

Высокомолекулярные соединения (серия А), 2019, T. 61, № 1, стр. 33-43
ПАРАМЕТРЫ ФЛОРИ–ХАГГИНСА ГУАРА, КСАНТАНА, АГАРОЗЫ И ГЕЛЛАНА В ВОДНЫХ РАСТВОРАХ
А. П. Сафронов a, b, Л. В. Адамова a, *, Г. В. Курляндская a, c
a Уральский федеральный университет им. Б.Н. Ельцина. Институт естественных наук и математики
620006 Екатеринбург, пр. Мира, 19, Россия
b Институт электрофизики Уральского отделения Российской академии наук
620016 Екатеринбург, ул. Амундсена, 106, Россия
c Университет Страны Басков. Факультет науки и технологий
48940 Лейоа, Испания
* E-mail: Lidia.adamova@urfu.ru
Поступила в редакцию 18.04.2018
После доработки 16.07.2018
Принята к публикации 30.07.2018
Аннотация
Исследована термодинамика взаимодействия с водой гуара, ксантана, агарозы и геллана. Методом изотермической интервальной сорбции получены изотермы сорбции воды полисахаридами при 25°С, которые имеют S-образный вид, типичный для стеклообразных полимеров. Значения энергии Гиббса смешения полисахаридов с водой, определенные из изотерм, отрицательны. Рассчитанные параметры Флори–Хаггинса для растворов полисахаридов практически одинаковы и равны в среднем 0.82. Методом изотермической микрокалориметрии измерены концентрационные зависимости энтальпии разбавления водных растворов полисахаридов. Найденные из них величины энтальпийного параметра Флори–Хаггинса для всех полисахаридов близки к нулю. В то же время интегральная энтальпия растворения принимает большие отрицательные значения, которые целиком обусловлены релаксацией стеклообразного состояния полимеров при растворении. Рассчитанный энтропийный параметр Флори–Хаггинса принимает большие положительные значения. Это свидетельствует об упорядочении в растворах полисахаридов, проявляющемся в сильном агрегировании макромолекул.
ВВЕДЕНИЕ
Ввиду низкой стоимости, нетоксичности, способности к биоразложению, хорошей биосовместимости, растворимости в воде и высокой вязкости растворов во многих областях промышленности, науки, медицины и биотехнологии используются природные полисахариды различного происхождения. В частности, они применяются как стабилизаторы и загустители в косметической и пищевой промышленности, для проклейки отделочных материалов в текстильной и бумажной промышленности, в качестве добавки к жидкости для гидроразрыва пласта при нефтедобыче, для контролируемого высвобождения лекарственных препаратов в медицине, в качестве среды для разделения белковых смесей; также они используются как компоненты субстратов для выращивания клеток в биотехнологии, как биосовместимые стабилизаторы водных суспензий наночастиц для биоинженерии, для борьбы с повреждениями растений в садоводстве, а в некоторых случаях бактерицидные свойства полисахаридов выступают как альтернатива антибиотикам [1–9]. К наиболее широко применяемым полисахаридам относятся гуар (структура а), ксантан (б), агароза (в) и геллан (г). Их макромолекулярная цепь состоит из циклических моносахаридных звеньев, отличающихся друг от друга числом и пространственным расположением гидроксильных групп, а также наличием заместителей другого типа:
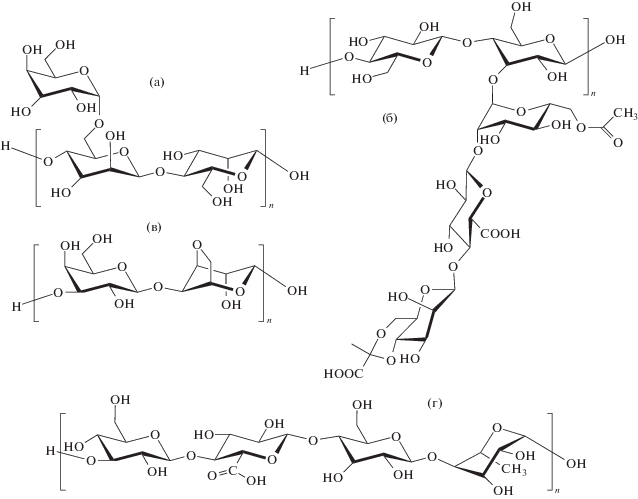
Макромолекулы гуара и ксантана имеют разветвленное строение. Гуар получают экстракцией из семян горохового дерева Cyamopsis tetragonalobus. Главная цепь макромолекулы гуара состоит из остатков D-маннозы, соединенных β(1 → 4)-связями. D-галактозные остатки привиты к основной маннозной цепи через α(1 → 6)-связи (а) [2]. Ксантан представляет собой внеклеточный полисахарид, вырабатываемый бактерией Xanthomonas Campestris. Главная цепь макромолекулы ксантана идентична молекуле целлюлозы (β(1 → 4)-глюкопираноза), к каждому второму звену которой привит заряженный трисахарид, состоящий из β-D-маннозы, β-D-глюкуроновой кислоты и α-D-маннозы (б) [10].
