

Высокомолекулярные соединения (серия А), 2019, T. 61, № 6, стр. 559-568
ВЛИЯНИЕ ЭЛЕКТРОСТАТИЧЕСКОГО ДАЛЬНОДЕЙСТВИЯ И БЛИЗКОДЕЙСТВИЯ НА КОНФОРМАЦИЮ МАКРОМОЛЕКУЛ ГИБКОЦЕПНЫХ ЛИНЕЙНЫХ ПОЛИЭЛЕКТРОЛИТОВ С РАЗЛИЧНОЙ ПЛОТНОСТЬЮ ЗАРЯДА В ВОДНЫХ БЕССОЛЕВЫХ РАСТВОРАХ
Г. М. Павлов a, *, О. А. Доммес a, О. В. Окатова a, И. И. Гаврилова a, Е. Ф. Панарин a, b
a Институт высокомолекулярных соединений Российской академии наук
199004 Санкт-Петербург, Большой пр., 31, Россия
b Санкт-Петербургский государственный политехнический университет Петра Великого
195251 Санкт-Петербург, Политехническая ул., 29, Россия
* E-mail: georges.pavlov@mail.ru
Поступила в редакцию 24.05.2019
После доработки 21.06.2019
Принята к публикации 05.07.2019
Аннотация
Синтезирован и исследован ряд сополимеров N-метил-N-винилацетамида и N-метил-N-виниламина гидрохлорида со средним содержанием заряженных групп (4.4 ± 0.2) мол. % в интервале молекулярных масс 22 < M × 10–3, г/моль <630. Фракции охарактеризованы методами седиментационно-диффузионного анализа в 0.2 моль/л NaCl при практически подавленных полиэлектролитных эффектах. В бессолевых водных растворах изучено их вязкое течение и определена характеристическая вязкость как [η] ≡ lim(∂lnηr/∂c) при c → 0. Результаты сопоставлены с полученными ранее данными для сополимера с (15.7 ± 0.5) мол. % заряженных групп и гомополимера. Показано, что вид зависимости характеристической вязкости от молекулярной массы сополимера при содержании заряженных групп 4.4 мол. % типичен для цепей, проявляющих внутрицепные объемные взаимодействия, а при содержании заряженных групп 15.7 мол. % – типичен для цепей, проявляющих внутрицепные эффекты близкодействия, а значит цепи ведут себя как жесткоцепные, протекаемые. Определена длина статистических сегментов макромолекул сополимеров в бессолевых растворах, а также в 0.2 моль/л NaCl. Данные проанализированы в терминах нормированных построений Куна–Марка–Хаувинка–Сакурады.
ВВЕДЕНИЕ
Жизнь и функционирование живых объектов на нашей планете обеспечиваются полиэлектролитами (нуклеиновые кислоты, белки, полисахариды и другие), которые выполняют различные функции. В настоящее время полиэлектролиты и материалы на их основе, обладающие широким спектром структурных и функциональных свойств, находят применение во всевозможных областях технологий и биотехнологий [1–5]. Полиэлектролиты, как и нейтральные полимеры, включают в себя макромолекулы разных топологий, которые исследуются как теоретически, так и экспериментально [6–11]. Большое внимание уделяется изучению межцепных взаимодействий и комплексов, образуемых противоположно заряженными полиэлектролитами, а также комплексов полиэлектролитов с биологически активными макромолекулами [12–16].
Исследования заряженных макромолекул и макромолекулярных систем на их основе ведутся уже более семи десятилетий [17, 18]. Описание поведения заряженных цепей является и сегодня самой сложной задачей среди всех биологических и химических систем, так называемых “soft matter”, многие аспекты поведения линейных полиэлектролитных молекул остаются не до конца выясненными [1]. С экспериментальной точки зрения, это объясняется тем, что поведение полиэлектролитов контролируется многими переменными, которые необходимо учитывать. Так, для линейных полиэлектролитов на основе исходно гибких цепей до сих пор нет четкого представления, как соотносятся электростатическое набухание и электростатическое ужестчение цепей [1, 19, 20].
Цель настоящего исследования – установление количественных критериев разделения вкладов электростатического дальнодействия и близкодействия в размеры линейных полиионов при минимальной ионной силе растворов. Поиск ответа на этот вопрос необходимо вести в области малых и средних степеней заряженности цепей. При этом ряды сополимеров должны быть синтезированы на основе водорастворимого гомополимера для обеспечения полной растворимости в воде сравниваемых сополимеров.
