

Высокомолекулярные соединения (серия А), 2019, T. 61, № 3, стр. 272-282
Определение супрамолекулярной структуры фторсополимера методом обращенной газовой хроматографии
Ф. А. Шумилов a, *, Е. Е. Щадилова a, b, А. П. Возняковский a
a Институт синтетического каучука им. С.В. Лебедева
198035 Санкт-Петербург, ул. Гапсальская, 1, Россия
b Санкт-Петербургский государственный технологический институт (Технологический Университет)
190013 Санкт-Петербург, Московский пр., 26, Россия
* E-mail: Itachi16@mail.ru
Поступила в редакцию 17.10.2018
После доработки 25.12.2018
Принята к публикации 09.01.2019
Аннотация
Молекулярная организация сополимеров этилена с перфторированными эфирами изучена методом обращенной газовой хроматографии. Рассчитаны термодинамические параметры сорбции низкомолекулярных сорбатов сополимерами (коэффициент активности, парциальные молярные величины смешения) в широком диапазоне температур. Исследовано влияние архитектуры макроцепей сополимеров на сорбционные параметры их тонких пленок. Показано, что, несмотря на чередующийся характер распределения мономеров по макроцепи сополимера, метиленовые группы формируют углеводородные кластеры в перфторированной дисперсионной среде. Микрогетерогенная структура исследованных сополимеров обусловливает проницаемость тонких пленок сополимеров низкомолекулярными жидкими сорбатами (как растворителями, так и нерастворителями). Высказано предположение, что сополимеры такого типа перспективны для использования в области мембранных технологий.
ВВЕДЕНИЕ
Обладая высокой стойкостью к агрессивным средам вплоть до высоких температур, пленкообразующие фторэластомеры являются естественными кандидатами для использования в качестве основы протекторных покрытий. Следует, однако, отметить, что ультратонкие пленки фторполимеров также применяются и в качестве селективно проницаемых мембран. Конкретная область применения может быть достаточно обосновано спрогнозирована на основе систематического анализа корреляционной связи структуры звена полимера с сорбционными характеристиками последнего [1–7].
Отметим, что по реологическим параметрам фторполимеры могут быть разделены на два класса – фторопласты и фторэластомеры.
Фторопласты по своим реологическим характеристикам являются термопластами. Они характеризуются наименьшей диффузной проницаемостью, что и обусловливает многочисленные попытки их использования в качестве протекторных покрытий.
Фторэластомеры в отличие от фторопластов содержат в макроцепях структурные элементы, обеспечивающие им эластические свойства. По архитектуре макроцепей фторэластомеры в основном представляют собой сополимеры винилиденфторида с перфторированными алкенами. По химической стойкости сшитые фторэластомеры практически не уступают фторопластам. С точки зрения протекторных покрытий преимущество фторэластомеров – возможность формирования пленок из растворов в летучих растворителях. При этом физико-механические свойства сшитых пленок фторэластомеров заметно выигрывают по сравнению со свойствами пленок фторопластов.
Однако фторэластомеры на основе винилиденфторида характеризуются низкой устойчивостью в полярных и нуклеофильных средах, что является существенным недостатком.
Такого недостатка лишены сополимеры этилена с перфторированными алкил-виниловыми эфирами (ЭПФЭ) [8]. Сочетание в линейной цепи сополимеров термодинамически несовместимых углеводородных и перфторированных последовательностей приводит к организации в них молекулярного порядка отличного от молекулярного порядка перфторированных аналогов, а также аналогов на основе винилиденфторида. Структура пленочных материалов в основном контролируется архитектурой полимерных макромолекул. Соответственно можно ожидать и появления нового комплекса физико-химических параметров у этих сополимеров.
Цель настоящей работы – изучение сорбционных свойств тонких пленок сополимеров этилена с перфторированными эфирами в связи с особенностями химической структуры макромолекул сополимеров.
При выборе метода исследования следует принимать во внимание вероятное значительное отличие свойств полимера в виде тонкой или ультратонкой пленки от свойств и структуры полимера в блочном состоянии. В связи с этим в качестве метода исследования был выбран динамический вариант сорбционного метода исследования полимеров – метод обращенной газовой хроматографии [9].
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы
Сополимеры этилена с перфторированными алкилвиниловыми эфирами получали из этилена и перфторированных эфиров путем эмульсионной полимеризации с использованием персульфата калия в качестве инициатора. Перфторированные простые эфиры были приобретены в “Sigma Aldrich Co. Ltd.” Более подробно метод синтеза описан в работе [10].
Полный список изученных фторэластомеров и их химических структур представлен в табл. 1. Большинство экспериментов проведено с образцами 5–9. Чтобы установить более глубокое понимание отношения структура–свойства также были проанализированы некоторые стандартные фторэластомеры (образцы 1–4).
Таблица 1.
Структурные звенья исследуемых сополимеров
| Образец, № | Химическая структура полимера | Название |
|---|---|---|
| 1 | 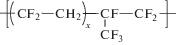 |
Сополимер винилиденфторида с гексафторпропиленом, где x ≈ 1.3 |
| 2 | 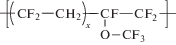 |
Сополимер винилиденфторида с перфтрометилвиниловым эфиром, где x ≈ 1.3 |
| 3 |  |
Сополимер винилиденфторида с перфтропропилвиниловым эфиром, где x ≈ 1.3 |
| 4 | 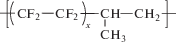 |
Сополимер тетрафторэтилена с пропиленом, где x ≈ 1.3 |
| 5 | 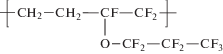 |
Сополимер этилена с перфторпропилвиниловым эфиром |
| 6 | 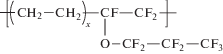 |
Сополимер этилена с перфторпропилвиниловым эфиром повышенной блочности, где 2 ≤ x ≤ 3 |
| 7 | 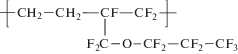 |
Сополимер этилена с перфторпропилаллиловым эфиром |
| 8 | 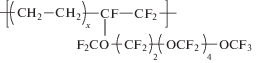 |
Сополимер этилена с перфтор- 4,7,9,11,13,15-(гексаоксогексадеценом-1), где 2 ≤ x ≤ 3 |
| 9 | 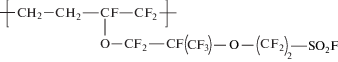 |
Сополимер этилена с перфтор-(5-метил-3,6-диоксооктен-7)-сульфофторидом |
Полимерами для сравнения служили натуральный каучук (NR-RSS-1, Malaysia), полидиметилсилоксан (OV-1) как довольно популярный материл для селективных мембран, и некоторые стандартные сополимеры винилиденфторида.
