

Высокомолекулярные соединения (серия А), 2019, T. 61, № 2, стр. 163-171
НАНОПОРИСТЫЕ ПОЛИМЕРНЫЕ СЕТКИ N-ВИНИЛПИРРОЛИДОНА С ДИМЕТАКРИЛАТОМ ТРИЭТИЛЕНГЛИКОЛЯ И СОРБЦИЯ МАКРОМОЛЕКУЛ
С. В. Курмаз a, *, Н. В. Фадеева a, А. А. Грищук a, Е. И. Кнерельман a, Г. И. Давыдова a
a Институт проблем химической физики Российской академии наук
142432 Московская обл., Черноголовка, пр. ак. Семенова, 1, Россия
* E-mail: skurmaz@icp.ac.ru
Поступила в редакцию 03.10.2018
После доработки 19.10.2018
Принята к публикации 02.11.2018
Аннотация
Методом трехмерной радикальной сополимеризации N-винилпирролидона c диметакрилатом триэтиленгликоля в присутствии разветвленного сополимера как макромолекулярного темплата с функцией порогена получены нанопористые полимерные сетки различного состава. Изучены процессы адсорбции поливинилпирролидона, полиметилметакрилата и полистирола из их смесей в хлороформе нанопористыми полимерными сетками N-винилпирролидона и их десорбции. С помощью гель-проникающей хроматографии и ИК-спектроскопии исследовано молекулярно-массовое распределение и состав смесей ПВП–ПММА и ПВП–ПС до и после адсорбции, а также макрообъектов после десорбции. Показана высокая избирательность нанопористой полимерной сетки адсорбировать поливинилпирролидон из его смеси с неполярным полистиролом.
ВВЕДЕНИЕ
Полимеры и сополимеры N-винилпирролидона (ВП) вследствие высокой биосовместимости, гидрофильности, способности к комплексообразованию и сорбционной активности успешно используют в качестве носителей и модификаторов лекарственных препаратов [1–4]. Для этих целей также перспективными могут быть полимерные сетки ВП с развитой удельной поверхностью и пористой структурой. На примере рибофлавина нами показана возможность их применения в качестве носителей, обеспечивающих пролонгированное выделение витамина [5]. Эффективный способ формирования мезопористых полимерных сеток ВП без применения “плохих” растворителей как порогенов и модификации специально вводимых полимерных наноматериалов в качестве заготовок пор предложен в работах [6–9], где для организации пор в трехмерной матрице были использованы растворимые в смеси мономеров сополимеры ВП разветвленной топологической структуры.
Протокол приготовления полимерных сеток ВП (полимеризация, приводящая к сетчатым полимерным матрицам, содержащим макромолекулярные порогены, их удаление с помощью растворителя, наличие пор) формально соответствовал требованиям трехмерного макромолекулярного импринтинга и способствовал созданию полимеров, которые могли повторно распознавать импринт-молекулы или молекулы близкого к ним строения, формы и размера [10–13]. Более того, на стадии приготовления мономер-полимерной смеси потенциальный пороген, находясь в гидратном состоянии, связывается с полярным мономером ВП с образованием комплекса ВП–(Н2О)х–разветвленный сополимер [9]. Как известно [10–13], формирование комплексов между функциональным мономером и импринт-молекулой является ключевым условием при создании молекулярно-импринтированных полимеров. С учетом этих фактов логично было исследовать способность пористых полимерных сеток ВП адсорбировать макромолекулы, близкие по размеру и химической природе к использованному при их синтезе макромолекулярному порогену.
Цель настоящей работы – изучить способности нанопористых полимерных сеток N-винилпирролидона с диметакрилатом триэтиленгликоля различного состава адсорбировать макрообъекты наноразмерного масштаба – ПВП, ПММА и ПС из их смесей в хлороформе; ПВП выступал в качестве модели исходного темплата-порогена, т.е. разветвленного сополимера.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Мономер N-винилпирролидон (“Alfa Aesar”) очищали перегонкой в вакууме от ингибитора NaOH. Сомономер – диметакрилат триэтиленгликоля (ДМТЭГ) производства “Aldrich” и передатчик цепи – 1-декантиол (ДТ) производства “Alfa Aesar” использовали без дополнительной очистки.