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ И КОНЦЕПЦИИ
Важной задачей при исследовании полиэлектролитов является изучение свойств индивидуальных макромолекул при предельно малых значениях ионной силы, т.е. в бессолевых растворах. Количественное объяснение больших размеров линейных полиэлектролитов при предельно малых значениях ионной силы остается одной из фундаментальных проблем в молекулярной биофизике и физике полимеров. Наличие зарядов в цепи приводит к внутри- и межмолекулярным электростатическим взаимодействиям. Электростатические взаимодействия проявляются намного сильнее и распространяются на бóльшие расстояния, чем внутри- и межцепные взаимодействия в незаряженных макромолекулах. Первые теории [17, 21] объясняли увеличение размеров полиэлектролитов исключительно дальнодействующей природой электростатических взаимодействий. Однако в линейной макромолекуле, несущей заряды, сильные электростатические внутрицепные взаимодействия могут проявляться как на длинных, так и на коротких участках, что определяется линейной плотностью заряда. Более поздние теории учитывали также силы электростатического близкодействия – взаимодействия близко расположенных вдоль по цепи зарядов. T. Odijk, J. Skolnick, M. Fixman [22–24] предположили, что электростатические взаимодействия между заряженными звеньями, расположенными на расстояниях, меньших длины экранирования Дебая, приводят к дополнительному ужестчению цепи (теория ОШФ), т.е. к вкладу электростатической составляющей в персистентную длину (длину статистического сегмента). Первоначальный вывод был получен для полугибких цепей и основан на оценке изменения электростатической энергии цепи при отклонении конформации цепи от жесткого стержня. В случае гибких сильно заряженных полиэлектролитов электростатический вклад в персистентную длину цепи – это основной фактор, контролирующий изгибную жесткость цепи. Таким образом, для полиэлектролитных цепей общая длина статистического сегмента Atot является суммой структурной Abare и электростатической Ael составляющих [22–24]:
Учет электростатического близкодействия [22–24] приводит к тому, что электростатическая составляющая длины сегмента пропорциональна квадрату длины Дебая–Хюккеля λDH= (8πλBμ)–1/2 (λB – длина Бьеррума, μ – ионная сила раствора). Для слабо заряженных макромолекул при lqq > λB:
для сильно заряженных макромолекул (lqq < λB): где lqq – среднее расстояние между ближайшими по цепи зарядами.Позднее Баррат, Джоанни и Добрынин [25–28] использовали вариационный подход с пробной функцией, описывающей полиэлектролитную цепь при натяжении. В этой модели учитывалась дополнительная изгибная жесткость цепи, и было получено [27] следующее соотношение для электростатической составляющей статистического сегмента:
(4)
${{A}_{{el}}}(l) = 2{{\lambda }_{{\text{B}}}}\lambda {{\lambda }_{{{\text{DH}}}}}l{\text{/}}l_{{qq}}^{2}(3 + (4\lambda l{\text{/}}{{\lambda }_{{{\text{DH}}}}})).$Здесь l – дополнительный безразмерный изменяемый параметр, т.е. число рассматриваемых связей, на котором наблюдают корреляцию; λ – проекция мономерного звена на направление основной цепи.
В этой модели величина электростатической составляющей статистического сегмента Ael зависит от выбранной шкалы l. Когда $\lambda l \gg 3{{\lambda }_{{{\text{DH}}}}}{\text{/}}4$, то ${{A}_{{el}}} = {{\lambda }_{{\text{B}}}}\lambda _{{{\text{DH}}}}^{2}{\text{/}}2l_{{qq}}^{2}$, что эквивалентно результату ОШФ (выражение (2)); в другом предельном случае при $\lambda l \ll 3{{\lambda }_{{{\text{DH}}}}}{\text{/}}4$, получаем
(5)
${{A}_{{el}}}(l) = 2{{\lambda }_{{\text{B}}}}\lambda {{\lambda }_{{{\text{DH}}}}}l{\text{/}}3l_{{qq}}^{2},$Дальнейшие исследования методами компьютерного моделирования [29–31] приводят к результатам, которые возможно представить в общем виде: Ael ~ (λDH)ν, где показатель 1 ≤ ν ≤ 2 может быть нецелым числом, в зависимости от используемой модели.
Стратегия экспериментальных исследований данной работы состояла из следующих этапов.
Синтез рядов заряженных сополимеров в широком интервале молекулярных масс.
Определение молекулярных масс полиионов одним из абсолютных методов – седиментационно-диффузионным анализом при подавленных электростатических взаимодействиях в 0.2 моль/л NaCl.
Анализ скейлинговых соотношений между молекулярной массой и гидродинамическими характеристиками и определение длины статистического сегмента в 0.2 моль/л NaCl.
Исследование вязкого течения разбавленных растворов во всем интервале ионных сил, и особенно в бессолевых водных растворах при экстремально малой ионной силе.
Установление корреляций между средней плотностью заряда линейной цепи и гидродинамическим объемом, длиной статистического сегмента и(или) коэффициентом набухания полиэлектролитной цепи в бессолевом растворе.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Для решения поставленных задач синтезировали ряд водорастворимых сополимеров N-метил-N-винилацетамида и N-метил-N-виниламина гидрохлорида (МВАА–ГМВА) с широким диапазоном молекулярных масс (22–630) × 103 заданного состава путем управляемого частичного гидролиза нейтрального поли-N-метил-N-винилацетамида (ПМВАА). Синтезированные сополимеры МВАА–ГМВА растворяются в воде во всем интервале линейной плотности заряда, включая малую:
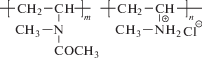
Исследования, проведенные ранее [32–34] на сериях образцов сополимеров МВАА–ГМВА с приблизительно постоянной контурной длиной, но в широком интервале средних степеней заряда (1.5 ≤ n ≤ 39, мол. %) показали, что в зависимости гидродинамического объема от степени заряженности цепей можно выделить три области. При малых степенях заряженности n < 5 мол. % наблюдается слабый рост объема полиэлектролитного клубка, в интервале 5 < n < 15, мол. % прослеживается его резкий рост, а при n > 15, мол. % – наступает практичекое насыщение зависимости, т.е. размеры клубка достигают своего максимума. Предположительно, в первой области проявляется электростатическое дальнодействие, в третьей – электростатическое близкодействие, а в центральной области проявляют себя оба эффекта. Для количественного изучения этого вопроса целесообразно изучить и сопоставить ряды сополимеров с содержанием заряженных групп n ≤ 5 и n ∼ 16, мол. %.
