. 2013. V. 51. № 6. P. 696.
Афанасьев Ю.И., Юрина Н.А., Котовский Е.Ф. и др. Гистология, эмбриология, цитология. М.: ГЭОТАР-Медиа. 2016. 800 с.
Woodley D.T. // Dermatologic Clinics. 2017. V. 35. № 1. P. 95.
Eklouh-Molinier C., Happillon T., Bouland N. et al. // Analyst. 2015. V. 140. № 18. P. 6260.
Sriram G., Bigliardi P.L., Bigliardi-Qi M. // Eur. J. Cell Biol. 2015. V. 94. № 11. P. 483.
Tracy L.E., Minasian R.A., Caterson E.J. // Adv.Wound Care. 2016. V. 5. № 3. P. 119.
Kehlet S.N., Willumsen N., Armbrecht G. et al. // PLOS One. 2018. V. 13. № 3.
Kular J.K., Basu S., Sharma R.I. // J. Tissue Eng. 2014. V. 5. P. 2041731414557112.
Halper J., Kjaer M. // Progress in heritable soft connective tissue diseases. 2014. P. 31.
Rodrigues M. et al. // Physiol. Rev. 2019. V. 99. № 1. P. 665.
Pastar I., Stojadinovic O., Yin N.C. et al. // Adv. Wound Care. 2014. V. 3. № 7. P. 445.
Krampert M., Bloch W., Sasaki T. et al. // Mol. Biol. Cell. 2004. V. 15. № 12. P. 5242.
Lynch M.D., Watt F.M. // J. Clin. Investig. 2018. V. 128. № 1. P. 26.
Stunova A., Vistejnova L. // Cytokine Growth Factor Rev. 2018. V. 39. P. 137.
Olczyk P., Mencner Ł., Komosinska-Vassev K. // BioMed Res. Int. 2014. V. 747584. P. 8.
Zou M.L., Teng Y.Y., Wu J.J. et al. // Front. Cell Dev. Biol. 2021. V. 9. P. 713605.
Satoshi K., Natsuko K., Kenji S., Kenji K. // Ann. Plast. Surg. 2013. V. 71. № 2. P. 219.
Varkey M., Ding J., Tredget E.E. // Tissue Eng. A. 2013. № 131017062434004.
Fisher G.J., Quan T., Purohit T. et al. // Am. J. Pathol. 2009. V. 174. № 1. P. 101.
Rippa A.L., Kalabusheva E.P., Vorotelyak E.A. // Cells. V. 2019. № 8. P. 607.
Sorrell J.M., Baber M.A., Caplan A.I. // J. Cell Physiol. 2004. V. 200. P. 134.
Mahmoudi S., Mancini E., Xu L. et al. // Nature. 2019. V. 574. P. 553.
Bentov I., Damodarasamy M., Plymate S. et al. // Biogerontology. 2014. V. 15. P. 329.
Левченко В.М. Сравнительная характеристика морфофункциональных свойств фибробластов сельскохозяйственных животных. Ставропольский государственный аграрный университет. 2016. 114 с.
Des Jardins-Park H.E., Foster D.S., Longaker M.T. // Regen. Med. 2018. V. 13. № 5.
Wallace H.A., Basehore B.M., Zito P.M. Wound Healing Phases. StatPearls Publishing. 2017.
Xue M., Jackson.C.J. // Adv. Wound Care. 2015. V. 4. № 3. P. 119.
Desmoulière A., Chaponnier C., Gabbiani G. // Wound Repair Regen. 2005. V. 13. № 1. P. 7.
Eming S.A., Martin P., Tomic-Canic M. // Sci. Transl. Med. 2014. V. 6. № 265. P. 265.
Darby I.A., Laverdet B., Bonté F., Desmoulière A. // Clin. Cosmet. Investig Dermatology. 2022. V. 7. № 2014. P. 301.
Bennett N.T., Schultz G.S. // Am. J. Surg. 1993. V. 165. № 6. P. 728.
Schurch W., Seemayer T.A., Hinz B., Gabbiani G. // Am. J. Pathol. 2007. V. 170. № 6. P. 1807.