Разветвленный сополимер ВП с диметакрилатом триэтиленгликоля получали трехмерной радикальной сополимеризацией при мольном соотношении реагентов [ВП] : [ДМТЭГ] : [ДТ] = 100 : : 12 : 12 в толуоле (75 об. %). Высокомолекулярную фракцию после сополимеризации выделяли из смеси осаждением в 10-кратный избыток гексана. Сополимер сушили до постоянной массы в вакууме от толуола и гексана.
Молекулярные массы разветвленного сополимера определяли методом ГПХ на приборе “Waters GPCV 2000” (2 колонки PL-gel, 5 мкм, MIXED-C, 300 × 7.5 мм) по методике [6]. Для анализа использовали два детектора – рефрактометрический и светорассеяния “WYATT DAWN HELEOS II”, λ = 658 нм, программное обеспечение “Empower Pro” и “Astra” (версия 5.3.2.20). Динамическую вязкость сополимера в N-винилпирролидоне и смеси ВП–ДМТЭГ измеряли на вискозиметре Штабингера SVM-3000 при 25°С.
Разветвленный сополимер (20 мас. %) растворяли в смеси мономеров ВП–ДМТЭГ состава 40 : 60 и 80 : 20 мас. %. Полученные мономер-полимерные смеси помещали в стеклянные ампулы (диаметр 3 мм), вакуумировали и запаивали. Трехмерную радикальную полимеризацию в массе, инициированную ДАК (0.2 мас. %), в присутствии разветвленного сополимера осуществляли при 60°С и получали полимерные композиты различного мономерного состава.
Проводили золь-гель-анализ полученных полимерных композитов с предельной конверсией связей С=С в аппарате Сокслетта в течение 5 ч при температуре кипения растворителя – изопропанола. После экстракции золей (растворимых продуктов) получали густосшитые полимерные сетки 1 и 2, которые сушили до постоянной массы в вакууме. Они представляли собой блочные образцы с диаметром 2.5 и высотой 5–6 мм.
Молекулярные массы линейных ПММА, ПВП и ПС определяли методом ГПХ, используя детектор-рефрактометр. Методом динамического рассеяния света исследовали 1%-ные растворы полимеров и их смесей в хлороформе на угле детектирования 90° с использованием установки “Photocor Compact” (“Photocor Instruments Inc.”, США), оснащенной диодным лазером, работающим на λ = 654 нм. Предварительно растворы полимеров фильтровали, используя фильтр с диаметром пор 0.45 мкм. Перед измерением виалы с раствором термостатировали в течение ~20 мин. Автокорреляционные функции рассеянного света, определенные в режиме динамического светорассеяния, обрабатывали с помощью программы “DynаLS”, что позволяло анализировать функции распределения частиц, находящихся в растворе, по временам релаксации tc и коэффициентам диффузии D. Используя уравнение Эйнштейна–Стокса D = kT/6πηR (k – константа Больцмана; T – абсолютная температура; η – вязкость среды, в которой взвешены дисперсные частицы), рассчитывали величину гидродинамического радиуса Rh полимеров. Строили зависимости распределения интенсивности рассеяния света по гидродинамическим радиусам Rh.
Пористость полимерной сетки 1 определяли методом низкотемпературной адсорбции азота на приборе “Autosorb-1” (“Quantachome”, США). Удельную поверхность полимерных сеток 1 и 2 оценивали также из данных по адсорбции гидрофильного красителя – бенгальского розового из водного раствора [5].
Для исследования процессов адсорбции макромолекулярных объектов пористыми сетками 1 и 2 применяли линейные ПВП и ПММА, полученные радикальной полимеризацией в режиме передачи цепи, и ПС. Хлороформ выбрали в качестве общего растворителя, не склонного к межмолекулярным взаимодействиям с полимерными объектами. Готовили 10%-ные растворы ПВП и ПММА в хлороформе, а также 5%-ные растворы линейных ПВП и ПС в хлороформе. Смешивали растворы ПВП с ПММА и ПВП с ПС в соотношении 50 : 50 об. %. Полимерные сетки 1 и 2 помещали в бюкс, содержащий смесь полимеров ПВП–ПММА и ПВП–ПС в хлороформе, и выдерживали при 25°С. Рассчитывали значение адсорбции Sадc по формуле
где m – значение массы образца после адсорбции, m0 – исходная масса образца. Затем образцы, высушенные при комнатной температуре, помещали в хлороформ и изучали процесс десорбции. Значение относительной десорбции Sдес рассчитывали по формулеЗдесь mадс – значение массы образца после адсорбции, mдес – масса образца после десорбции.