Синтез сополимеров и определение их молекулярных масс
Сополимеры МВАА–ГМВА получали в лаборатории гидрофильных полимеров Института высокомолекулярных соединений РАН путем частичного гидролиза поли-N-метил-N-винилацетамида, расфракционированного на восемь фракций [35] (в частности, сополимер 4.4 мол. % синтезировали гидролизом фракций ПМВАА в 1.8 н HCl при температуре 100–110°C в течение 14 ч). Структуру сополимеров подтверждали с помощью ИК-спектроскопии на спектрометре “Vertek-70” фирмы “Brucker”. Состав сополимеров МВАА–ГМВА находили при помощи аргентометрического титрования: определяли концентрацию противоионов Cl, которая соответствует мольному проценту заряженных групп в полимере МВАА–ГМВА, его среднее значение для фракций сополимера составило (4.4 ± 0.2) мол. %.
Сополимеры изучали и характеризовали методами молекулярной гидродинамики (скоростная седиментация, поступательная диффузия и вискозиметрия разбавленных растворов) в 0.2 моль/л водном растворе NaCl при практически подавленных полиэлектролитных эффектах. Установки и способы обработки первичных гидродинамических результатов были изложены в предыдущих публикациях [36–39].
Таким образом, определили молекулярные массы сополимеров, рассчитали гидродинамические инварианты и установили согласующиеся корреляции типа Марка–Куна–Хаувинка–Сакурады.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Вискозиметрия разбавленных растворов полиэлектролитов при предельно малых значениях ионной силы
Наибольший интерес представляют экспериментальные данные, полученные при изучении бессолевых водных растворов полиэлектролитов, где электростатические взаимодействия проявляются в наибольшей степени, которые и являются предметом обсуждения в настоящей работе. Так, было предложено [34, 40] интерпретировать вискозиметрические данные разбавленных растворов заряженных макромолекул, используя исходные определения величины характеристической вязкости [η] полимеров, представленные Штаудингером (limc→0(∂ηsp/∂c) ≡ [η]) [41] и Кремером (limc→0(∂lnηr/∂c) ≡ [η]) [42], где ηr – относительная вязкость, равная отношению динамических вязкостей раствора и растворителя (ηr = η/η0), ηsp – удельная вязкость (ηsp = ηr – 1), c – концентрация раствора.
Следует отметить, что эти базовые определения не получили распространения в полимерном сообществе при оценке величин характеристической вязкости незаряженных полимеров. Альтернативой для нахождения [η] незаряженных полимеров служат линейные в области разбавленных растворов интерпретационные зависимости ηsp/c = f1(c) и lnηr/c = f2(c), известные как построения Хаггинса и Кремера. Величина характеристической вязкости в этом случае определяется как отсекаемый на оси ординат отрезок при c = 0. Для заряженных цепей эти построения становятся нелинейными и не могут быть использованы для нахождения величины [η]. Важно, что зависимости lnηr = f(c) и lnηr/c = f2(c) (рис. 1а, 1б), так же как пара ηsp = f '(c) и ηsp/c = f1(c), имеют совершенно разный вид. Зависимости lnηr/c и ηsp/c от c кардинально отличаются для нейтральных и заряженных полимеров, тогда как зависимости lnηr и ηsp от c имеют схожий вид для нейтральных и заряженных полимеров, различающийся только крутизной наклона в области малых значений концентрации, которая значительно больше для заряженных цепей. Значения характеристической вязкости [η] получены из начального наклона зависимости логарифма относительной вязкости lnηr от c. Наклон был определен путем построения квадратичной или линейной зависимости в области малых значений концентрации, когда ηr меньше ηr ≤ 1.4 (рис. 1в).
Рис. 1.
Cтандартное построение Крэмера (а) и зависимость логарифма относительной вязкости от концентрации (б) для сополимеров МВАА–ГМВА с содержанием ГМВА 4.4 мол. % в бессолевом водном растворе; номера точек соответствуют номерам образцов в табл. 1. в – Часть зависимости, представленной на рис. 1б, в области малых значений концентрации; сплошные линии – результаты экстраполяции линейной (1, 4) и квадратичной (6, 8) зависимости.
Сопоставление гидродинамического объема сополимеров МВАА–ГМВА разного состава и гомополимера ПМВАА в бессолевых растворах
Проведем сравнение результатов вискозиметрических исследований (табл. 1), полученных в бессолевых растворах для сополимеров с (4.4 ± ± 0.2), мол. % (далее сополимер I), а также данных, представленных ранее, для сополимера с (15.7 ± 0.5), мол. % заряженных звеньев (сополимер II) [43] и исходного гомополимера [44].
Таблица 1.