Hinz B. // Curr. Res. Transl. Med. 2016. V. 64. № 4. P. 171.
Darby I., Skalli O., Crabbiani G. // Lab. Invest. 1990. V. 63. P. 21.
Hinz B., Celetta G., Tomasek J.J. et al. // Mol. Biol. Cell. 2001. V. 12. № 9. P. 2730.
Desmoulière A., Geinoz A., Gabbiani F., Gabbiani G. // J. Cell Biol. 1993. V. 122. № 1. P. 103.
Wang Y., Mack J.A., Maytin E.V. // J. Biol. Chem. 2019. V. 294. P. 12779.
Midgley A.C., Rogers M., Hallett M.B. et al. // J. Biol. Chem. 2013. V. 288. P. 14824.
Kumawat K., Koopmans T., Menzen M.H. et al. // Am. J. Physiol. Lung Cell Mol. Physiol. 2016. V. 311. № 3. P. 529.
Bainbridge P. // J. Wound Care. 2013. V. 22. № 8. P. 407.
Hinz B. // J. Invest. Dermatol. 2007. V. 127. № 3. P. 526.
Gumbiner B.M. // Nat. Rev. Mol. Cell Biol. 2005. V. 6. P. 622.
Ornitz D.M., Itoh N. // WIREs Mechanisms of Disease. 2022. V. 14. № 4.
Shin J.W., Kwon S.H., Choi J.Y. et al. // Int. J. Mol. Sci. 2019. V. 20. № 9. P. 2126.
Du H.C., Jiang L., Geng W.X. et al. // Biomed Res. Int. 2017. № 2578017.
Newman A.C., Nakatsu M.N., Chou W. et al. // Mol. Biol. Cell. 2011. V. 22. № 20. P. 3791.
Frantz C., Stewart K.M. // J. Cell Sci. 2010. V. 123. № 24. P. 4195.
Myllyharju J. // Matrix Biol. 2003. V. 22. № 1. P. 15.
Yamauchi M., Srocholpech M. // Essays Biochem. 2012. V. 52. P. 113.
Province P., Griguer C.E., Han X. et al. // Evolution of the Molecular Biology of Brain Tumors and the Therapeutic Implications 2. 2017. P. 106.
Thisse B., Thisse C. // Dev. Biol. 2005. V. 287. № 2. P. 390.
Xie Y., Su N., Yang J. et al. // Sig. Transduct. Target Ther. 2020. V. 5. № 181.
Barrientos S., Stojadinovic O., Golinko M.S. et al. // Wound Repair Regen. 2008. V. 16. № 5. P. 585.
Schultz G.S., Wysocki A. // Wound Repair Regen. 2009. V. 17. № 2. P. 153.
Yamakawa S., Hayashida K. // Burns Trauma. 2019. V. 7. P. 10.
Wilgus T.A. // Adv. Wound Care. 2012. V. 1. № 6. P. 249.
Park S. // Biogerontology. 2022. V. 23. P. 275.
Xu J., Zgheib C., Hobges M. et al. // Physiol Genomics. 2017. V. 26.
Gao W., Zhang Y., Yuan L. et al. // Photochem. Photobiol. 2023. V. P. 1.
Wells J.M., Gaggar A., Blalock J.E. // Matrix Biol. 2015. V. 44. P. 122.
Guillemin Y., Le Broc D., Segalen C. et al. // J. Wound Care2. 2016. V. 25. № 7. P. 406.
Mendez M.V., Stanley A., Phillips T. et al. // J. Vasc. Surg. 1998. V. 28. № 6. P. 1040.
Raffetto J.D., Mendez M.V., Marien B.J. et al. // J. Vasc. Surg. 2001. V. 33. № 6. P. 1233.
Mendez M.V., Raffetto J.D., Phillips T. et al. // J. Vasc. Surg. 1999. V. 30. № 4. P. 734.
Herrick S.E., Sloan P., McGurk M. et al. // Am. J. Pathol. 1992. V. 141. № 5. P. 1085.
Cook H., Stephens Ph., Davies K.J. et al. // J. Invest. Dermatol. 2000. V. 115. № 2. P. 225.