ЯМР
Спектры ядерного магнитного резонанса получали на аппарате “Bruker Spectrospin AM-50” (Германия) с рабочим диапазоном частот 270 и 254 МГц для ЯМР 1H и ЯМР 19F соответственно.
Спектральный анализ ЯМР синтезированных ЭПФЭ-5 и ЭПФЭ-6 показывает, что (со структурной точки зрения) они являются в основном альтернатными сополимерами углеводородных и перфторированных последовательностей. Однако около 15% этиленовых единиц образуют диады и менее 0.01% – триады.
В образцах ЭПФЭ-7 и ЭПФЭ-8 содержание диад составляло не менее 80%, триад – примерно 20%.
ТМА, ДСК
Температуру стеклования и теплоемкость образцов определяли методом дифференциальной сканирующей калориметрии (DSC 8000, “Perkin Elmer”) и методом термомеханического анализа (TMA/SВЕ 1 LN600).
Согласно термомеханическому анализу и данным ДСК, для диапазона температур –40…+150°С был зарегистрирован только один температурный переход, соответствующий Tg = = –27°C.
Хроматографическая установка
В работе использовали хроматографическую установку, конструктивные особенности которой дали возможность снизить погрешность определения параметров, вносящих наибольший вклад в погрешность измерения удерживаемого объема (объемной скорости потока газа-носителя, температуры потока газа-носителя, давления, температуры колонки, массы полимера в колонке), до значений, позволяющих проводить физикохимические измерения [9]. Установка была оснащена детектором по теплопроводности (газ-носитель гелий).
При приготовлении насадки хроматографической колонки навеску ЭПФЭ растворяли в гексафторбензоле. В раствор при постоянном перемешивании небольшими порциями добавляли расчетное количество твердого носителя (Chromaton-N-Super). Полученную суспензию помещали в эксикатор, который присоединяли к водоструйному насосу, и дегазировали суспензию при пониженном давлении 2–3 мин. Затем суспензию переносили в вытяжной шкаф и выпаривали при постоянном тщательном перемешивании. Окончательно полученную таким образом насадку досушивали до постоянной массы в вакууме при 60°C.
Для работы использовали спиральные стеклянные колонны (длина 1.5 м, диаметр спирали 65 мм, внутренний диаметр 4 мм). Приготовленную насадку помещали в продуваемую сухим аргоном колбу, снабженную двухходовым краном. К колбе подсоединяли пустую колонку и передавали в нее током сухого аргона часть насадки. Количество последней в колонке определяли по весу колбы до и после заполнения колонки.
Содержание образцов 5–9 в колонке составляло 8.9, 5.7, 4.5, 8.9 и 7.0% соответственно.
Сорбатами служили н-алканы (C6…C10), бензол, циклогексан и гексафторбензол. Все используемые сорбаты по степени подготовки были хроматографически чистыми.
Расчеты в ОГХ [9, 11, 12]. Значения для конкретных объемов удерживания $V_{g}^{0}$ могут быть рассчитаны по уравнению
где tr = (t – t0); t – время удерживания, соответствующее максимуму пика для сорбата; t0 – время удерживания несорбирующегося газа;(2)
$F = \{ {{F}_{t}}({{P}_{{flw}}} - {{P}_{w}}){\text{/}}{{P}_{0}}\} (273.2{\text{/}}{{T}_{{flw}}}),$(3)
$J_{3}^{2} = (3{\text{/}}2)\{ [{{({{P}_{{inp}}}{\text{/}}{{P}_{{out}}})}^{2}} - 1]{\text{/}}[{{({{P}_{{inp}}}{\text{/}}{{P}_{{out}}})}^{3}} - 1]\} .$Согласно уравнению (1), выражение для значения относительной ошибки имеет вид:
(4)
$\Delta V_{g}^{0}{\text{/}}V_{g}^{0} = \Delta t{\text{/}}t + \Delta F{\text{/}}F + f(P)\Delta P{\text{/}}P + \Delta g{\text{/}}g$Погрешность измерения температуры колонки и скорости потока газа-носителя в условиях нашего эксперимента составляла ±0.1°С и ±0.5%, соответственно. Так, общая погрешность первых трех слагаемых уравнения (4) не превышает 1%. Основной вклад в ошибку в величине $V_{g}^{0}$ вносит ошибка в определении количества полимера в колонке [13]. Мы использовали аналитические весы ADV200 (погрешность взвешивания ±1 мг). С учетом того факта, что масса твердого носителя, содержащегося в колонке, находилась в диапазоне от 2 до 10 г, а масса нанесенного полимера составляла 5–15% от массы твердого носителя, погрешность измерения количества полимера не превышала 1%.
Исправленное время удерживания рассчитывали по значениям общего времени удерживания максимума пика сорбата и времени удерживания максимума пика несорбируемого в условиях эксперимента вещества (воздуха), записанным программно-аппаратным комплексом МультиХром 1.7x с погрешностью ±0.05 с.
Количество полимера в колонке находили следующим образом. Взвешенную часть приготовленной упаковки трижды промывали избытком растворителя, помещали в муфельную печь на нескольких часов и нагревали до 800°С. Количество сгоревшего полимера рассчитывали с учетом потери масы при прокаливании исходного инертного носителя и пересчитывали на 1 г насадки.