Коэффициенты седиментации s0 и диффузии D0, молекулярная масса MSD и характеристическая вязкость [η] сополимеров МВАА–ГМВА с содержанием ГМВА 4.4 мол. % в растворе NaCl с концентрацией 0.2 моль/л и в бессолевом растворе
| № п/п | s0 × 1013 (0.2 моль/л NaCl) | D0 × 107, см2/с | MSD × 10–3 | [η], см3/г | |
|---|---|---|---|---|---|
| 0.2 моль/л NaCl | Н2O | ||||
| 1 | 4.0 | 0.98 | 630 | 250 | 1840 |
| 2 | 3.35 | 1.15 | 450 | 194 | 1000 |
| 3 | 2.83 | 1.39 | 315 | 175 | 800 |
| 4 | 2.60 | 1.54 | 260 | 141 | 430 |
| 5 | 2.25 | 2.20 | 160 | 91 | 250 |
| 6 | 1.70 | 3.06 | 85 | 57 | 80 |
| 7 | 1.17 | 4.81 | 38 | 37 | 44 |
| 8 | 0.93 | 6.4 | 22 | 22 | 36 |
Размеры и конформация полииона в водных растворах в общем случае зависят от линейной плотности заряда в цепи, ионной силы раствора, химической структуры цепи. В рассматриваемом случае ионная сила является одинаковой и минимальной (∼10–6 моль/л); структуру основных цепей в сравниваемых сополимерах и гомополимере можно полагать одинаковой. Ключевым параметром, от которого зависит уровень проявления полиэлектролитных свойств, можно назвать среднее расстояние между ближайшими по цепи зарядами lqq ≡ L/p = λ/(p/Ν), где L – контурная длина цепи, N – общее число мономерных звеньев, λ – проекция мономерного звена на направление основной цепи, p – число заряженных мономерных звеньев в цепи, p/Ν – мольная доля заряженных мономерных звеньев (табл. 2).
Таблица 2.
Расстояние между соседними зарядами lqq, его соотношение с длиной сегмента незаряженного аналога ПМВАА Abare и длиной Бьеррума λB, молекулярная масса единицы длины ML сополимеров МВАА–ГМВА и гомополимера ПМВАА
| Содержание ГМВА, мол. % | lqq, нм | lqq/Abare* | λB/lqq | ML**, г/(моль нм) |
|---|---|---|---|---|
| 4.4 | 5.73 | 2.6 | 0.12 | 386 |
| 15.7 | 1.58 | 0.7 | 0.45 | 367 |
| 0 | – | – | – | 393 |
Из табл. 2 видно, что в сополимере I среднее расстояние между ближайшими по цепи зарядами почти в 4 раза превышает таковое в сополимере II. Целесообразно сопоставить эти показатели расстояния с длиной статистического сегмента незаряженного исходного гомополимера Abare = = 2.2 нм [44], т.е. измерить расстояние между ближайшими зарядами в числе статистических сегментов незаряженного гомополимера: lqq/Abare (табл. 2).
Наиболее чувствительной к размерам и конформациям линейных макромолекул гидродинамической величиной является их характеристическая вязкость [η], поскольку она пропорциональна кубу размеров линейной макромолекулы в соответствии с соотношением Флори–Фокса [45]:
где ${{\langle {{h}^{2}}\rangle }^{{1/2}}}$ – среднеквадратичное расстояние между концами цепи, Φ – вискозиметрический параметр Флори.Сведения, которые можно извлечь, сопоставляя величины характеристической вязкости с соответствующими значениями молекулярной массы, тем достоверней и информативней, чем шире интервал молекулярных масс изученных фракций (образцов). Обычно, интерпретация начинается с установления соотношения Куна–Марка–Хаувинка–Сакурады (КМХС):
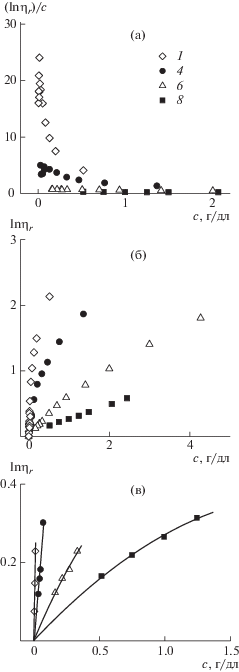
т.е. определения коэффициентов Kη и bη. Важно отметить, что в общем случае при изучении фракций (образцов) линейных полимеров в достаточно широком интервале молекулярных масс коэффициент bη не сохраняет свое постоянство во всем интервале молекулярных масс. В этом случае анализ зависимостей дает дополнительную информацию о характере внутриклубковых взаимодействий. Эффекты дальнодействия в цепях, хорошее термодинамическое качество растворителя приводят к тому, что коэффициент bη увеличивается с 0.50 до 0.6–0.8 при переходе из области малых молекулярных масс в области больших. Обратная тенденция наблюдается при изучении жесткоцепных линейных полимеров, макромолекулы которых являются рыхлыми, протекаемыми для молекул растворителя, что связано с высокими значениями персистентной длины, которая обеспечивается большими эффектами близкодействия. Для таких систем коэффициент bη максимален в области малых ММ (bη ∼ 1.8) и уменьшается при переходе в область больших молекулярных масс с теоретическим пределом при M → ∞ bη = 0.50.Исходя из этих позиций, проанализируем вискозиметрические данные, полученные в бессолевых водных растворах для рядов сополимеров I и II, а также гомополимера в координатах нормированного построения КМХС (рис. 2).