Mason R.M., Wahab N.A. // J. Am. Soc. Nephrol. 2003. V. 14. P. 1358.
Salvatori M., Katari R., Patel T. et al. // J. Diabetes Sci. Technol. 2014. V. 8. № 1. P. 159.
McGarry K., Sefat E., Suh T.C. et al. // Biomimetics. 2023. V. 8. № 1. P. 99.
Зорин В.Л., Зорина А.И., Петракова О.С., Черка-сов В.Р. // Клеточная трансплантология и тканевая инженерия. 2009. Т. 4. № 4. С. 26.
Theobald V.A., Lauer J.D., Kaplan F.A. et al. // Transplantation. 1993. V. 55. № 1. P. 128.
Morimoto N., Saso Y., Tomihata K. et al. // J. Surg. Res. 2005. V. 125. № 1. P. 56.
Kazemi-Darabadi S., Sarrafzadeh-Rezaei F., Far-shid A.A., Dalir-Naghadeh B. // Int. J. Surg. 2014. V. 12. № 8. P. 751.
Burke J.F., Yannas I.V., Quinby Jr. et al. // Ann. Surg. 1981. V. 194. № 4. P. 413.
Tsao C.T., Leung M., Chang et al. // J. Mater. Chem. B. 2014. V. 2. № 32. P. 5256.
Jingyue G., Jiaxing H., Shaojin L. et al. // ACS Biomater. Sci. Eng. 2023. V. 9. № 2. P. 844.
Farris A.L., Rindone A.N., Grayson W.L. // J. Mater. Chem. B. 2016. № 20.
Wu D., Potluri N., Lu J. et al. // 2015. V. 524. № 7565. P. 303.
Semenza G.L., Jiang B.H., Leung S.W. et al. // J. Biol. Chem. 1996. V. 271. № 51. P. 32529.
Ziello J.E., Jovin I.S., Huang Y. // Yale J. Biol. Med. 2007. V. 80. P. 51.
Huang L.E., Arany Z., Livingston D.M., Bunn H.F. // J. Biol. Chem. 1996. V. 271. № 50. P. 32253.
Huang L.E., Gu J., Schau M., Bunn H.F. // Proc. Natl. Acad. Sci. 1998. V. 95. № 14. P. 7987.
Marxsen J.H., Stengel P., Doege K. et al. // Biochem J. 2004. V. 381. P. 761.
Rasheduzzaman C., Ignacio C.L.J., Chan M.C. et al. // ACS Chem. Biol. 2013. V. 8. № 7. P. 1488.
Tanimoto K., Yoshiga K., Eguchi H. et al. // Carcinogenesis. 2003. V. 24. № 11. P. 1779.
Chowdhury R., Candela-Lena J.I., Chan M.C. et al. // ACS Chem. Biol. 2013. V. 8. № 7. P. 1488.
Aprelikova O., Chandramouli G.V., Wood M. et al. // J. Cell Biochem. 2004. V. 92. № 3. P. 491.
Qutub A.A., Popel A.S. // J. Cell Sci. 2007. V. 119. № 16. P. 3467.
Cockman M.E., Masson N., Mole D.R. et al. // J. Biol. Chem. 2000. V. 275. № 33. P. 25733.
Kosyna F.K., Nagel M., Kluxen L. et al. // Biol. Chem. 2015. V. 396. № 12.
Fabian Z. The Signaling Nature of Cellular Metabolism: The Hypoxia Signaling. Cell Signalling — Thermodynamics and Molecular Control. 2019.
Huffman J.L., Mokashi A., Bächinger H.P., Bren-nan R.G. // J. Biol. Chem. 2001. V. 276. № 44. P. 40537.
Greijer A.E., van der Wall E. // J. Clin. Pathol. 2004. V. 57. № 10. P. 1009.
Peyssonnaux C., Boutin A.T., Zinkernagel A.S. et al. // J. Invest. Dermatol. 2008. V. 128. № 8. P. 1964.
Lee J.W., Ko J., Ju C., Eltzschig H.K. // Exp. Mol. Med. 2019. V. 51. № 6.
Deng W., Feng X., Li X. et al. // Cell. Immunol. 2016. V. 303. P. 7.