Молекулярно-массовое распределение и состав смесей ПВП–ПММА и ПВП–ПС до и после сорбции, а также макрообъектов после десорбции исследовали с помощью ГПХ (детектор-рефрактометр) и ИК-спектроскопии. Записывали ИК-спектры пленок на стеклах KBr, высушенных до полного удаления растворителя.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Характеристики макромолекулярного порогена и пористых полимерных сеток
Макромолекулярный пороген – разветвленный сополимер получен с помощью радикальной сополимеризации в толуоле в условиях передачи цепи на 1-декантиол [7]. По данным элементного анализа, содержание CHNS составляло 59.9, 9.1, 8.2, 1.7%. Из этих данных был рассчитан состав сополимера: содержание звеньев ВП, ДМТЭГ и ДТ составляло 80.5, 12.4 и 7.1 мол. %. Его относительная и абсолютная средневесовая Mw и среднечисловая Mn молекулярная масса составляла (18 и 20) × 103 и (3.9 и 11.1) × 103 соответственно. Фактор Зимма g', рассчитанный как отношение среднеквадратичного радиуса инерции макромолекул с Mw ~ 104 и линейного ПВП, был равен 0.6–0.7 и свидетельствовал об их разветвленности [8].
Разветвленный сополимер ВП, несмотря на включение в его состав звеньев ДМТЭГ и неполярных остатков ДТ, сохранял высокую гидрофильность и аналогично ПВП [14] содержал сорбированную воду, которая образовывала с С=О-группами ВП-звеньев комплексы (Н2О)х–разветвленный сополимер различной прочности [9]. С помощью ИК-спектроскопии показано, что при растворении гидратированного разветвленного сополимера в смеси мономеров появлялся комплекс ВП–(Н2О)х–сополимер [9], в котором С=О-группы ВП также связывались водородной связью с сополимером.
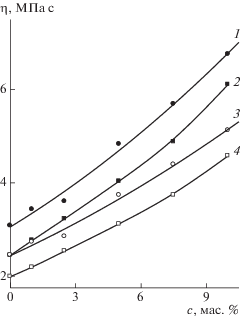
Другой особенностью амфифильного разветвленного сополимера ВП является его существование в полярных средах в виде мономолекулярных мицелл и их агрегатов [8, 9]. Так, например, в этаноле в рассеяние света основной вклад вносят отдельные макромолекулы с Rh ~ 4 нм, при этом наблюдаются крупные частицы с Rh ~ 102 и более. В изопропиловом спирте пики агрегатов сдвигаются в область меньших значений Rh. В смеси ВП–ДМТЭГ состава 80 : 20 мас. % разветвленный сополимер (1 мас. %) существует в виде отдельных макромолекул и не агрегирует [9]. Однако в концентрированных растворах, как следует из зависимостей динамической вязкости от концентрации сополимера в ВП и в смеси ВП–ДМТЭГ (рис. 1), разветвленный сополимер, по-видимому, образует агрегаты. На зависимости динамическая вязкость–концентрация наблюдается отклонение от линейности при концентрации сополимера в ВП свыше 6 мас. %. В смеси мономеров оно имеет место при более низкой концентрации разветвленного сополимера (~2.5 мас. %). Снижение полярности среды в смеси ВП–ДМТЭГ приводит к усилению процессов агрегации амфифильных макромолекул. При содержании разветвленного сополимера 20% в реакционных смесях ВП–ДМТЭГ его макромолекулы присутствуют, скорее всего, в форме агрегатов, которые наряду с отдельными макромолекулами представляют собой заготовки пор.
Рис. 1.