Отметим следующие обстоятельства. Во-первых, система данных для сополимеров располагается тем выше по оси lg[η] по сравнению с гомополимером, чем больше линейная плотность заряда. Во-вторых, наблюдается одинаковый характер изменения кривизны зависимостей, аппроксимирующих точки, соответствующие гомополимеру и сополимеру I, а именно, увеличение наклона при переходе в область больших молекулярных масс. Такое изменение наклона характерно для систем, проявляющих внутриклубковые объемные взаимодействия, т.е. эффекты дальнодействия. Вместе с тем, увеличение наклона для заряженного сополимера значительно больше, чем для незаряженного гомополимера. В-третьих, зависимость, соответствующая системе точек сополимера II, проявляет тенденцию с обратным характером кривизны, который характерен для линейных систем с высокой равновесной жесткостью, т.е. с большим внутримолекулярным близкодействием. В табл. 3 суммированы наклоны участков зависимостей, соответствующие областям малых и больших молекулярных масс сравниваемых систем.
Таблица 3.
Наклоны зависимостей КМХС в разных областях контурной длины гомополимера и сополимеров
| Содержание ГМВА, мол. % | Интервал контурной длины L, нм | bη | Тип внутримолекулярных эффектов |
|---|---|---|---|
| 0 | 10–70 | 0.52 ± 0.07 | объемные |
| 140–1400 | 0.76 ± 0.02 | ||
| 4.4 | 60–220 | 0.6 ± 0.1 | объемные |
| 400–1600 | 1.4 ± 0.1 | ||
| 15.7 | 200–540 | 1.9 ± 0.4 | протекание; большая жесткость цепей |
| 540–1500 | 1.0 ± 0.4 |
Таким образом, в сополимере I расстояние по цепи между двумя ближайшими зарядами равное 2.6 Abare обеспечивает для этих цепей тип зависимости, характерный для эффектов дальнодействия (рис. 2), тогда как в сополимере II соседние заряды находятся на расстоянии меньшем, чем Abare, это по определению означает, что взаимодействия между такими зарядами относятся к классу близкодействия. Все это приводит к значительному возрастанию размеров цепи за счет увеличения суммарной длины статистического сегмента Atot.
Объемные эффекты в слабо заряженных цепях линейных полиэлектролитов
Сравним эффекты дальнодействия в цепях гомополимера и сополимера I. Для этого рассчитаем значения вискозиметрических коэффициентов набухания αη =([η]/[η]θ)1/3 для цепей сополимеров МВАА–ГМВА и гомополимера в бессолевом растворе. Здесь [η] – характеристическая вязкость макромолекулы в бессолевом растворе, [η]θ – характеристическая вязкость макромолекулы при ее невозмущенных размерах. Поскольку в этом подходе полагается, что равновесная жесткость заряженных цепей такая же, как у незаряженного гомолога, то величины [η]θ могут быть оценены из анализа вискозиметрических данных для гомологической серии ПМВА [44].
Для оценки [η]θ используем построение Бурхарда–Штокмайера– Фиксмана [46, 47]:
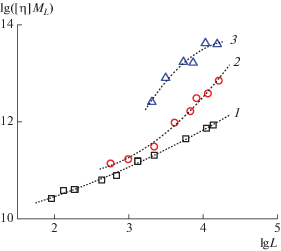
где Kθ – так называемый параметр невозмущенных размеров макромолекулы, Ф0 – гидродинамический параметр Флори, B – параметр термодинамического взаимодействия системы полимер–растворитель. Когда система термодинамически идеальная, то B = 0 и уравнение (8) приобретает видХарактеристическую вязкость в θ-условиях [η]θ рассчитываем на основании данных, полученных для незаряженного гомополимера ПМВАА в бессолевом водном растворе [44] и построения (рис. 3), из которого определяем величину Kθ = = 0.097 ± 0.005. Характеристическую вязкость в θ-условиях [η]θ для сополимеров рассчитываем по соотношению (9), используя Kθ и величины молекулярной массы.
Рис. 3.
Построение Бурхарда–Штокмайера–Фиксмана для незаряженного гомополимера ПМВАА в воде по данным работы [44].
На рис. 4 приведена зависимость коэффициента набухания от молекулярной массы в логарифмическом масштабе для сополимера I в сравнении с гомополимером ПМВАА. Резкое возрастание коэффициента набухания наблюдается для сополимера I при молекулярных массах M > 85 × × 103 г/моль, а для гомополимера при M > 30 × 103. Размеры заряженных цепей не только больше размеров соответствующих незаряженных цепей, но, начиная с некоторой контурной длины цепи, они растут значительно быстрее, чем размеры нейтральных цепей, что отражается в величине соответствующих скейлинговых индексов (рис. 4, кривые 1–4). Это есть проявление изменения наклона зависимости КМХС для гибких линейных макромолекул в термодинамически хороших растворителях. Для коротких макромолекул объемные взаимодействия почти не проявляются.