Агароза и геллан – это полимеры преимущественно линейного строения. Агарозу в промышленном масштабе выделяют из некоторых видов красных водорослей. Макромолекулярная цепь ее построена из чередующихся остатков D-галактозы и 3,6-ангидро-L-лактозы, соединенных попеременно β(1 → 4)- и α(1 → 3)-связями (в) [11]. Геллан – это внеклеточный гетерополисахарид, вырабатываемый бактериями Pseudomonas elodea. Его цепь включает звенья 1,3-β,D-глюкозы, 1,4-β,D-глюкуроновой кислоты, 1,4-α,D-глюкозы и 1,4-α,L-рамнозы (г) [9].
В основе применения гуара, ксантана, агарозы и геллана лежит общая для них способность к гелеобразованию в водной среде за счет формирования физической сетки, узлы которой представляют собой упорядоченные цепи или их участки, находящиеся в конформации спиралей, стабилизированных водородными связями. В литературе представлено большое число работ, посвященных изучению реологических свойств и структурных особенностей строения растворов полисахаридов [12–18]. Вполне понятно, что на молекулярном уровне структура растворов и их реологические характеристики определяются особенностями взаимодействий между звеньями макромолекул полисахарида и молекулами воды. Это обусловливает фундаментальный интерес к познанию различных видов молекулярных взаимодействий в растворах полисахаридов. Одним из основных подходов является оценка изменения термодинамических функций состояния системы – энтальпии, энтропии, энергии Гиббса в ходе протекающих в ней различных процессов.
Термодинамические исследования водных растворов полисахаридов в основном посвящены изучению конформационного перехода клубок–спираль, который лежит в основе структурных изменений растворов, протекающих при гелеобразовании. Так, методом дифференциальной сканирующей калориметрии установлено [16], что данный переход сопровождается экзотермическим тепловым эффектом, величина которого и положение на оси температуры зависят от природы полисахарида, концентрации раствора, его ионной силы и присутствия низкомолекулярных неэлектролитов.
В существенно меньшей степени изучена термодинамика гидратации полисахаридов, т.е. взаимодействие звеньев полисахаридов с молекулами воды. В физической химии полимеров его принято описывать параметром Флори–Хаггинса, представляющим собой нормированную по температуре свободную энергию взаимообмена для парных контактов звеньев полимера и молекул растворителя [19]. Значения параметра Флори–Хаггинса для водных растворов полисахаридов в литературе практически отсутствуют. Это связано с тем, что наиболее распространенные способы измерения этого параметра – светорассеяние и осмометрия, успешно используемые для растворов синтетических полимеров, оказываются неприменимыми для водных растворов полисахаридов вследствие процессов агрегирования и структурирования. Единственно возможными методами определения параметра Флори–Хаггинса для таких систем оказываются термодинамические методы, основанные на сорбционных и калориметрических измерениях энергии Гиббса и энтальпии смешения.
Цель настоящей работы состояла в получении сорбционных и калориметрических данных для процесса гидратации гуара, ксантана, агарозы, геллана и их использовании для расчета параметра Флори–Хаггинса для водных растворов указанных полисахаридов. Ниже представлены изотермы сорбции паров воды образцами полисахаридов и зависимости теплоты разбавления растворов от состава при 25°С, на основании которых будут определены значения параметра Флори–Хаггинса и его энтальпийной и энтропийной компонент для данных систем.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве объектов исследования взяли агарозу Agarose hohe-EEO-high производства “SERVA” (“Feinbiochemica“, Германия), геллан Phytagel, гуар Guar gum и ксантан Xanthan gum from Xanthomonas campestris – все производства “Sigma-Aldrich” (США). Образцы полисахаридов представляли собой мелкодисперсные порошки белого или желтоватого цвета. Полимеры использовали без дополнительной очистки. Молекулярную массу гуара (М = 1.6 × 106), ксантана (2.2 × 106) и агарозы (2.7 × 104) определяли методом вискозиметрии с помощью имеющихся в литературе констант Марка–Хаувинка [20–22]: для гуара K = = 3.7 × 10–4, a = 0.74, вода, Т = 25°С; для ксантана K = 1.7 × 10–6, a = 1.14, 0.1 моля/л водный раствор NaCl, Т = 25°С; для агарозы K = 7.0 × 10–4, a = = 0.72, 0.75 моля/л водный раствор KSCN, Т = = 35°С.
Для геллана константы Марка–Хаувинка отсутствуют. Характеристическая вязкость его водного раствора при 25°С составила 25 дл/г, что близко к характеристической вязкости раствора ксантана (29 дл/г).
Образцы полисахаридов перед измерениями предварительно высушивали на воздухе до постоянной массы при 75°С; поэтому все термодинамические данные относятся к исходной структуре полисахаридов в порошкообразном состоянии.
Порошкообразные образцы гуара, ксантана и геллана самопроизвольно растворялись в воде при комнатной температуре с образованием агрегированных опалесцирующих растворов, типичных для водорастворимых полисахаридов. Образец агарозы не растворялся, а лишь набухал. Степень набухания агарозы, оцененная весовым методом, составила порядка 300%, что отвечает ~30% концентрации агарозы в набухшем состоянии. В этой связи, термодинамические данные, полученные для агарозы, ограничены диапазоном концентрации от 30 до 100%.