Зависимость динамической вязкости смеси ВП–ДМТЭГ (80 : 20 мас. %)–разветвленный сополимер (1, 3) и смеси ВП–разветвленный сополимер (2, 4) от концентрации разветвленного сополимера при T = 20 (1, 2) и 30°С (3, 4).
Составы мономер-полимерных смесей, из которых сначала формировали полимерные композиты 1 и 2, приведены в табл. 1. После сополимеризации получали монолитные (блочные) образцы с предельной конверсией связей С=С, которые заметно опалесцировали и характеризовались двумя значениями температуры стеклования [8, 9]. Так, по данным ДСК, у полимерного композита 1 наблюдаются α-переходы, обусловленные размораживанием молекулярной подвижности сегментов макромолекулярного порогена и полимерной матрицы при 58 и 125°С соответственно. В ходе трехмерной радикальной сополимеризации, очевидно, имело место микрофазовое разделение, в результате которого макромолекулярные добавки выделялись в отдельные области различного размера, отличающиеся от полимерной матрицы показателем преломления и температурой стеклования.
Таблица 1.
Составы мономер-полимерных смесей и конверсия двойных связей в полимерных композитах ВП–ДМТЭГ–разветвленный сополимер
| Полимерный композит | Состав смеси ВП–ДМТЭГ, мас. % | [Разветвленный сополимер], мас. % | Конверсия, % |
|---|---|---|---|
| 1 | 40 : 60 | 20 | 77.0 |
| 2 | 80 : 20 | 20 | 68.0 |
Из полимерных композитов экстрагировали с помощью изопропилового спирта растворимые полимерные продукты, среди которых идентифицировали фракции разветвленного сополимера с высоким содержанием ВП-звеньев и олигомерный ВП [8, 9]. В результате получали монолитные полимерные сетки с иными физико-химическими характеристиками, чем полимерные композиты. Так, полимерная сетка 1 характеризовалась температурой стеклования в высокотемпературной области [8]. Ее модуль упругости E практически не изменялся по сравнению с полимерным композитом и составлял 1.1 × 103 МПа [8]. Полимерные сетки 1 и 2, образованные мономерами с различной реакционной способностью, представляли собой густосшитые химические сетки с концевыми цепями, состоящими из ВП-звеньев. В них равномерно распределена дисперсная фаза, образованная макромолекулами темплата-порогена, которая экстрагируется изопропиловым спиртом.
Специальные исследования показали, что кипящий хлороформ для экстракции порогена и получения полимерных сеток с бóльшей пористостью менее эффективен, чем изопропиловый спирт. В мягких условиях (при комнатной температуре) оба растворителя не обеспечивают достаточное для вымывания порогена объемное набухание и, как следствие, полимерные сетки обладают примерно одинаковой пористостью, которая существенно ниже, чем у сеток, экстрагированных кипящими растворителями. Высокая степень сшивания полимерной сетки обеспечивает жесткость, механическую стабильность и слабое набухание в растворителях различной полярности [8, 9].
Морфология поверхности сеток, по данным CЭМ (см. рис. 1 в электронном виде; Дополнительный материал), является пористой; они имеют открытые поры и каналы, пронизывающие полимерное тело. Методом низкотемпературной адсорбции азота в полимерной сетке 1 определены значения удельной площади поверхности Sуд – 26.2 м2/г и объема пор Vпор – 0.21 см3/г [8]. Изотермы адсорбции-десорбции азота для полимерных сеток 1 и 2 показаны на рис. 2 в электронном виде (Дополнительный материал). Следует отметить, что сополимер, полученный в отсутствие макромолекулярного порогена, имел значение Sуд менее 1 м2/г и был непористым. Форма кривой распределения пор по размерам указывала на наличие в полимерной сетке 1 пор различного размера, причем большинство из них относится к мезопорам с диаметром от 2 до 50 нм [8]. Кроме них, имелись микропоры, мелкие мезопоры или субмикропоры. Метод низкотемпературной адсорбции азота оказался неприемлем для оценки удельной поверхности и пористости полимерной сетки 2, так как неполярные молекулы азота слабо взаимодействовали с полярной поверхностью полимерной сетки данного состава [5]. Как следствие, получали сильно заниженные значения Sуд и Vпор, противоречащие данным СЭМ о развитой пористой структуре полимерной сетки 2. Результаты БЭТ для полимерной сетки 1 хорошо коррелировали с экспериментальной Sуд, определенной из данных по адсорбции бенгальского розового из водных растворов и значения эффективного радиуса его молекулы [15]. В связи с этим, с помощью гидрофильного красителя также была оценена удельная площадь поверхности пористого сополимера по мономерному составу аналогичного 2, которая составила ~33 м2/г [5]. Таким образом, полимерные сетки 1 и 2 обладали заметной поверхностью в отличие от традиционных сополимеров, полученных в отсутствие разветвленного сополимера как порогена.