Ранее нами было показано [14], что аппроксимирующее уравнение
(5)
$\ln V_{g}^{0} = {{A}_{1}}{{(\ln T)}^{2}} + {{B}_{1}}\ln T + {{C}_{1}}(1{\text{/}}T) + {{D}_{1}},$Расчет термодинамических параметров взаимодействия полимер–низкомолекулярное соединение. Мы исходили из возможностей, вытекающих из теории активности. В термодинамике Гиббса введение понятия активность формально учитывает все межмолекулярные взаимодействия в растворах, которые собственно и являются причиной различия идеальных и реальных растворов. Коэффициент активности вводится как
(a1 – активность, c1 – концентрация раствора). Таким образом, коэффициент активности является мерой отклонения раствора от идеальности. В случае растворов полимеров естественно ожидать его чувствительность к особенностям химической структуры макроцепей при сольватации последних молекулами низкомолекулярных веществ.При определении коэффициента активности полимеров используют фундаментальное выражение (a1/w1) – массовый коэффициент активности $\Omega _{1}^{\infty }$ [15, 16].
Расчет значений $\Omega _{1}^{\infty }$ проводили по уравнению
(7)
$\ln {{\Omega }^{\infty }} = \ln \frac{{273.15R}}{{V_{g}^{0}P_{1}^{0}{{M}_{1}}}} - \frac{{P_{1}^{0}}}{{RT}}({{B}_{{11}}} - {{V}_{1}}),$в котором $V_{g}^{0}$ – удельный удерживаемый объем сорбата; P1, V1, M1 – давление насыщенных паров, мольный объем и молекулярная масса сорбата соответственно; B11 – второй виральный коэффициент; R – универсальная газовая постоянная.
Избыточные термодинамические функции связаны со значением Ω1∞ известными выражениями
(9)
${\Delta }\bar {S}_{1}^{{E\infty }} = (R{\text{/}}T)d(\ln \Omega _{1}^{\infty }){\text{/}}d(T) - R\ln \Omega _{1}^{\infty },$При расчете значений ${\Delta }\bar {H}_{1}^{{E\infty }}$ предполагают, что ${\Delta }\bar {C}_{{P1}}^{{E\infty }}$ = const (const ≠0), т.е. температурная зависимость ${\Delta }\bar {H}_{1}^{{E\infty }}$ может быть экстраполирована линейной функцией [17]. При расчете коэффициента активности оптимальное выражение для экстраполирующей функции выглядит следующим образом [14]:
где A2, B2 и C2 – константы.Соответствующие выражения для ${\Delta }\bar {H}_{1}^{{E\infty }}$ и ${\Delta }\bar {S}_{1}^{{E\infty }}$, согласно уравнениям (7), (9) и (11), будут иметь вид
Значения коэффициентов A2, B2 и C2 могут быть найдены из уравнения (11) методом наименьших квадратов. При обработке данных (в нашем случае не менее 30 значений) ошибка расчетных коэффициентов, как правило, не превышала 0.5%, а ошибка в ${\Delta }\bar {H}_{1}^{{E\infty }}$ составляла не более 10%.
Парциальная молярная избыточная энтальпия. Парциальная молярная избыточная энтальпия смешения $\quad{\Delta }\bar {H}_{1}^{{E\infty }}$ является результирующей всех межмолекулярных взаимодействий в образующихся бесконечно разбавленных растворах [18]. А именно, она может быть определена как сумма
(14)
${\Delta }\bar {H}_{1}^{{E\infty }} = {\Delta }{{H}_{{{\text{BREAK}}}}} + {\Delta }{{H}_{{{\text{HOLE}}}}} + {\Delta }{{H}_{{{\text{DISP}}}}} + {\Delta }{{H}_{{{\text{SP}}}}}$Здесь ΔHBREAK – энтальпия разрыва связи между молекулами растворенного вещества, ΔHHOLE – энтальпия образования полости в структуре чистого растворителя (в методе ОГХ таковым является полимер), ΔHDISP – энтальпия неполярного взаимодействия, ΔHSP – энтальпия специфического взаимодействия. Таким образом, определение тепловых эффектов смешения позволяет получать важную информацию об особенностях как межмолекулярного взаимодействия в растворах, так и морфологии полимеров.
Парциальная мольная избыточная энтропия. Выражение для парциальной мольной энтропии н-алканов имеет вид:
где m – отношение твердоядерных объемов полимера и низкомолекулярного растворителя. Уравнение справедливо при выполнении ограничений теории на отсутствие межмолекулярных взаимодействий и постоянство свободного объема при смешении. В этих ограничениях уравнение (15) предсказывает уменьшение значений ${\Delta }\bar {S}_{1}^{{E\infty }}$ с увеличением мольного объема н-алкана.Коэффициент диффузии сорбата и проницаемость полимера. Важную информацию о структуре полимеров можно получить из данных по их диффузной проницаемости низкомолекулярными соединениями.
Коэффициент диффузии сорбата D через пленку полимера может быть из уравнения Ван-Дееметра [20, 21], связывающего высоту теоретической тарелки H с линейной скоростью газа-носителя U:
Коэффициент C3 из уравнения (16) связан с D как
(17)
${{C}_{3}} = (8{\text{/}}{{\pi }^{2}})({{\delta }^{2}}{\text{/}}D)[k{\text{/}}{{(1 - k)}^{2}}],$Температурная зависимость коэффициента диффузии определяется уравнением Аррениуса
Здесь D0 – предэкспоненциальный коэффициент диффузионного процесса, не зависящий от температуры; ΔED – энергия активации диффузионного соединения в данной полимерной матрице, необходимая для выхода из существующего окружения и перемещения в соседнее окружение; R – газовая постоянная; T – абсолютная температура.
Проницаемость рассчитывается по известному уравнению
в котором S – коэффициент растворимости, S = ${{\rho }_{L}}V_{g}^{0}$, и ρL – плотность полимера.РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Диаграммы удерживания
Анализ диаграмм удерживаний в методе ОГХ позволяет получить информацию о наличии каких-либо фазовых или температурных переходов в матрице полимера. Признаком температурного перехода в методе ОГХ служит, как правило, отклонение диаграммы удерживания от линейности (или, в общем случае, от монотонного хода) [22]. Диаграммы удерживания образцов ЭПФЭ-5–ЭПФЭ-8 для всех изученных сорбатов практически не отличились от линейной зависимости во всем исследованном интервале температур (рис. 1). Диаграммы удерживания сорбатов для ЭПФЭ-9 имели явный нелинейный характер. Ранее нами было установлено, что нелинейные диаграммы удерживания могут быть представлены в виде комбинации двух линейных функций, экстраполированных из низко- и высокотемпературных областей диаграммы удерживания [23]. Использование этого приема для ЭПФЭ-9 позволило установить, что диаграммы удерживания характеризуются изгибом в диапазоне температур около 75°C (рис. 2). Принимая во внимание тот факт, что температура изгиба диаграмм удерживания для ЭПФЭ-9 не зависит от природы сорбата, а также, что эта температура намного выше температуры стеклования полимера (–27°С), изгиб на диаграммах удерживания может быть связан с процессами типа порядок–беспорядок. А именно, с распадом кластеров CH2-последовательностей с повышением температуры. Эти структурные изменения приводят к скачкообразному изменению механизма сорбции, что в методе ОГХ регистрируется в виде излома диаграммы удерживания.