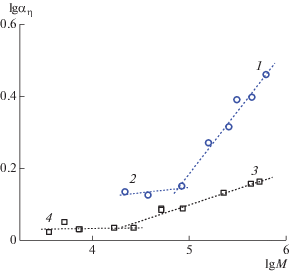
Равновесная жесткость линейных цепей с разной степенью заряда
Количественная оценка длины статистического сегмента была проведена на основе теории Грея–Блюмфельда–Хирста [48]. Получена зависимость коэффициента поступательного трения червеобразного ожерелья от молекулярной массы, учитывающая как эффекты внутримолекулярного протекания, так и эффекты исключенного объема. Эта зависимость может быть представлена в следующем виде:
(10)
$\begin{gathered} \text{[}s]{{P}_{0}}{{N}_{A}}{\text{/}}{{M}_{L}} = {{({{M}^{2}}{{\Phi }_{0}}{\text{/}}[\eta ])}^{{1/3}}}{\text{/}}{{M}_{L}} = \\ = (3{\text{/}}(1--\varepsilon )(3--\varepsilon )){{\left( {L{\text{/}}A} \right)}^{{(1--}}}^{{\varepsilon )/2}} + \\ \, + ({{P}_{0}}{\text{/}}3\pi )[{\text{ln}}(A{\text{/}}d)--(d{\text{/}}3A)--\varphi (\varepsilon )] \\ \end{gathered} $Здесь P0 и Φ0 – гидродинамические параметры Флори (P0 = 5.11, Φ0 = 2.86 × 1023 моль–1); NА – число Авогадро; ε – термодинамический параметр, характеризующий набухание цепей; d – гидродинамический диаметр цепи. Термодинамический параметр ε был рассчитан из соотношения ε = (2bη – 1)/3 [49]; φ(ε) = 1.431 + 2.64ε + 4.71ε2 [50].
Замена переменных (M2Φ0/[η])1/3 = [s]P0NA сделана в предположении, что размеры макромолекул одинаковы в явлениях поступательного и вращательного трения [38, 51].
На рис. 5 представлена зависимость для сополимера I, соответствующая уравнению (10). Из наклона зависимости определена длина статистического сегмента, из отсекаемого отрезка – гидродинамический диаметр. Параметр ε = 0.6 был рассчитан по скейлинговому индексу bη = 1.4 (табл. 3). Для сополимера II и гомополимера оценки A и d проведены аналогичным образом по результатам работ [43, 44]. В табл. 4 показано сравнение полученных оценок длины статистических сегментов сополимеров и гомополимера в растворах в бессолевом водном растворе и в 0.2 моль/л NaCl.
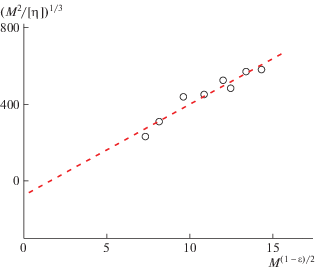
Таблица 4.
Равновесная жесткость и гидродинамический диаметр сополимеров МВАА–ГМВА и гомополимера ПМВАА в водных бессолевых растворах и в 0.2 моль/л NaCl
| Содержание ГМВА, мол. % | ε | Atot, нм | d, нм | ε | Atot, нм | d, нм |
|---|---|---|---|---|---|---|
| H2O | 0.2 моль/л NaCl | |||||
| 0 | 0.16 | 2.2 ± 0.1 | 0.35 ± 0.04 | 0.16 | 2.5 ± 0.2 | 0.3 ± 0.1 |
| 4.4 | 0.6 | 2.5 ± 1.0 | 0.5 ± 0.5 | 0.147 | 2.4 ± 0.1 | 0.4 ± 0.1 |
| 15.7 | 0 | 120 ± 50 | 2.3 ± 0.8 | 0.13 | 2.3 ± 0.2 | 0.6 ± 0.4 |
| 0.4 | 60 ± 40 | 5.0 ± 4.0 | – | – | – | |
Из табл. 4 видно, что оценки длины статистического сегмента, полученные для сополимеров I и II в 0.2 моль/л NaCl вместе с оценками этой величины для незаряженного гомополимера в H2O практически совпадают. При таких ионных силах электростатические взаимодействия не проявляются, и цепи сополимеров принимают такие же размеры, как и незаряженные цепи гомополимера с практически одинаковым уровнем внутриклубковых объемных взаимодействий. На построении КМХС нормированном на ML вискозиметрические данные сополимеров разного состава вместе с исходным гомополимером создают единую зависимость [η]ML = 5.75 × 104(M/ML)0.73 ± 0.02, что и отражает практически одинаковый уровень внутриклубковых объемных взаимодействий в их цепях.
Экспериментальные оценки длин статистического сегмента в бессолевых растворах сопоставим с теоретическими расчетами. Теоретическое соотношение для электростатической составляющей равновесной жесткости цепей, контурная длина которых удовлетворяет соотношению $L{\text{/}}{{\lambda }_{{{\text{DH}}}}} \gg 1$, было получено в теории ОШФ [22–24]. Прежде всего, в изученных рядах сополимеров только по четыре образца удовлетворяют простому условию L/λDH > 1. Учитывая, что радиус экранирования Дебая–Хюккеля в бессолевом растворе равен λDH = 300 нм, были получены следующие оценки электростатической составляющей равновесной жесткости Ael: для сополимера I lqq > λB, используя соотношение (2), Ael ∼ 980 нм. Для сополимера II выполняется условие lqq < λB и в соответствии с соотношением (3) Ael ∼ 13 000 нм. Эти величины на несколько порядков превосходят экспериментальные оценки Abare, а следовательно, и $A_{{el}}^{{exp}}$.