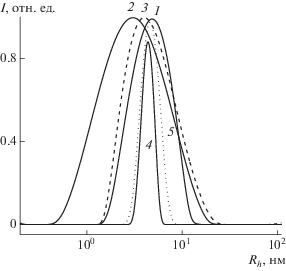
Исследование процессов адсорбции макромолекул пористыми полимерными сетками
Характеристики макромолекулярных объектов – линейных ПВП, ПММА и ПС, использованных для изучения сорбционных свойств пористых полимерных сеток 1 и 2, приведены в табл. 2. Видно, что ПВП и ПММА отличаются молекулярной массой, и значение Mw их смеси закономерно снижается. Методом динамического рассеяния света исследованы растворы отдельных полимеров и их смесей в хлороформе и были получены кривые распределения интенсивности I рассеяния света по размерам – гидродинамическим радиусам Rh для отдельных полимеров и их смеси (рис. 2). Кривые I(Rh) являются унимодальными и различаются значениями Rh в максимуме пиков (табл. 2). В процессе адсорбции большие макромолекулы не проникают в поры, только лишь макромолекулы с диаметром менее 50 нм вместе с растворителем диффундируют в мезопоры и могут быть адсорбированы полимерной сеткой, а в процессе десорбции растворитель будет вымывать их из пор.
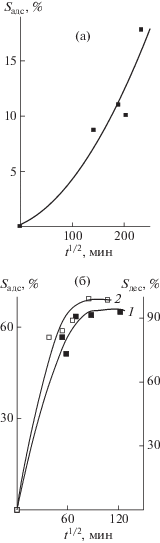
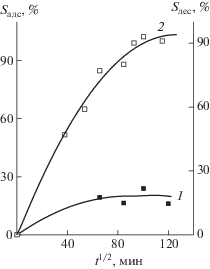
На рис. 3 приведены зависимости адсорбции макрообъектов полимерными сетками 1 и 2 из смеси ПВП–ПММА в хлороформе в координатах уравнения Фика. Из сравнения кривых 1 на рис. 3а и 3б следует, что скорость адсорбции выше в полимерной сетке 2, по-видимому, вследствие бóльшей ее пористости. За время наблюдения значение Sадс сополимером 1 составило ~18%, тогда как второй образец достигал насыщения и предельного значения Sадс ~ 65%.
Рис. 3.
Зависимости адсорбции макрообъектов из смеси ПВП–ПММА в хлороформе полимерной сеткой 1 (а), а также адсорбции (1) и десорбции (2) полимерной сеткой 2 (б) при 25°С.
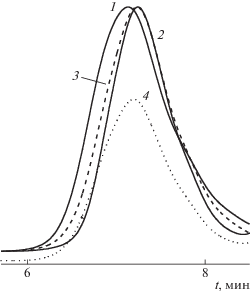
Анализировали данные ГПХ линейных ПВП, ПММА, их смеси ПВП–ПММА до и после адсорбции полимерной сеткой 1. На хроматограммах ПВП и ПММА, нормированных для сравнения на максимальную высоту пика (рис. 4), присутствуют унимодальные пики; времена их элюирования являются близкими и, как следствие, максимумы пиков плохо разделены. Хроматограмма смеси лежит в области меньших молекулярных масс относительно ПВП. После адсорбции интенсивность этого пика уменьшается, и он становится более узким. Как следствие, значение Mw смеси снижается (табл. 2). Таким образом, ММР смеси ПВП–ПММА изменяется, очевидно, в результате адсорбции макромолекул из раствора. Однако данные ГПХ не дают ответа об избирательности адсорбции из смеси ПВП–ПММА этой полимерной сеткой.