Сорбцию паров воды полисахаридами при 25°С изучали объемным методом на автоматическом анализаторе площади поверхности и пористости ASAP 2020 (“Micromeritics”) c приставкой для сорбции паров, которая позволяет измерять сорбцию паров воды при комнатной температуре. Принцип метода заключается в определении количества сорбированного вещества по уменьшению давления пара сорбата, находящегося в парообразной фазе и занимающего определенный заранее прокалиброванный объем.
Энтальпию разбавления растворов полисахаридов в воде при 25°С измеряли на микрокалориметре Кальве лабораторной конструкции с объемом ячеек 10 см3. В тонкостенную стеклянную калориметрическую ампулу помещали навеску полимера или раствора заданной концентрации с содержанием полимера ~0.01–0.02 г. Ампулы запаивали и помещали в ячейки калориметра, заполненные 7 мл воды. В процессе калориметрического опыта после установления термического равновесия ампулы разбивали внутри ячейки и регистрировали тепловой эффект растворения полимера или разбавления раствора до конечной концентрации (менее 0.3%). Абсолютная погрешность калориметрических измерений (по данным электрической калибровки прибора) ±0.02 Дж. Измеряемые тепловые эффекты составляли не менее 0.4 Дж, так что относительная погрешность измерений не превышала 5%. При малых значениях удельных тепловых эффектов пропорционально увеличивали навеску образца.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Изотермы сорбции паров воды образцами гуара, ксантана, агарозы и геллана при 25°С представлены на рис. 1 в виде зависимости массовой доли сорбированной воды от относительного давления пара P/PS. Изотермы имеют отчетливый S-образный вид с выпуклым начальным участком. Такая форма изотермы характерна для стеклообразных полимеров, у которых в начальной стадии сорбции одновременно протекают два процесса: заполнение молекулами растворителя имеющихся неплотностей упаковки и набухание полимера, сопровождающееся перестройкой структуры. Изотермы сорбции подобного вида ранее были получены для крахмала и производных целлюлозы [23, 24]. Значения сорбции воды для гуара и ксантана близки и находятся в пределах погрешности сорбционных измерений. Поэтому для них характерна единая изотерма (рис. 1а). Также единой кривой описывается другая пара: геллан–агароза (рис. 1б).
Рис. 1.
Изотермы сорбции воды полисахаридами при 25°С. Здесь и на рис. 2, 3 точки 1 и 2 – гуар и ксантан (а), 3 и 4 – геллан и агароза (б). Кривые отвечают интерполяции экспериментальных точек модельными зависимостями (уравнение (1)); на оси абсцисс отложено относительное давление пара.
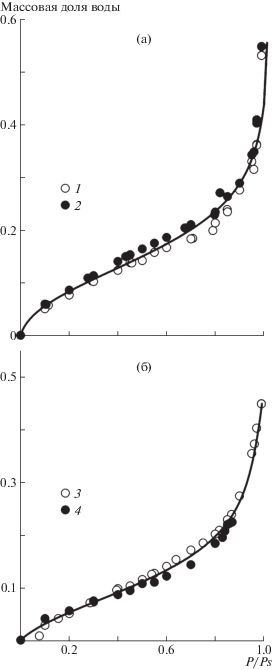
Несмотря на то что гуар, ксантан и геллан самопроизвольно растворяются в воде, изотермы сорбции ограничены массовой долей воды ~0.5. Это связано с большой погрешностью измерений при высоких значениях P/PS, когда разность давления пара сорбата над чистой жидкостью и над раствором становится очень маленькой.
На основании анализа изотерм сорбции по стандартной методике [19] были рассчитаны значения энергии Гиббса смешения полисахаридов с водой.
Изменение химического потенциала растворителя во всем диапазоне относительного давления пара было найдено по уравнению
где Δμ1 – разность удельных химических потенциалов растворителя, М1 – мольная масса растворителя.Изменение химического потенциала полимера рассчитано интегрированием уравнения Гиббса–Дюгема:
Здесь ω – массовая доля полимера в растворе. Нижний предел интегрирования определен как точка сплайн-интерполяции подынтегрального выражения к нулевому значению.
Удельная энергия Гиббса смешения раствора заданной концентрации была рассчитана как сумма изменений химических потенциалов растворителя и полимера по уравнению
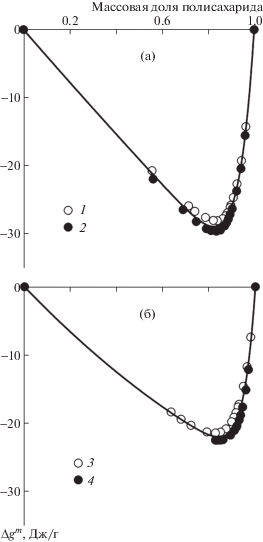
Концентрационные зависимости энергии Гиббса смешения водных растворов гуара, ксантана, агарозы и геллана в изученном диапазоне составов представлены на рис. 2. Ввиду того что сорбция воды образцами гуара и ксантана описывается единой изотермой, концентрационная зависимость энергии Гиббса смешения этих полисахаридов выражается единой кривой. То же относится и к энергии Гиббса смешения с водой геллана и агарозы. Видно, что энергия Гиббса смешения растворов для всех полисахаридов отрицательна во всей области составов, которой отвечают точки на изотерме сорбции. Это свидетельствует о наличии термодинамического сродства полисахаридов к воде. Гуар и ксантан характеризуются более сильным сродством к воде по сравнению с гелланом и агарозой.