Согласно теории Добрынина [26–28], Ael(l) = = $2{{\lambda }_{{\text{B}}}}\lambda {{\lambda }_{{{\text{DH}}}}}l{\text{/}}l_{{qq}}^{2}(3 + (4\lambda l{\text{/}}{{\lambda }_{{{\text{DH}}}}})$, где l – число рассматриваемых пробных зарядов. Если принять l = 1 (минимальное значение), то для сополимера I значение для электростатической составляющей равновесной жесткости Ael = 1.1 нм, что, учитывая погрешность определения сравниваемых величин, не противоречит экспериментально получаемому результату $A_{{el}}^{{exp}}$ =(Atot – Ahomo) ∼ 0.5 нм.
Для сополимера II, Ael = 58 нм при l = 4 и Ael = = 115 нм при l = 8. Таким образом, для сильно заряженной цепи путем подбора параметра l можно достичь соответствия с экспериментальными оценками величины электростатической составляющей длины сегмента (персистентной длины).
Конформация линейной цепной макромолекулы, заряженной или нейтральной, определяется, прежде всего, длиной статистического сегмента и ее контурной длиной. Полиэлектролитные цепи, сравниваемые в настоящей работе, покрывают существенный интервал значений длины сегмента Куна, известных для линейных цепей. Учет этой ключевой характеристики – длины статистического сегмента – следующий шаг в нормировании соотношений КМХС (скейлингового соотношения) (7). В результате двойной нормировки, учитывается как структурный параметр ML, так и длина статистического сегмента А. Фундаментальное уравнение Флори–Фокса (6) принимает следующий вид:
(11)
$[\eta ]{{M}_{L}}{\text{/}}{{A}^{2}} = \Phi (L{\text{/}}A,d{\text{/}}A,\varepsilon )(L{\text{/}}A),$Зависимость [η]ML/A2 от (L/A) представляет собой геометрическое место точек параметра Флори Φ(L/A, d/A, ε). Эта зависимость была проанализирована в работах [52, 53] на основе теории Ямакава–Фуджии, описывающей поведение величины характеристической вязкости для модели персистентного цилиндра в отсутствие внутрицепных объемных эффектов, а также на основе широкого спектра литературных экспериментальных данных для линейных полимеров разнообразных строений и конформаций. Так, были получены две огибающие кривые (I–0–II) и (III–0–IV), представленные на рис. 6 пунктирными линиями. Ветвь I–0–II характеризует поведение линейных цепей без объемных взаимодействий в интервале величин А = 1–500 нм при небольших значениях поперечного размера цепи d. Полуветвь 0–III описывает цепи в термодинамически хороших растворителях, а полуветвь 0–IV – цепи, у которых поперечный размер d сравним с длиной статистического сегмента. Область справа от точки 0 соответствует цепям с полностью сформировавшимся клубком, а область слева от точки 0 – цепям, в которых полимерный клубок еще не сформировался.
Рис. 6.
Зависимость [η]ML/A2 от (L/A) для гомополимера (1), статистических сополимера I (2, 5) и сополимера II (3, 4, 6) в бессолевых водных растворах (1–4) и 0.2 моль/л NaCl (5, 6). Кривые I–II и III–0–IV описывают поведение массива линейных макромолекул в отсутствие внутрицепных объемных взаимодействий (I–II), в цепях с внутрицепными объемными взаимодействиями (III–0) и (0–IV) – цепи с большим поперечным размером.
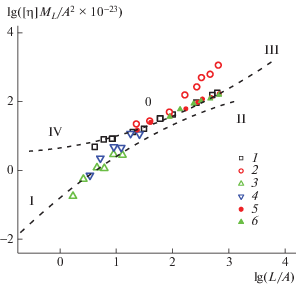
На рис. 6 точки отвечают величинам [η] для сополимеров I и II, а также для нейтрального гомополимера в бессолевых растворах и в растворах NaCl. Следует отметить, что данные, полученные для сополимеров I и II в 0.2 моль/л NaCl вместе с данными для незаряженного гомополимера в H2O, группируются около кривой III–0, характерной для незаряженных цепей с внутриклубковыми объемными взаимодействиями. Это означает, что в макромолекулах сополимеров I и II при ионной силе 0.2 моль/л NaCl практически подавлены полиэлектролитные эффекты. Данные для сополимера I в бессолевых растворах располагаются выше кривой, соответствующей незаряженным цепям с внутрицепными объемными взаимодействиями, что свидетельствует о значительно бóльших эффектах дальнодействия в слабо заряженных цепях. Результаты, полученные для сополимера II, обработаны как с максимальным значением Atot = 120 нм, так и с минимальным значением Atot = 60 нм (табл. 4), и относятся к ветви I–0, описывающей поведение жесткоцепных полимеров, т.е. цепей с большими эффектами близкодействия. В целом, экспериментальные точки удовлетворительно укладываются на соответствующие зависимости, общие для линейных цепей.