Рис. 4.
Хроматограмма ПВП (1), ПММА (2), смеси ПВП–ПММА до (3) и после сорбции (4) полимерной сеткой 1.
На рис. 5 приведены ИК-спектры исходных полимерных смесей, после адсорбции макрообъектов полимерными сетками 1 и 2 и их десорбции в области 1800–1600 см–1, в которой регистрируются характеристичные полосы валентных колебаний связей С=О в метакрильных и ВП-звеньях при ν ~ 1730 и 1660 см–1 соответственно. Отношение их оптических плотностей D может служить характеристикой изменения состава смеси ПВП–ПММА в ходе проведенных исследований. Судя по спектрам (рис. 5а), после десорбции из полимерной сетки 1 в хлороформе присутствуют оба полимера. Из приведенных данных в табл. 3 следует, что характерное для исходной смеси значение DПВП/DПММА после адсорбции снижается в ~1.4 раза. Из разности значений DПВП/DПММА смеси полимеров до и после адсорбции полимерной сеткой 1 следует, что значение DПВП/DПММА смеси ПВП–ПММА после десорбции должно составлять ~0.5. Экспериментальное значение DПВП/DПММА удовлетворительно согласуется с расчетным. Различия могут быть связаны не только с десорбцией из полимерной сетки макромолекул, но и с экстрагированием остатков порогена из высокоплотных областей густосшитой сетки. В пользу этого свидетельствует и относительное значение предельной десорбции ~106%. Таким образом, из полимерной сетки 1 высвобождаются и ПВП, и ПММА, а следовательно, она не обладает избирательностью, по-видимому, из-за сродства к обоим полимерам. Можно предположить, что адсорбируются не только отдельные макромолекулы, но и их агрегаты смешанного типа, представляющие собой физически связанные полимерные цепи, в которых имеются центры сильных межмолекулярных взаимодействий – сложноэфирные и амидные группы. Результаты десорбции свидетельствуют о более слабом взаимодействии макрообъектов с функциональными группами полимерной сетки 1 по сравнению с низкомолекулярными объектами, в частности рибофлавином [5], для которого относительное значение предельной десорбции составляет ~20%.
Рис. 5.
ИК-спектры исходной смеси ПВП–ПММА (1), после адсорбции полимеров (2) и их десорбции (3) полимерными сетками 1 (а) и 2 (б); исходной смеси ПВП–ПС (1), после адсорбции полимеров (2) и их десорбции (3) полимерной сеткой 1 (в).
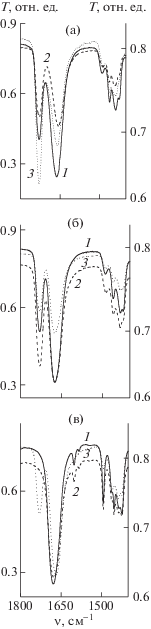
Таблица 3.
Отношение оптической плотности D характерных полос поглощения ПВП, ПММА и ПС в исходных смесях полимеров, полимеров после адсорбции полимерными сетками 1 и 2 и их десорбции
| Поли- мерные сетки | DПВП/DПММА (DПВП/DПС) | |||||
|---|---|---|---|---|---|---|
| исходная смесь ПВП–ПММА | ПВП–ПММА после адсорбции | ПВП–ПММА после десорбции | исходная смесь ПВП–ПС | ПВП–ПС после адсорбции | ПВП–ПС после десорбции | |
| 1 | 1.7 | 1.2 | 0.7 | 10.8 | 8.2 | 18.2 |
| 2 | 2.0 | 1.2 | 1.5 | – | – | – |
Были проанализированы ИК-спектры (рис. 5б) смеси ПВП–ПММА до и после адсорбции–десорбции полимерной сеткой 2, которые затем сравнивались с соответствующими значениями DПВП/DПММА (табл. 3). После адсорбции значение DПВП/DПММА падает в ~1.7 раза, очевидно, вследствие адсорбции макрообъектов из хлороформа. После десорбции из полимерной сетки 2 в ИК-спектре (рис. 5б, кривая 3) присутствуют полосы поглощения обоих полимеров – ПВП и ПММА. Таким образом, полимерная сетка данного состава также не обладает избирательностью относительно ПВП. Обращает на себя внимание значительное увеличение отношения DПВП/DПММА после десорбции. Оно оказалось в ~2 раза больше ожидаемого, исходя из разности DПВП/DПММА в смеси полимеров до и после адсорбции. В составе смеси полимеров после десорбции, по-видимому, имеется макромолекулярный пороген, который в силу диффузионных ограничений остается в полимерной сетке и медленно вымывается. Действительно, по данным гравиметрии, после десорбции имеет место незначительная потеря массы полимерным матриксом. Таким образом, пористые полимерные сетки 1 и 2 способны адсорбировать макрообъекты близкой химической природы из растворов, но не обладают избирательностью.