Рис. 2.
Концентрационные зависимости средней удельной энергии Гиббса смешения водных растворов полисахаридов при 25°С. Кривые отвечают результату расчета на основании теоретической изотермы (уравнение (1)) с использованием параметров, приведенных в таблице.
Для определения параметра Флори–Хаггинса гуара, ксантана, агарозы и геллана на основании анализа изотерм сорбции (см. рис. 1) мы воспользовались известной термодинамической моделью [25–27]. Модель рассматривает процесс сорбции как комбинацию процесса взаимодействия растворителя и полимера с процессом структурной перестройки, в ходе которой происходит релаксация неравновесной структуры стекла и ее переход в равновесное эластическое состояние раствора. Особенностью неравновесной структуры стекла является наличие в ней неплотностей упаковки – неравновесных вакансий, которые в процессе адсорбции заполняются молекулами растворителя. Согласно данной модели, зависимость относительного давления паров растворителя от объемной доли полимера в растворе φ имеет вид [25]
(1)
$\begin{gathered} \ln \frac{P}{{{{P}_{S}}}} = {\text{ln(1}} - \varphi {\text{)}} + \varphi + \chi {{\varphi }^{{\text{2}}}} + {{g}_{V}}\varphi \frac{{1 - \varphi _{V}^{0}}}{{\varphi _{V}^{0}}}{{\varphi }_{V}} - \\ - \;\frac{{{{\varphi }_{V}}}}{{\varphi _{V}^{0}}}[(1 - 2\varphi _{V}^{{}})\ln {{\varphi }_{V}} + 1 - \varphi _{V}^{0}] \\ {{\varphi }_{{\text{V}}}} = \varphi _{V}^{0}{{\varphi }^{{\frac{1}{{\varphi _{V}^{0}}} - 2}}} \\ \end{gathered} $Здесь χ – параметр Флори–Хаггинса, gV – избыточная свободная энергия размещения молекулы растворителя в вакансии, $\varphi _{V}^{0}$ – объемная доля неравновесных вакансий в исходной структуре стеклообразного полимера, φV – объемная доля неравновесных вакансий в растворе с заданной концентрацией полимера. Объемная доля полимера в растворе рассчитана по уравнению
(2)
$\varphi = \frac{{\omega {\text{/}}{{d}_{2}}}}{{\omega {\text{/}}{{d}_{2}} + (1 - \omega ){\text{/}}{{d}_{1}}}},$Результат аппроксимации экспериментальных изотерм сорбции воды уравнением (1) представлен линиями на рис. 1. Видно, что модельные уравнения хорошо описывают экспериментальные данные. Параметры модели χ, gV, $\varphi _{V}^{0}$ приведены в таблице 1.
Таблица 1.
Параметр Флори–Хаггинса и его компоненты для водных растворов полисахаридов и характеристики их стеклообразной структуры
| Полисахарид | ΔHdissol, Дж/г | ε22, кДж/моль | gVRT, кДж/моль | $\varphi _{V}^{0}$ (1) | $\varphi _{V}^{0}$ (2) | χ | χH | χS |
|---|---|---|---|---|---|---|---|---|
| Гуар | –117.8 | –30.2 | –8.7 | 0.085 | 0.079 | 0.82 | –0.08 | 0.90 |
| (±5.9) | (±1.6) | (±1.5) | (±0.009) | (±0.007) | (±0.04) | (±0.03) | ||
| Ксантан | –109.3 | –30.2 | –8.7 | 0.085 | 0.079 | 0.82 | –0.08 | 0.90 |
| (±5.5) | (±1.6) | (±1.5) | (±0.009) | (±0.007) | (±0.04) | (±0.03) | ||
| Агароза | –197.2 | –38.5 | –5.7 | 0.077 | 0.106 | 0.81 | –0.01 | 0.82 |
| (±9.9) | ±1.5 | ±1.0 | ±0.009 | ±0.004 | ±0.05 | ±0.03 | ||
| Геллан | –131.9 | –29.0 | –5.7 | 0.077 | 0.102 | 0.81 | 0.04 | 0.78 |
| (±6.6) | (±3.6) | (±1.0) | (±0.009) | (±0.013) | (±0.05) | (±0.07) |
Как следует из таблицы, параметр Флори–Хаггинса для всех исследованных полимеров близок к 0.82. Различия в величинах χ лежат в пределах погрешности их вычисления при интерполяции. Следует отметить, что данное значение параметра Флори–Хаггинса получено для концентрационного диапазона, отвечающего изотермам сорбции (см. структуры) и концентрационным зависимостям энергии Гиббса (рис. 2), т.е. растворам с массовой долей полисахарида выше 0.5. Поскольку более разбавленные растворы недоступны для измерений сорбционным методом, то использовать полученное значение χ для таких растворов следует с осторожностью.
Вполне понятно, что водные растворы вообще и водные растворы полисахаридов в частности имеют заведомо более сложную структуру, чем растворы гибкоцепных полимеров в органических растворителях, для которых, собственно, и была сформулирована теория Флори–Хаггинса. С этой точки зрения, параметр Флори–Хаггинса следует рассматривать как некую эффективную величину, вбирающую в себя баланс всех видов взаимодействий различной природы, имеющихся в водных растворах полисахаридов. Полученное значение χ = 0.82 свидетельствует о том, что данный баланс соответствует плохому термодинамическому качеству воды как растворителя по отношению к полисахаридам.