Рис. 1.
Диаграммы удерживания. Сорбат – н-октан. 1 – ЭПФЭ-5; 2 – ЭПФЭ-8; 3 – ЭПФЭ-9. Сплошными линиями показаны теоретические кривые, рассчитанные по уравнению (5): $R_{{{\text{Э П Ф Э - 5}}}}^{2}$ = 0.998, $R_{{{\text{Э П Ф Э - 6}}}}^{2}$ = 0.998, $R_{{{\text{Э П Ф Э - 7}}}}^{2}$ = 0.997, $R_{{{\text{Э П Ф Э - 8}}}}^{2}$ = 0.998, $R_{{{\text{Э П Ф Э - 9}}}}^{2}$ = 0.988.
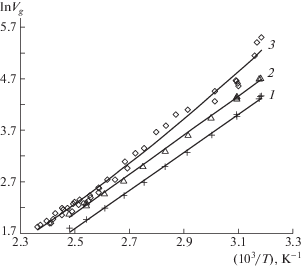
Рис. 2.
Диаграмма удерживания для ЭПФЭ-9. 1 – циклогексан, 2 – бензол, 3 – н-гептан, 4 – н-октан. Сплошными линиями показаны теоретические кривые, рассчитанные по уравнению (5): $R_{1}^{2}$ = 0.991, $R_{2}^{2}$ = = 0.990, $R_{3}^{2}$ = 0.996, $R_{4}^{2}$ = 0.988. Диапазон температур, где происходит внутренняя перестройка, выделен.
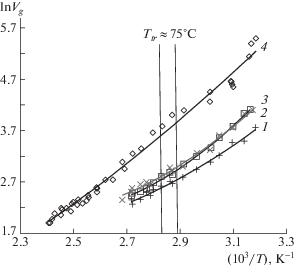
В связи с такой особенностью хроматографического поведения ЭПФЭ-9 представлялось целесообразным рассмотреть его сорбционные характеристики отдельно.
Термодинамические параметры сорбции сополимеров
Расчет и анализ значений коэффициента активности. Значения коэффициентов активности для систем полимер–сорбат, рассчитанные нами согласно уравнению (7), представлены в табл. 2. Как видно, $\Omega _{1}^{\infty }$ действительно довольно чувствительны как к химическим, так и к стереометрическим параметрам полимерного звена. Так, появление эфирного кислорода в боковой подвеске образца 2 приводит к повышению $\Omega _{1}^{\infty }$. Кроме того, увеличение длины боковой подвески (образец 3) способствует снижению $\Omega _{1}^{\infty }$. Замена перфторированного сомономера на углеводородный (образцы 1 и 4) вызывает резкое снижение $\Omega _{1}^{\infty }$.
Таблица 2.
Коэффициент активности сорбатов при 60°С
| Сорбат | Коэффициент активности $\Omega _{1}^{\infty }$ для образцов⁎ | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 7 | 6 | 8 | 9 | НК | ПДМС** | |
| Пентан | – | – | – | 16.84 | 32.71 | – | 22.65 | 25.92 | – | – | 5.88 |
| Гексан | – | 72.62 | 61.59 | 17.07 | 36.73 | 22.47 | 22.66 | 28.09 | – | 6.79 | 5.85 |
| Гептан | 51.98 | 92.86 | 70.40 | 17.86 | 42.77 | 19.59 | 25.69 | 31.10 | 28.72 | 6.50 | 5.84 |
| Октан | 64.38 | 117.55 | 82.50 | 18.08 | 50.11 | 28.36 | 28.45 | 36.22 | 24.04 | 6.14 | 5.98 |
| Нонан | 71.57 | 142.16 | 96.21 | 19.03 | 59.44 | 31.69 | 31.93 | 41.47 | – | 6.01 | 6.19 |
| Декан | 80.88 | 173.43 | 115.22 | 21.16 | 70.67 | 35.96 | 36.15 | 49.00 | – | 5.93 | 6.42 |
| Бензол | 15.41 | 22.93 | 20.57 | 11.77 | 28.52 | 17.77 | 14.82 | 22.06 | 19.28 | 4.68 | 5.80 |
| Циклогексан | 45.29 | 53.78 | 56.11 | 16.06 | 34.68 | 21.10 | 21.05 | 26.11 | 24.57 | 4.78 | 5.02 |
| Гексафторбензол | 5.72 | 4.72 | 4.29 | 3.05 | 4.28 | 3.41 | 3.19 | 3.70 | 3.31 | – | 4.21 |
Данные табл. 2 свидетельствуют о том, что, несмотря на повышенное содержание метиленовых последовательностей (по сравнению с сополимерами на основе винилиденфторида) углеводороды являются термодинамически плохими растворителями для синтезированных сополимеров ($\Omega _{1}^{\infty }$ > 20 для углеводородов и $\Omega _{1}^{\infty }$ = 3–4 для хорошего растворителя гексафторбензола).
Прослеживая тенденции изменения значений $\Omega _{1}^{\infty }$ с увеличением длины цепи сорбата в ряду н-алканов, следует отметить ухудшение термодинамики сорбции (рост значений $\Omega _{1}^{\infty }$) для всех рассматриваемых фторполимеров.