ПВП и ПС сильно различались по полярности и термодинамическому сродству относительно полимерной сетки 1. При этом неполярный полимер характеризовался более высокой молекулярной массой. Распределение интенсивности рассеяния света по размерам частиц в хлороформе является узким (рис. 2); значение Rh в максимуме пика составляло ~4 нм. В смеси ПВП–ПС кривая I(Rh) уширялась, и значение в максимуме пика смещалось в сторону более высоких значений Rh. Таким образом, мезопористая полимерная сетка может адсорбировать из смеси полимеров лишь макромолекулы с диаметром ~3–16 нм. Большие макромолекулы не проникают в поры данного размера и, очевидно, вымываются с поверхности полимерной сетки растворителем.
Кривые адсорбции макромолекул из смеси ПВП–ПС и их десорбции в координатах уравнения Фика приведены на рис. 6. Видно, что их адсорбция протекает с более высокой скоростью, чем из смеси ПВП–ПММА (рис. 3) и достигает предельного значения Sадс ~ 20%. Причиной этого, скорее всего, является высокое термодинамическое сродство ПВП к полимерной сетке 2. Процесс десорбции также протекает с высокой скоростью, и полимер высвобождается из полимерной сетки.
Рис. 6.
Зависимость адсорбции (1) макрообъектов из смеси ПВП–ПС в хлороформе полимерной сеткой 1 и их десорбции (2).
О составе смеси полимеров ПВП–ПС после адсорбции–десорбции судили на основе данных ИК-спектроскопии. В спектре исходной смеси ПВП–ПС в области 1800–1400 см–1 наблюдаются характеристические полосы, соответствующие валентным колебаниям групп С=О в лактамном цикле ПВП при ν ~ 1680 см–1, а также колебаниям ароматического кольца при частоте ~1600, 1580, 1500 и 1450 см–1 [16]. Из ИК-спектров полимеров в исходной смеси и после адсорбции–десорбции было определено отношение оптических плотностей полосы поглощения связи С=О в ПВП при 1680 см–1 и полосы поглощения ПС при 1600 см–1DПВП/DПС (табл. 3). Судя по ИК-спектрам и уменьшению значения DПВП/DПС, макрообъекты адсорбируются полимерной сеткой; данные по величине предельной адсорбции и ИК-спектроскопии о количестве адсорбированных полимеров из смеси (~24%) хорошо коррелируют. Из ИК-спектра (рис. 5в, кривая 2) видно, что после адсорбции в смеси полимеров остается ПС. Однако в ИК-спектре (рис. 5в, кривая 3) полимеров, экстрагированных из полимерной сетки 1, полоса поглощения скелетных колебаний бензольного кольца при ν ~ 1600 см–1 практически отсутствует, что позволяет сделать вывод о малом содержании ПС в анализируемом продукте. Кроме того, в ИК-спектре (рис. 5в, кривая 3) появляется новая полоса поглощения при ν ~ 1730 см–1, соответствующая валентным колебаниям связи С=О в метакрилатной группе, а частота связи С=О в лактамном цикле ВП-звеньев понижается до 1665 см–1. Очевидно, что в процессе исследования из полимерной сетки 1 продолжает экстрагироваться пороген с высоким содержанием диметакрилата. В пользу этого свидетельствуют данные гравиметрии о потере веса полимерной сетки на ~7%. В процессе адсорбции и десорбции в полимерной сетке 1, по-видимому, возникают новые каналы и поры открытого типа, размеры которых позволяют высвобождаться макромолекулам. Таким образом, полимерная сетка 1 демонстрирует избирательность при адсорбции из хлороформа двух сильно различающихся по полярности макрообъектов: преимущественно адсорбируется ПВП.