Что же касается параметров, характеризующих неравновесность стеклообразной структуры, то доли неравновесных вакансий для всех исследованных полисахаридов также близки и находятся в пределах погрешности расчета; среднее значение $\varphi _{V}^{0}$ составляет ~0.081. Отличия между агарозой и гелланом, с одной стороны, гуаром и ксантаном, с другой, наблюдаются в свободной энергии размещения молекул воды в вакансии gV. Величины gV отрицательны во всех случаях, что указывает на термодинамическую выгодность локализации молекул воды в вакансии. В случае гуара и ксантана такой процесс выгоднее, на что указывают более отрицательные значения gV, чем в случае агарозы и геллана. Возможно, обнаруженное различие связано с более рыхлой разветвленной структурой гуара и ксантана, обусловленной наличием боковых заместителей основной цепи (см. структуры).
Учитывая достаточно большую положительную величину эффективного параметра Флори–Хаггинса для полисахаридов, представляло интерес провести анализ его энтальпийной и энтропийной компонент. Для этого были выполнены микрокалориметрические исследования растворения гуара, ксантана, геллана и набухания агарозы в воде при 25°С.
Интегральные энтальпии растворения гуара, ксантана, геллана и энтальпия набухания агарозы в воде при 25°С (ΔHdissol) приведены в таблице, из данных которой видно, что энтальпия растворения (набухания) принимает большие отрицательные значения для всех полисахаридов. Она в несколько раз превышает типичные значения ΔHdissol для синтетических полимеров в неводных растворителях [25] и энтальпию растворения в воде для таких полимеров как полиакриловая кислота [28]. Из таблицы также видно, что значения энтальпии растворения гуара, ксантана и геллана близки; разница между ними находится в пределах 10–20%. Энтальпия набухания агарозы существенно отрицательнее.
Энтальпия растворения или набухания по своему смыслу представляет собой разность энтальпии предельно разбавленного раствора и энтальпий его индивидуальных компонентов. (В реальности, это о растворе конечной малой концентрации). Отсюда можно было бы сделать вывод о том, что при растворении полисахаридов в воде энергия образующихся связей существенно выше, чем энергия связей в воде и полисахариде. Поскольку в водных системах доминирующими являются водородные связи, это означает, что звенья полисахаридов образуют существенно более прочные водородные связи с молекулами воды, чем молекулы воды между собой и звенья полисахарида между собой. Но справедлив ли данный вывод?
При анализе интегральной энтальпии растворения полимера следует учитывать, что ее величина помимо собственно энтальпии взаимодействия, обусловленной межмолекулярными контактами звеньев и молекул растворителя, включает и структурные вклады, обусловленные как фазовым, так и релаксационным состоянием полимера. Разделение указанных вкладов может быть успешно проведено на основании анализа концентрационных зависимостей энтальпии разбавления растворов [29].
Зависимости энтальпии разбавления растворов полимера избытком воды представлены на рис. 3. Абсцисса точек, приведенных на графиках, отвечает составу предварительно приготовленного раствора, который был разбавлен избытком воды; ордината точек отвечает тепловому эффекту (изменению энтальпии) процесса разбавления. Для последующего использования формализма теории Флори–Хаггинса массовая доля полисахарида в разбавляемом растворе была пересчитана в объемную долю по уравнению (2), а экспериментальные энтальпии разбавления ΔHdil, выраженные в единицах Дж/г полимера, пересчитаны на моль узлов решетки, соответствующих объему молекулы воды, по уравнению
Рис. 3.
Концентрационные зависимости энтальпии разбавления водных растворов полисахаридов при 25°C. Кривые отвечают интерполяции экспериментальных точек модельными зависимостями (уравнение (3)).
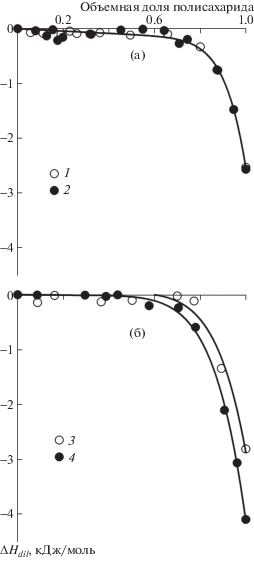
На рис. 3 видно, что концентрационная зависимость энтальпии разбавления для всех полисахаридов выглядит однотипно. Параметр ΔHdil принимает близкие к нулю отрицательные значения в диапазоне концентрации до 80% и резко уменьшается при повышении концентрации от 80 до 100%. Энтальпии разбавления водных растворов гуара и ксантана близки, и их концентрационные зависимости описаны единой кривой (рис. 3а). Энтальпии разбавления водных растворов геллана и агарозы несколько различаются (рис. 3б).