При этом чем выше содержание метиленовых групп в макромолекуле, тем менее выражена отмеченная тенденция (образцы ЭПФЭ-4, ЭПФЭ-6, ЭПФЭ-7 и ЭПФЭ-8). Можно предположить, что вероятная причина такого характера сорбции н-алканов фторполимерами заключается в образовании областей с повышенным содержанием метиленовых групп (углеводородных кластеров) в объеме полимера. Количество углеводородных кластеров тем больше, чем выше количество метиленовых групп в макроцепи. Увеличение размера клубка н-алкана затрудняет доступ его молекул к углеводородным кластерам в объеме полимера и ухудшает термодинамику смешения. Однако необходимо учитывать, что основной механизм сольватации н-алканов – дисперсионные взаимодействия [24]. Более того, это взаимодействие усиливается с увеличением размера молекулярного клубка. Последнее обстоятельство частично компенсирует стерические затруднения распределения молекул н-алкана в случае полимеров с повышенным содержанием углеводородных кластеров, и соответственно улучшается термодинамика смешения.
Представляет интерес сравнить полученные тенденции сорбции н-алканов фторполимерами и полимерами, для которых выбранные сорбаты являлись бы хорошими растворителями. В качестве примера мы выбрали углеводородный карбоцепной полимер – натуральный каучук и представителя элементорганических полимеров – полидиметилсилоксан (табл. 2).
В соответствии с данными табл. 2, дисперсионные силы для НК возрастают с увеличением длины цепи н-алкана, что способствует улучшению термодинамики сорбции.
В случае ПДМС дисперсионные силы не столь эффективны. Здесь термодинамика смешения обусловлена высокой гибкостью силоксановых цепей и их низкой энергией когезии. Эти обстоятельства отражаются и на тенденции изменения термодинамики сорбции н-алканов. В начале, рост дисперсионных сил с увеличением числа атомов углерода в молекуле н-алкана приводит к некоторому улучшению термодинамики смешения, а затем стерические затруднения смешения с увеличением длины цепи меняют направление тенденции. Те же тенденции характерны и для изменения термодинамических параметров.
Циклические сорбаты не дают заметного выигрыша в термодинамике их сорбции. Так, значения $\Omega _{1}^{\infty }$ для циклогексана лишь несколько ниже, чем для н-гексана. Однако появление подвижных π-электронов у бензола может способствовать значительному улучшению термодинамики взаимодействия полимер–сорбат [25]. Действительно, значения $\Omega _{1}^{\infty }$ для бензола уменьшаются обратно пропорционально содержанию метиленовых групп в полимере по сравнению с н-алканами и циклогексаном. Сольватация реализуется по характерному для ароматического кольца механизму донорного присоединения к метиленовой группе (соседней с CF2) с частично положительным зарядом на атоме водорода.
Данные табл. 2 позволяют также проследить влияние длины боковых цепей на термодинамику взаимодействия в системе полимер–сорбат. Рост длины боковой цепи (образцы 2 и 3, а также 5 и 7) приводит к улучшению термодинамики взаимодействия в системе полимер–сорбат. Однако увеличение длины боковой цепи свыше 10 атомов не приводит к улучшению термодинамики смешения (ср. образцы ЭПФЭ-5, ЭПФЭ-7 и ЭПФЭ-8). Улучшение термодинамики смешения в системе полимер–сорбат с увеличением длины боковой цепи может быть связано с соответствующим увеличением свободного объема и диффузионной проницаемости полимера. Принимая во внимание значительную длину подвижных боковых цепей (nc > 10), мы, по-видимому, имеем гребнеобразный полимер, для которых значения свободного объема невысоки [26].
Парциальная молярная избыточная энтальпия. Значения ${\Delta }\bar {H}_{1}^{{E\infty }}$, рассчитанные по уравнению (12), представлены в табл. 3. Значения ${\Delta }\bar {H}_{1}^{{E\infty }}$ положительны и значительно больше единицы для всех сорбатов, кроме гексафторбензола.
Таблица 3.
Значения избыточных параметров взаимодействия ${\Delta }\bar {H}_{1}^{{E\infty }}$ (кДж/моль) и $ - {\Delta }\bar {S}_{1}^{{E\infty }}$ (Дж/ (моль К)) при 60°C
| Сорбат | ${\Delta }\bar {H}_{1}^{{E\infty }}$ | $ - {\Delta }\bar {S}_{1}^{{E\infty }}$ | ${\Delta }\bar {H}_{1}^{{E\infty }}$ | $ - {\Delta }\bar {S}_{1}^{{E\infty }}$ | ${\Delta }\bar {H}_{1}^{{E\infty }}$ | $ - {\Delta }\bar {S}_{1}^{{E\infty }}$ | ${\Delta }\bar {H}_{1}^{{E\infty }}$ | $ - {\Delta }\bar {S}_{1}^{{E\infty }}$ | ${\Delta }\bar {H}_{1}^{{E\infty }}$ | $ - {\Delta }\bar {S}_{1}^{{E\infty }}$ | ${\Delta }\bar {H}_{1}^{{E\infty }}$ | $ - {\Delta }\bar {S}_{1}^{{E\infty }}$ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ЭПФЭ-5 | ЭПФЭ-6 | ЭПФЭ-7 | ЭПФЭ-8 | ЭПФЭ-9 | НК | |||||||
| Пентан | 4.06 | 16.80 | 1.38 | 21.79 | 2.13 | 19.31 | 3.70 | 15.92 | – | – | – | – |
| Гексан | 6.50 | 10.44 | 5.08 | 10.70 | 3.54 | 15.24 | 4.34 | 14.69 | – | – | 0.85 | 13.35 |
| Гептан | 8.08 | 6.97 | 6.57 | 7.26 | 2.51 | 17.19 | 5.28 | 12.71 | 0.05 | 27.76 | 0.76 | 13.26 |
| Октан | 8.59 | 6.73 | 7.48 | 5.37 | 5.79 | 10.42 | 6.57 | 10.12 | 1.01 | 23.40 | 0.04 | 14.97 |
| Нонан | 9.99 | 3.95 | 8.53 | 3.17 | 6.98 | 7.76 | 7.24 | 9.23 | – | – | –0.04 | 15.03 |
| Декан | 11.06 | 2.20 | 9.73 | 0.61 | 7.77 | 6.46 | 8.37 | 7.21 | 11.70 | –1.92 | 0.66 | 12.83 |
| Бензол | 7.96 | 5.58 | 6.47 | 5.89 | 4.41 | 12.10 | 6.26 | 8.31 | 4.55 | 12.94 | 0.67 | 10.99 |
| Циклогексан | 7.23 | 6.15 | 4.61 | 8.58 | 3.36 | 13.83 | 5.19 | 10.12 | 0.42 | 23.32 | 1.05 | 9.68 |
| Гексафторбензол | 0.66 | 10.11 | –0.04 | 9.75 | 0.58 | 8.44 | –1.16 | 14.36 | –0.94 | 12.74 | – | – |
Рассматривая природу сил сольватации гексафторбензола макромолекул ЭПФЭ, отметим его склонность к реакциям электрофильного присоединения, что обусловлено значительно повышенной по сравнению с бензолом плотностью электронов π-системы [27]. Кроме того, гексафторбензол, как и другие перфторароматические соединения, активен к действию нуклеофильных групп. Таким образом, гексафторбензол способен к сольватации как метиленовых групп с частично положительным зарядом на атоме водорода, так и перфторметиленовых групп линейной цепи. Совокупность этих двух симбатно протекающих процессов и обеспечивает повышенное термодинамическое сродство гексафторбензола к полифторированным сополимерам.