В результате проведенных исследований в работе показано, что нанопористые полимерные сетки ВП с диметакрилатом триэтиленгликоля способны адсорбировать макрообъекты нанометрового размера из хлороформа, их сорбционная способность определяется мономерным составом, размерами пор и удельной площадью поверхности, доступной для взаимодействия с адсорбатом. При адсорбции двух сильно различающихся по полярности макрообъектов из хлороформа полимерная сетка 1 демонстрирует избирательность к полимеру, близкому по природе и размеру к макромолекуле темплата-порогена. Однако вопрос о характере связывания полимерных сеток ВП–ДМТЭГ и ПВП остается открытым и требует дополнительного исследования. С учетом полученных экспериментальных данных по избирательной адсорбции ПВП и ранее установленного факта – образования Н-связи между макромолекулярным порогеном и мономером ВП можно сделать предположение о перспективности полимерных сеток ВП–ДМТЭГ в качестве молекулярно-импринтированных полимеров для природных полимеров с амидными группами.
Работа выполнена по теме Госзадания (гос. регистрация № 01201055328) с использованием оборудования Центра коллективного пользования “Новые нефтехимические процессы, полимерные композиты и адгезивы” (№ 77601).
Список литературы
Panarin E.F. // Russ. Chem. Bull. 2015. V. 64. № 1. P. 15.
Panarin E.F. // Russ. Chem. Bull. 2017. V. 66. № 10. P. 1812.
Solovskii M.V., Borisenko M.S., Ershov A.Yu., Zakharova N.V., Tarabukina E.B. // Russ. J. Gen. Chem. V. 87. № 2. P. 276.
Borovikova L.N., Titova A.V., Kipper A.I., Pisarev O.A. // Russ. J. Gen. Chem. V. 87. № 5. P. 1031.
Kurmaz S.V., Fadeeva N.V., Knerelman E.I., Davydova G.I. // Russ. J. Appl. Chem. 2017. V. 91. № 1. P. 105.
Kurmaz S.V., Grubenko G.A., Knerelman E.I., Davydova G.I., Torbov V.I., Dremova N.N. // Mendeleev Commun. 2014. V. 24. № 2. P. 125.
Fadeeva N.V., Kurmaz S.V., Knerelman E.I., Davydova G.I., Torbov V.I., Dremova N.N. // Russ. Chem. Bull. 2016. V. 65. № 8. P. 2089.
Fadeeva N.V., Kurmaz S.V., Knerelman E.I., Davydova G.I., Torbov V.I., Dremova N.N. // Polymer Science B. 2017. V. 59. № 3. P. 257.
Fadeeva N.V., Kurmaz S.V., Knerelman E.I., Davydova G.I. // Polymer Science B. 2018. V. 60. № 2. P. 195.
Lisichkin G.V., Krutyakov Yu.A. // Russ. Chem. Rev. 2006. V. 75. № 10. P. 901.
Гендриксон О.Д., Жердев А.В., Дзантиев Б.Б. // Усп. биол. хим. 2006. Т. 46. С. 149.
Dmitrienko E.V., Pyshnaya I.A., Martyanov O.N., Pyshnyi D.V. // Russ. Chem. Rev. 2016. V. 85. № 5. P. 513.
Li S., Cao S., Whitcombe M., Piletsky S. // Prog. Polym. Sci. 2014. V. 39. № 1. P. 145.
Lebedeva T.L., Feldstein M.M., Kuptsov S.A., Plate N.A. // Polymer Science A. 2000. V. 42. № 9. P. 989.
Vlasova I.M., Polyansky D.V., Vlasov A.A., Saletsky A.M. // Moscow Univ. Phys. Bull. 2013. V. 68. № 3. P. 231.
Дехант И., Данц Р., Киммер В., Шмольке Р. Инфракрасная спектроскопия полимеров. М.: Химия, 1976.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия А)