В целом, вид концентрационных зависимостей энтальпии разбавления обусловлен суперпозицией различных энергетических вкладов, связанных как с установлением и разрушением молекулярных контактов, так и со структурными перестройками в системе. Представленный на рис. 3 вид зависимости энтальпии разбавления от концентрации полисахарида является типичным для полимеров, находящихся в стеклообразном состоянии [27, 30]. Так, в области концентрированных растворов добавление растворителя приводит к релаксации неравновесной стеклообразной структуры (пластификации), сопровождаемой выделением большого количества теплоты. Известно, что аморфные полисахариды при комнатной температуре находятся в стеклообразном состоянии. Для частично-кристаллических полисахаридов стеклообразной считается их аморфная часть. Наличие неравновесных стеклообразных участков в структуре полисахаридов с необходимостью приводит к большим по величине отрицательным значениям энтальпии разбавления в области концентрированных растворов.
Вклады межмолекулярного взаимодействия звеньев полимера с молекулами растворителя и вклад релаксации неравновесной структуры полимера в процессе растворения были разделены с помощью теоретических оценок данных вкладов, полученных в работах [25, 26]. Кривые на рис. 3 отвечают аппроксимации экспериментальной зависимости уравнением
(3)
$\Delta {{H}_{{dil}}} = {{\chi }_{H}}RT(\varphi - \varphi *) + {{\varepsilon }_{{22}}}\varphi _{V}^{0}{{\varphi }^{{\frac{1}{{\varphi _{V}^{0}}} - 2}}}$Здесь χH – энтальпийный параметр Флори–Хаггинса, φ – объемная доля полимера в растворе, φ* – конечная объемная доля полимера при разбавлении, ε22 – энергия когезии полимера, $\varphi _{V}^{0}$ – объемная доля метастабильных вакансий в исходной структуре полимера.
Значения параметров модели χH, ε22, $\varphi _{V}^{0}$ приведены в таблице. Из таблицы видно, что энтальпийный параметр Флори–Хаггинса для водных растворов всех полисахаридов близок к нулю. Наиболее отрицательными значениями χН (около –0.1) характеризуются гуар и ксантан. Энтальпийный параметр Флори–Хаггинса для растворов геллана и агарозы равен нулю в пределах экспериментальной погрешности. Следовательно, при взаимодействии данных полисахаридов с водой выигрыша в энергии не происходит. Иными словами, в рассматриваемых системах взаимодействия звеньев между собой, звеньев с молекулами воды и молекул воды между собой близки друг к другу по энергии.
Все исследуемые в работе полимеры имеют близкие значения параметров стеклообразной структуры. Энергия когезии гуара, ксантана и геллана одинакова в пределах экспериментальной погрешности. Энергия когезии агарозы принимает более отрицательные значения. Для гуара и ксантана доля неравновесных вакансий составляет ~0.08, для геллана и агарозы она близка к 0.10. В общем, доли неравновесных вакансий, найденные из анализа изотерм сорбции и кривых энтальпии разбавления, удовлетворительно согласуются между собой. Имеющиеся отличия, вероятно, обусловлены тем, что в первом случае в неравновесную стеклообразную структуру попадают молекулы из паровой фазы, а во втором случае – из жидкой. Следует отметить, что параметры gV, ε22, $\varphi _{V}^{0}$, представленные в таблице, описывают особенности той конкретной стеклообразной структуры, которая имелась в исследованных образцах полисахаридов. Эти параметры изменятся, если другой будет предыстория получения образцов.
Таким образом, калориметрические данные позволяют более полно описать термодинамику взаимодействия полисахаридов с водой. Как было отмечено выше, параметр Флори–Хаггинса составляет ~0.8, однако это не является следствием плохого энергетического взаимодействия. Напротив, с точки зрения энтальпии взаимодействия, растворы полисахаридов очень близки к атермическим. По-видимому, плохое качество растворителя может быть обусловлено только энтропийными причинами. В таблице приведены значения энтропийного параметра Флори–Хаггинса, рассчитанные из соотношения [19]:
Видно, что параметр χS для всех полисахаридов является существенно положительным. Положительным значениям χS отвечают отрицательные значения некомбинаторной энтропии смешения раствора, что свидетельствует об упорядочении в его структуре. На рис. 4 сопоставлены величины параметра Флори–Хаггинса и его компонент.
Рис. 4.
Параметр Флори–Хаггинса χ (показан серым цветом), его энтальпийная χH (поверхность темного цвета) и энтропийная χS (заштрихованная поверхность) компоненты для водных растворов полисахаридов при 25°С. Штриховой линией отмечено пороговое значение χ = 0.5.
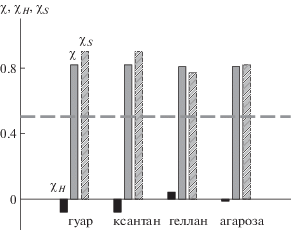
Полученные термодинамические данные проясняют картину поведения гуара, ксантана, агарозы и геллана в водных растворах. Хорошо известно [16–18], что в водных растворах полисахаридов наблюдается сильное агрегирование. Это согласуется с приведенными выше значениями параметра Флори–Хаггинса 0.82, которые свидетельствуют о преобладании контактов однородных молекул над контактами разнородных молекул. Обычно для синтетических полимеров плохое качество растворителя обусловлено тем, что взаимодействие молекул растворителя и звеньев полимера энергетически не выгодно и сопровождается положительным изменением энтальпии. Данное обстоятельство приводит к образованию преимущественных контактов однородных молекул, агрегированию и в конечном итоге к фазовому расслоению. Классической системой такого рода являются растворы полисахаридов в циклогексане, термодинамика смешения которых хорошо изучена в работах [30, 31]. Для них плохое качество растворителя имеет энтальпийную природу.