Анализируя данные табл. 3, предварительно отметим, что положительные значения ${\Delta }\bar {H}_{1}^{{E\infty }}$ указывают на предпочтительное взаимодействие молекул сорбата между собой, в то время как отрицательные – на предпочтительное взаимодействие полимер–сорбат. Согласно данным табл. 3, ЭПФЭ-6 и ЭПФЭ-8, т.е. образцы с повышенной долей метиленовых последовательностей, обладают отрицательными значениями ${\Delta }\bar {H}_{1}^{{E\infty }}$ при сорбции гексафторбензола. Вероятно, при более коротких последовательностях метиленовых звеньев стерические затруднения (экранирование соседними перфторированными группами) выводят часть метиленовых групп из взаимодействия. Принимая во внимание также меньшую локализацию δ+-заряда на соседнем с атомом фтора углеродном атоме (по сравнению с сополимерами на основе винилиденфторида, образцы 1–3), можно ожидать преобладание сольватации по перфторированным группам. Последнее обстоятельство должно обеспечивать стойкость исследуемых сополимеров в полярных растворителях.
Рассмотрим тенденции изменения ${\Delta }\bar {H}_{1}^{{E\infty }}$с длиной цепи н-алкана.
ЭПФЭ-5. Можно выделить начальный участок роста (н-С5…н-С7), плато (н-С7…н-С8) и второй участок роста (н-С8…н-С10). Увеличение длины цепи н-алкана затрудняет процесс его абсорбции полимером. Это отражает рост значений ${\Delta }\bar {H}_{1}^{{E\infty }}$ по причине повышения вклада энтальпии образования полости (ΔHHOLE) в полимере. Рост длины цепи н-алкана вместе с тем приводит к увеличению дисперсионных сил, улучшающих сольватацию макроцепей, что частично компенсирует увеличение ΔHHOLE (плато на зависимости). При дальнейшем росте цепи алкана увеличение дисперсионных сил не компенсирует рост ΔHHOLE (второй участок роста).
ЭПФЭ-6. Тенденции изменения термодинамических параметров сорбции с числом атомов углерода в молекуле н-алкана аналогичны ЭПФЭ-5. А именно, увеличение размера молекулы н-алкана приводит к повышению значения ${\Delta }\bar {H}_{1}^{{E\infty }}$.
Падение абсолютных значений ${\Delta }\bar {H}_{1}^{{E\infty }}$ ЭПФЭ-6 относительно ЭПФЭ-5 можно объяснить увеличением длины последовательностей метиленовых групп в среднем в 2 раза по сравнению с ЭПФЭ-5. Кривая ${\Delta }\bar {H}_{1}^{{E\infty }} = f({{n}_{c}})$ не имеет плато. Данные отличия могут быть связаны с увеличением в этом сополимере числа углеводородных кластеров. Это обуславливает снижение вклада ΔHHOLE углеводородов и определяющее влияние дисперсионных взаимодействий на характер изменения ${\Delta }\bar {H}_{1}^{{E\infty }}$ с молекулярным размером н-алкана.
ЭПФЭ-7. Абсолютные значения ${\Delta }\bar {H}_{1}^{{E\infty }}$ для рассматриваемого образца несколько ниже, чем для образца ЭПФЭ-5. Это можно объяснить следующим: увеличение бокового радикала на группу ${\text{CF}}_{2}^{ - }$ приводит к росту стерических ограничений пространственного расположения макроцепей, что проявляется в соответствующем росте свободного объема. Соответственно затраты энергии, связанные с энтальпией образования полости (ΔHHOLE) в полимере, также уменьшаются.
В то же время абсолютные значения ${\Delta }\bar {H}_{1}^{{E\infty }}$ для сорбатов ЭПФЭ-6 (за исключением пентана) больше, чем соответствующие значения для ЭПФЭ-7 из-за более коротких боковых радикалов.
Образование локального минимума на кривой ${\Delta }\bar {H}_{1}^{{E\infty }}$ = f(nC) позволяет соотнести среднее значение элементов свободного объема в ЭПФЭ-7 с молекулярным размером н-гептана.
ЭПФЭ-8. Для ЭПФЭ-8 на зависимости $\Delta \bar {H}_{1}^{{E\infty }}$ = f(nC) нет плато. Такой характер зависимости может быть связан с ростом в этом сополимере числа углеводородных кластеров, что обуславливает снижение вклада ΔHHOLE и определяющее влияние дисперсионных взаимодействий на характер изменения $\Delta \bar {H}_{1}^{{E\infty }}$с молекулярным размером н-алкана.