Rosenberger C., Solovan C., Rosenberger A.D. et al. // J. Invest. Dermatol. 2007. V. 127. № 10. P. 2445.
Abdou A.G., Farag A.G.A., Hammam M. et al. // J. Immunoassay Immunochem. 2018. V. 39. № 3. P. 249.
Marina M.E., Roman I.I., Constantin A.M. et al. // Clujul Med. 2015. V. 88. P. 247.
Torales-Cardeńa A., Martínez-Torres I., Rodríguez-Martínez S. et al. // Mediators Inflamm. 2015. № 607363.
Masoud G.N., Li W. // Acta Pharm. Sin. B. 2015. V. 5. № 5. P. 378.
Shih J.W., Chiang W.F., Wu A. et al. // Nat. Commun. 2017. V. 8. № 15874.
Yang H., Zhang H., Yang Y. et al. // Theranostics. 2020. V. 10. № 18. P. 8211.
Goel H.L., Mercurio A.M. // Nat. Rev. Cancer. 2013. V. 13. P. 871.
Kieran M.W., Kalluri R., Cho Y.J. // Cold Spring Harb Perspect Med. 2012. V. 2. № 12.
Carmeliet P. // Oncology. 2005. V. 69 № 3. P. 4.
Keith B., Celeste S.M. // Cell. 2007. V. 129. № 3. P. 465.
Gardner V., Madu C.O., Lu Y. // Physiologic and Pathologic Angiogenesis / Ed. Simionescu D. IntechOpen. 2017. P. 1380.
Trédan O., Lacroix-Triki M., Guiu S. et al. // Targeted Oncology. 2015. V. 10. № 2. P. 189.
Cantor S.B., Elting L.S., Hudson Jr., D.V., Ruben-stein E.B. // Cancer. 2003. V. 97. № 12. P. 3099.
Onnis B., Fer N., Rapisarda A. et al. // J. Clin. Invest. 2013. V. 123. № 4. P. 1615.
Steinbrech D.S., Gittes G.K. // J. Surg. Res. 1999. V. 84. № 2. P. 127.
Nauta T., van Hinsbergh V., Koolwijk P. // Int. J. Mol. Sci. 2014. V. 15. № 11. P. 19791.
D’Alessandro S., Magnavacca A., Perego F. et al. // BioMed Res. Int. 2019. P. 1.
McKay T.B., Hjortdal J., Priyadarsini S., Karami-chos D. // PLOS One. 2017. V. 12. № 4. e0176017.
Hong W.X., Hu M.S., Esquivel M. et al.// Adv. Wound Care. 2014. V. 3. № 5. P. 390.
Liu S.S., Wang H.Y., Tang J.M., Zhou X.M. // Int. J. Mol. Sci. 2013. V. 14. № 12. P. 24029.
Ruthenborg R.J., Ban J.J., Wazir A. et al. // Mol. Cells. 2014. V. 37. № 9. P. 637.
Yue B. // J. Glaucoma. 2014. P. 1.
Deppe J., Popp T., Egea V. et al. // Arch. Toxicol. 2015. V. 90. № 5. P. 1141.
Gilkes D.M., Bajpa S., Chaturvedi P. et al. // J. Biol. Chem. 2013. V. 288. № 15. P. 10819.
Kukkola L., Hieta R., Kivirikko K.I., Myllyharju J. // J. Biol. Chem. 2003. V. 278. № 48. P. 47685.
Valtavaara M., Szpirer C., Szpirer J., Myllylä R. // J. Biol. Chem. 1998. V. 273. № 21. P. 12881.
Shuo Q., Yachao J., Yunchu S. et al. // J. Diabetes Res. 2019. Art. 1897174.
Zhang X., Yan X., Cheng L. et al. // PLoS One. 2013. V. 8. № 12.
Dallas A., Trotsyuk A., Ilves H. et al. // Tissue Eng. A. 2018.
Zhao B., Guan H., Liu J.Q. et al. // Int. J. Mol. Med. 2017. V. 39. № 1. P. 153.
Дополнительные материалы отсутствуют.
Инструменты
Вестник Военного инновационного технополиса «ЭРА»