В случае водных растворов гуара, ксантана, агарозы, геллана наблюдается другая картина, в чем-то парадоксальная, – их взаимодействие с водой можно назвать практически атермическим, поскольку энтальпийный параметр Флори–Хаггинса очень близок к нулю. Это означает, что образование молекулярных контактов звеньев между собой, молекул воды между собой и звеньев с молекулами воды энергетически равноценно. Следует отметить, что упоминание здесь идет об образовании достаточно сильных водородных связей гидроксильных групп и молекул воды в разных комбинациях. Равноценность таких связей не является чем-то удивительным. Известно [32], что энтальпия смешения с водой низкомолекулярных спиртов (начиная с пропилового) является знакопеременной. Другими словами, в разных областях состава в таких системах наблюдается как преобладание контактов разнородных молекул (экзотермическое смешение), так и преобладание контактов однородных молекул (эндотермическое смешение). Близость к нулю эффективного параметра Флори–Хаггинса для полисахаридов можно трактовать как проявление динамического равновесия разных видов водородных связей.
Поскольку в водных растворах полисахаридов водородные связи между однородными и разнородными молекулами сбалансированы по энергии, агрегирование в водном растворе не может быть связано с выигрышем в энтальпии при образовании контактов однородных молекул. Энтальпийный фактор здесь не является движущей силой агрегирования, которое тем не менее сопровождается большими положительными значениями энтропийного параметра Флори–Хаггинса (таблица; рис. 4). Это означает, что растворение гуара, ксантана, геллана и набухание агарозы при комнатной температуре сопровождается уменьшением некомбинаторной энтропии, несмотря на то что энтальпия взаимодействия при этом практически не изменяется. В этой связи необходимо отметить следующее.
Во-первых, упоминается лишь некомбинаторная часть энтропии, а не изменение энтропии в целом. Отрицательная некомбинаторная часть, связанная с положительной величиной χS, является лишь добавкой к положительной комбинаторной энтропии смешения, которая и обеспечивает распределение как макромолекул, так и их агрегатов в объеме системы.
Во-вторых, значения χS рассчитаны лишь для диапазона концентрации, в котором был определен интегральный параметр Флори–Хаггинса, т.е. при массовой доле полисахарида выше 0.5. Значения χS для более разбавленных растворов следует применять с осторожностью. В то же время параметр χH близок к нулю в широком диапазоне концентрации, в том числе и в разбавленных растворах.
В-третьих, сочетание отрицательной некомбинаторной энтропии, являющейся признаком агрегирования, и близкой к нулю энтальпии взаимодействия можно назвать следствием принципиальной ограниченности теории Флори–Хаггинса по отношению к сильно структурированным водным растворам. В исходную модель теории была заложена очень простая идея, в которой предполагалась единственная возможность для структурирования системы – существенное ухудшение взаимодействия разнородных молекул. Иные механизмы структурирования в теории просто отсутствуют. Поэтому из того, что полученные термодинамические данные для водных растворов полисахаридов хорошо соответствуют формализму теории Флори–Хаггинса, не следует, что структура растворов полисахаридов описывается простой квази-кристаллической моделью, заложенной в ее основе.
Конкретный механизм структурирования в водных растворах полисахаридов не может быть получен из термодинамических данных, которые задают лишь рамки для рассмотрения возможных причин агрегирования. В водных растворах действует многообразие факторов, которые способствуют процессу агрегирования. Многие из них носят кооперативный характер. Можно упомянуть конформационные переходы макромолекул, в частности спирализацию, агрегирование отдельных спиралей с образованием физической сетки геля, гидрофобную гидратацию, связанную с упорядочением воды вокруг гидрофобных участков макромолекул и т.д. Эти процессы сопровождаются уменьшением некомбинаторной энтропии и обусловливают положительные значения параметров χS и χ. В то же время, существенным результатом является то, что в водных растворах полисахаридов агрегирование происходит при поддержании общего энергетического баланса различных видов водородных связей, присутствующих в системе. Характерный масштаб изменения энтальпии гидратации в ходе процессов образования и разрушения структур в водных растворах исследованных полисахаридов задается величиной параметра χH, которая составляет в среднем –0.1, что отвечает энергии, на порядок меньшей средней энергии теплового движения при 25°С (2.5 кДж/моль) и на два порядка ниже средней энергии водородных связей в воде (20 кДж/моль).
Данные термодинамические особенности растворения полисахаридов в воде, вероятно, биологически обусловлены. Близость термодинамических параметров гидратации для полисахаридов разного химического строения, возможно, отражает тот факт, что вне зависимости от их природного источника они выполняют однотипную функцию структурообразующих полимеров. Близость энтальпийного параметра Флори–Хаггинса к нулю, скорее всего, создает благоприятные условия для реализации обратимой гидратации и дегидратации полисахаридов в ходе структурообразования в их молекулярном окружении при незначительном изменении внешних условий.