Парциальная молярная избыточная энтропия. Полученные по уравнению (13) значения ${\Delta }\bar {S}_{1}^{{E\infty }}$ для изученных сополимеров представлены в табл. 3. Значения ${\Delta }\bar {S}_{1}^{{E\infty }}$ н-алканов уменьшаются с увеличением их молярного объема для всех изученных ЭПФЭ. Это соответствует динамике изменения прогнозируемых значений по уравнению (15). Следует отметить, что уравнение (15) не выполняется для многих эластомеров [28], и более типичным является отсутствие зависимости ${\Delta }\bar {S}_{1}^{{E\infty }}$ от молекулярного объема н-алкана. Снижение ${\Delta }\bar {S}_{1}^{{E\infty }}$ с молекулярным объемом н-алкана в случае ЭПФЭ может быть связано с механизмом сорбции, обусловленным внедрением молекул н-алкана во внутриассоциативные пустоты (элементы свободного объема) эластомера, что характерно для сорбции ассоциированными жидкостями [29].
Температурная зависимость ${\mathbf{\Delta }}\bar {H}_{1}^{{{\mathbf{E}}\infty }}$. Повышение температуры сопровождается увеличением сегментальной подвижности цепей. Следовательно, вероятность изменения плотности молекулярной упаковки ЭПФЭ высока. В связи с этим представлялось обоснованным проследить зависимость значений ${\Delta }\bar {H}_{1}^{{E\infty }}$ н-алканов от температуры для изученных образцов ЭПФЭ. Полученные данные представлены на рис. 3. Как видно, с ростом температуры значения ${\Delta }\bar {H}_{1}^{{E\infty }}$ уменьшаются для всех алканов.
Рис. 3.
Влияние температуры на $\Delta \bar {H}_{1}^{{E\infty }}$ для систем н-алкан–ЭПФЭ-5 (а) и н-алкан–ЭПФЭ-7 (б). Т = = 40 (1), 60 (2), 80 (3), 100 (4) и 120°C (5).
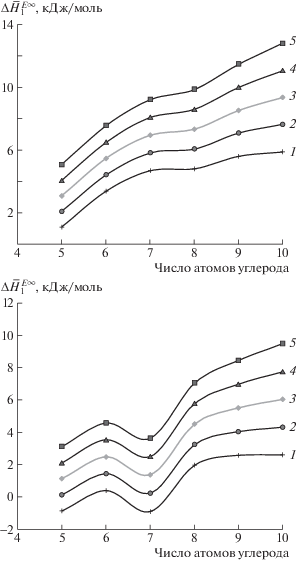
Также необходимо отметить, что, как следует из рисунков 2 и 3а, общий вид зависимости ${\Delta }\bar {H}_{1}^{{E\infty }}$ от длины цепи н-алкана с температурой меняется незначительно. Таким образом, можно сделать вывод, что механизм сорбции не изменяется с температурой. Исходя из этого, можно предположить, что конформационные переходы в исследуемых ЭПФЭ отсутствуют.
Рассмотрим образец ЭПФЭ-9 более подробно.
ЭПФЭ-9. Для ЭПФЭ-9 в отличие от образцов ЭПФЭ-5 и ЭПФЭ-7 в области температур ниже 50°C наблюдается зависимость значений $V_{g}^{0}$ сорбатов от скорости потока газа-носителя и величины пробы как для плохих растворителей (углеводороды), так и для хорошего растворителя (гексафторбензол). Тщательно проведенные исследования не показали зависимости значений $V_{g}^{0}$ от скорости потока газа-носителя и толщины полимерной пленки. Такой характер сорбции указывает на установление равновесия в системе полимер–сорбат в данной области температур, протекающее, однако, заметно медленней, чем в случае исследованных ранее образцов ЭПФЭ. По-видимому, отмеченное различие может быть объяснено присутствием в структуре ЭПФЭ-9 областей с ограниченной подвижностью цепей, в которых диффузия сорбатов замедленна по сравнению с остальным объемом полимера. Такие области могут быть образованы в результате взаимной ориентации боковых радикалов, содержащих группы –SO2F2. С повышением температуры увеличивается сегментальная подвижность цепей, что может привести к нарушению взаимной ориентации боковых радикалов и росту скорости диффузии молекул сорбатов в объем полимера. Действительно, как следует из рис. 2, диаграммы удерживания сорбатов, включая гексафторбензол, имеют излом в районе Ttr ≈ 75°С. Независимость Ttr от природы сорбата указывает на наличие некоторого конформационного перехода в исследуемом полимере. Можно предположить, что это переход типа порядок–беспорядок, обусловленный разрушением ассоциатов групп SO2F2 с повышением температуры. Энергия разупорядочивания (ΔHTR = = 4.4 ± 1.0 кДж/моль) ассоциатов была рассчитана из разности наклонов диаграмм удерживания до и после конформационного перехода. Полученное значение ΔHTR по порядку величины совпадает со значением ΔHTR, обусловленным изменением конформации макроцепей, ориентированных относительно друг друга вследствие образования ассоциатов боковых групп.
Коэффициент диффузии и проницаемость
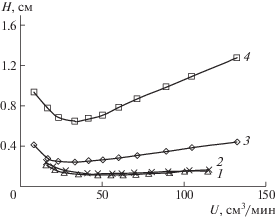
В табл. 4 представлены значения коэффициентов диффузии и проницаемости для гексафторбензола (хороший растворитель) и циклогексана (плохой растворитель), которые были рассчитаны для ЭПФЭ-5 и ПДМС (для сравнения) по уравнениям (16) и (20). Зависимость величины теоретической тарелки от линейной скорости газа-носителя приведена на рис. 4.
Таблица 4.
Значения коэффициентов диффузии D (см2/с) и проницаемости P (см3 м / (см2 с атм)) для ЭПФЭ-5 и ПДМС при 20°С
| Сорбат | D × 108 | P × 108 | D × 108 | P × 108 |
|---|---|---|---|---|
| ЭПФЭ-5 | ПДМС | |||
| Гексафторбензол | 0.082 | 3.6 | 0.473 | 9.3 |
| Циклогексан | 0.127 | 1.2 | 0.278 | 10.6 |
Рис. 4.
Зависимость высоты теоретической тарелки H от линейной скорости газа-носителя U. 1 – ПДМС–гексафторбензол, 2 – ПДМС–циклогексан, 3 – ЭПФЭ-5–гексафторбензол, 4 – ЭПФЭ-5–циклогексан. Содержание неподвижной фазы ПДМС 2.38%, ЭПФЭ-5 2%; T = 20°С.
Математическая обработка температурных зависимостей D показала, что в исследуемой температурной области коэффициенты диффузии как термодинамически хорошего, так и плохого растворителей изменяются с температурой в соответствии с уравнением Аррениуса. Таким образом, можно сделать вывод о механизме диффузии сорбатов в данных сополимерах как активационном процессе.
Найдены энергии активации EA, рассчитанные по зависимостям D(T). Они составляют 35.2 и 62.0 кДж/моль для гексафторбензола и циклогексан, соответственно. Как следует из табл. 4, величины D сорбатов в ЭПФЭ-5 и ПДМС имеют сопоставимые значения. Данный факт, учитывая отсутствие такого соответствия для термодинамических параметров сорбции этих же полимеров (табл. 2), свидетельствует о значительном свободном объеме в объеме полимерной матрицы. Образование большого свободного объема, по-видимому, в первую очередь обусловлено стерическими затруднениями достижения плотной упаковки цепей сополимеров вследствие наличия боковых радикалов. Безусловно, образованию большого свободного объема способствует также наличие углеводородных участков цепей, термодинамически несовместимых с перфторуглеродными участками.
Абсорбционный механизм взаимодействия с полимерами позволяет сделать вывод о том, что использование таких сополимеров в качестве материала для селективно проницаемых мембран является более перспективным, чем для защитных покрытий.
ЗАКЛЮЧЕНИЕ
Проведенный в работе анализ сорбционных свойств сополимеров углеводородного и перфторированных сомономеров показал, что даже при альтернантной архитектуре цепи возможны процессы самоорганизации, приводящие к формированию углеводородных кластеров в перфторированной дисперсионной среде.
Абсорбционный механизм взаимодействия сополимеров этилена с перфторированными эфирами даже с термодинамически очень плохими растворителями позволяет сделать вывод о перспективности использования таких сополимеров в качестве материала для селективно-проницаемых мембран.
При выборе областей практического применения полимеров подобной структуры следует учитывать, что даже практически полное отсутствие растворимости полимеров в агрессивных средах не может гарантировать их успешное применение в качестве протекторного покрытия.
Список литературы
Guizarda C., Boutevin B., Guida F., Ratsimihety A., Amblard P., Lasserre J.C., Naiglin S. // Separ. Purif. Technol. 2001. V. 22. P. 23.
Norsten T.B., Guiver M.D., Murphy J., Astill T., Navessin T., Holdcroft S., Frankamp B.L., Rotello V.M., Ding J. // Adv. Funct. Mater. 2006. V. 16. № 14. P. 1814.
Rastogi A.C., Desu S.B. // Polymer. 2005. V. 46. № 10. P. 3440.
Carr J.M., Mackey M., Flandin L., Hiltner A., Baer E. // Polymer. 2013. V. 54. № 6. P. 1679.
Drobny J.G. Technology of fluoropolymers. Boca Raton: CRC Press, 2008.
Masaaki Y. // Proc. Macromol. Symposia. Basel, 1992. P. 11.
Valencia Bel F., Caranielli F. // Proc. 4 Int. Spacecraft Propulsion Conference. Cagliari, Italy, 2004. P. 555.
Voznyakovsky A.P., Sokolov Yu.P., Lovchikov K.V., Krivoruchko E.M. // Proc. 2 Int. Conf. “Material and Coatings for Extreme Performances: Investigations, Applications, Ecologically Safe Technologies for Their Production and Utilization”. Katsiveli, Ukraine, 2002. P. 63.
Нестеров А.Е. Обращенная газовая хроматография полимеров. Киев: Наукова думка, 1988.
Sokolov Yu.P., Voznyakovskii A.P. // Russ. J. Appl. Chem. 2002. V. 75. № 1. P. 142.
Mohammadi-Jam S., Waters K.E. // Adv. Colloid Interface Sci. 2014. V. 212. P. 21.
Belov N.A., Safronov A.P., Yampolskii Yu.P. // Polymer Science A. 2012. V. 54. № 11. P. 859.
Roth M. // Macromolecules. 1990. V. 23. № 6. P. 1696.
Genkin A.N., Voznyakovskii A.P., Krivoruchko E.M. // Russ. J. Phys. Chem. 1998. V. 72. № 7. P. 1174.
Patterson D., Tewari Y.B., Schreiber H.P., Guillet J.E. // Macromolecules. 1971. V. 4. № 3. P. 356.
Yampolskii Y., Belov N. // Macromolecules. 2015. V. 48. № 19. P. 6752.
Gray D.G., Guillet J.E. // Macromolecules. 1972. V. 5. № 3. P. 319.
Белоусов В.П., Морачевский А.Г. Теплоты смешения жидкостей. Л.: Химия, 1970.
Prausnitz J.M. Molecular Thermodynamics of Fluid-Phase Equilibria. New Jersey: Prentice-Hall Inc., 1969.
Van Deemter J.J., Zuiderweg F.J., Klinkenberg A. // Chem. Eng. Sci. 1956. V. 5. № 6. P. 278.
Wang D., Li J., Zeng C., Chen J., Chen C. // J. Chem. Eng. Data. 2007. V. 52. № 2. P. 370.
Nastasovic A.B., Onjia A.E. // J. Chromatogr. A. 2008. V. 1195. № 1–2. P. 8.
Возняковский А.П., Генкин А.Н. // Высокомолек. соед. А. 1986. Т. 28. № 5. P. 941.
Smith P.E., Matteoli E., O’Connell P. Fluctuation Theory of Solutions: Applications in Chemistry, Chemical Engineering, and Biophysics. Boca Raton: CRC Press, 2013.
Prigogine I. The Molecular Theory of Solution. Amsterdam: North-Holland, 1957.
Plate N.A., Shibaev V.P. Comb-Shaped Polymers and Liquid Crystals. New York: Plenum Press, 1987.
Sheppard W.A., Sharts C.M. Organic Fluorine Chemistry. New York: Benjamin, 1969.
Roth M., Novak J. // Macromolecules. 1986. V. 19. № 2. P. 367.
Yampolskii Yu.P., Kaluzhnyi N.E., Durgar’jan S.G. // Macromolecules. 1986. V. 19. № 3. P. 850.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия А)