ЗАКЛЮЧЕНИЕ
Сорбционным и микрокалориметрическим методами проведены прямые термодинамические измерения изотерм сорбции и концентрационных зависимостей энтальпии разбавления для водных растворов природных полисахаридов гуара, ксантана, агарозы и геллана при 25°С. На основании этих данных определены значения параметра Флори–Хаггинса, его энтальпийной и энтропийной компонент. Показано, что параметр Флори–Хаггинса для всех четырех полисахаридов совпадает в пределах экспериментальной погрешности и равен 0.82 ± 0.05. Интересной особенностью растворов гуара, ксантана, агарозы и геллана является то, что причиной плохого термодинамического сродства к воде является сильное понижение некомбинаторной энтропии при растворении, которое проявляется в положительных значениях энтропийного параметра Флори–Хаггинса, лежащих в диапазоне 0.8–0.9, что согласуется с сильным агрегированием, наблюдаемым в растворах полисахаридов. Вместе с тем, энтальпийный параметр Флори–Хаггинса близок к нулю и составляет в среднем –0.1, что свидетельствует об энергетическом балансе взаимодействий между однородными и разнородными молекулами в данных системах.
Исследование выполнено за счет гранта Российского научного фонда (проект № 18-19-00090).
Список литературы
Thombare N., Jha U., Mishra S., Siddiqui M.Z. // Int. J. Biological Macromol. 2016. V. 88. P. 361.
Kono H., Otaka F., Ozaki M. // Carbohydr. Polym. 2014. V. 111. P. 830.
Jana S., Gandh A., Sen K.K., Basu S.K. // J. Pharma Sci. Tech. 2011. V. 1. № 1. P. 16.
Mudgil D., Barak S., Khatkar B.S. // J. Food Sci. Tech. 2014. V. 51. № 3. P. 409.
Chang C., Zhang L. // Carbohydr. Polym. 2011. V. 84. № 1. P. 40.
Simkovic I. // Carbohydr. Polym. 2013. V. 95. № 2. P. 697.
Guilherme M.R., Auada F.A., Fadjardo A.R., Martins A.F., Paulino A.T., Davi M.F.T., Rubira A.F., Muniz E.C. // Eur. Polym. J. 2015. V. 72. № 1. P. 365.
Гринберг Т.А., Смоляр С.И., Малашенко Ю.Р., Капиловская Е.Д. // Микробиол. журн. 1991. Т. 53. № 5. С. 82.
Iurciuc C., Savin A., Lungu C., Martin P., Popa M. // Cellulose Chem. Tech. 2016. V. 50. № 1. P. 1.
Abbaszadeh A., Lad M., Janin M., Morris G.A. // Food Hydrocolloids. 2015. V. 44. № 1. P. 162.
Guenet M.J. Polymer-Solvent Molecular Compounds. Oxford: Elsevier Ltd., 2008.
Chandrasekaran R., Millane R.P., Arnott S., Atkins E.D.T. // Carbohydr. Res. 1988. V. 175. № 1. P. 15.
Norton I.T., Goodall S.A., Frangou S.A., Morris E.R., Rees D.A. // J. Mol. Biol. 1984. V. 175. № 3. P. 371.
Ma X., Pawlik M. // Carbohydr. Polym. 2007. V. 70. № 1. P. 15.
Moreira L.R.S., Filho E.X.F. // Appl. Microbiol. Biotechnol. 2008. V. 79. № 1. P. 165.
Morris E.R., Katsuyoshi N., Rinaudo M. // Food Hydrocoll. 2012. V. 28. № 1. P. 373.
Tako M. // Adv. Biosci. Biotechnol. 2015. V. 6. № 1. P. 22.
Carbohydrates. Structures, Synthesis and Dynamics / Ed. by P. Finch. Dordrecht: Springer, 1999.
Тагер А.А. Физикохимия полимеров. М.: Научный мир, 2007.
Picout D.R., Ross-Murphy S.B. // Carbohydr. Polym. 2007. V. 70. № 2. P. 145.
Brunchi C.E., Morariu S., Bercea M. // Coll. Surf. B. 2014. V. 122. № 10. P. 512.
Rochas C., Lahaye M. // Carbohydr. Polym. 1989. V. 10. № 4. P. 289.
Suvorova A.I., Safronov A.P., Melnikova O.A. // Polymer Science A. 2002. V. 44. № 1. P. 84.
Vshivkov S.A., Adamova L.V., Liriva B.I. // Polymer Science A. 2012. V. 54. № 11. P. 821.
Safronov A.P., Adamova L.V. // Polymer. 2002. V. 43. № 9. P. 2653.
Safronov A.P., Adamova L.V. // Polymer Science A. 2002. V. 44. № 4. P. 408.
Adamova L.V., Safronov A.P., Terziyan T.V., Shabad-rov P.A., Klyukina A.V. // Polymer Science A. 2018. V. 60. № 2. P. 190.
Сафронов А.П., Тагер А.А., Клюжин Е.С., Адамова Л.В. // Высокомолек. соед. А. 1993. Т. 35. № 6. С. 700.
Safronov A.P., Terziyan T.V. // Polymer Science A. 2008. V. 50. № 7. P. 733.
Сафронов А.П., Тагер А.А. // Высокомолек. соед. А. 1991. Т. 33. № 10. С. 2198.
Jenckel E., Gorke K.Z. // Electrochem. 1956. V. 60. P. 579.
Lama R.F., Lu B.C.-Y. // J. Chem. Eng. Data. 1965. V. 10. № 3. P. 216.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия А)