ЗАКЛЮЧЕНИЕ
Характер и соотношение электростатических взаимодействий в линейных цепях полиэлектролитов определяется плотностью зарядов в цепи и ионной силой растворов. В наибольшей мере электростатические взаимодействия проявляют себя в бессолевых водных растворах, т.е. при минимальной ионной силе. Для изучения цепей с широким спектром плотности заряда, включая самые малые, необходимо, чтобы основная цепь была гидрофильной. Сополимеры N-метил-N-винилацетамида и N-метил-N-виниламина гидрохлорида являются удобным объектом для таких исследований. Сопоставление характеристической вязкости, определенной в бессолевых водных растворах этих сополимеров с разным средним содержанием заряженных групп, а также с характеристической вязкостью незаряженного гибкоцепного гомополимера, позволяет установить: когда параметр lqq/Abare > 2.5, то в цепях полииона проявляются электростатические эффекты дальнодействия, а когда lqq/Abare < 1.0, то в цепях полииона преобладают электростатические эффекты близкодействия. В области 2.5 > > lqq/Abare > 1.0 в цепях полииона будут проявляться как электростатическое дальнодействие, так и близкодействие. Роль последнего будет возрастать по мере увеличения плотности заряда цепи. Оказывается, что характеристическая вязкость цепей с внутрицепными электростатическими взаимодействиями единообразно интерпретируется с характеристическими вязкостями всего массива линейных цепных структур, независимо от механизма реализации их равновесной жесткости (свободное заторможенное вращение вокруг валентных связей, термодинамические взаимодействия, многоцепные спиральные структуры).
Работа выполнена в рамках Госзадания (№ АААА-А 16-116071450049-0 и АААА-Ф 16-116071450046-9).
Список литературы
Muthukumar M. // Macromolecules. 2017. V. 50. P. 9528.
Dobrynin A.V., Rubinstein M. // Prog. Polym. Sci. 2005. V. 30. № 11. P. 1049.
Dobrynin A.V. // Polymer Science: a Comprehensive Reference / Ed. by K. Matyjaszewski, M. Möller. Amsterdam: Elsevier, 2012. P. 81. V. 1.
Mandel M. // Polyelectrolytes: Science and Technology / Ed. by M. Hara. New York: Marcel Dekker, 1993.
Dautzenberg H., Jaeger W., Koetz J., Philipp B., Seidel C., Stscherbina D. // Polyelectrolytes: Formation, Characterization, and Application. Munich: Hanser-Gardner Publ., 1994. P. 343.
Kabanov V.A., Zezin A.B., Rogacheva V.B., Gulyaeva Zh.G., Zansochova M.F., Joosten J.G.H., Brackman J. // Macromolecules. 1998. V. 31. P. 5142.
Uhlík F., Košovan P., Limpouchová Z., Procházka K., Borisov O.V., Leermakers F.A.M. // Macromolecules. 2014. V. 47. № 12. P. 4004.
Wolterink J.K., Leermakers F.A.M., Fleer G.J., Koopal L.K., Zhulina E.B., Borisov O.V. // Macromolecules. 1999. V. 32. № 7. P. 2365.
Liu L., Chen W., Chen J. // Macromolecules. 2017. V. 50. № 17. P. 6659.
Polyelectrolytes with Defined Molecular Architecture I / Ed. by M. Schmidt. Berlin: Springer, 2004.
Polyelectrolytes with Defined Molecular Architecture II / Ed. by M. Schmidt. Berlin: Springer, 2010.
Изумрудов В.A., Зезин A.Б. // Высокомолек. соед. А. 1976. Т. 18. № 11. С. 2840.
Zezin A.B., Kabanov V.A. // Russ. Chem. Rev. 1982. T. 51. № 9. P. 833.
Izumrudov V.A., Zezin A.B., Kabanov V.A. // Russ. Chem. Rev. 1991. V. 60. № 7. P. 1570.
Pergushov D.V., Remizova E.V., Feldthusen J., Zezin A.B., Mu1ller A.H.E., Kabanov V.A. // J. Phys. Chem. B. 2003. V. 107. P. 8093.
Penott-Chang E.K., Pergushov D.V., Zezin A.B., Mueller A.H.E. // Langmuir. 2010. V. 26. № 11. P. 7813.
Kuhn W., Kunzle O., Katchalsky A. // Helv. Chim. Acta. 1948. V. 31. № 7. P. 1994.
Каргин В.A., Мирлина С.Я., Антипина A.Д. // Высокомолек. соед. А. 1960. Т. 1. № 3. С. 584.
Förster S., Schmidt M. // Adv. Polym. Sci. 1995. V. 120. P. 51.
Beer M., Schmidt M., Muthukumar M. // Macromolecules. 1997. V. 30. P. 8375.
Птицын О.Б. // Высокомолек. соед. А. 1962. Т. 3. № 1. С. 197.
Odijk T. // J. Polym. Sci., Polym. Phys. 1977. V. 15. № 3. P. 477.
Odijk T., Houwaart A.C. // J. Polym. Sci., Polym. Phys. 1978. V. 16. № 4. P. 627.
Skolnick J., Fixman M. // Macromolecules. 1977. V. 10. № 5. P. 944.
Barrat J.L., Joanny J.F. // Europhys. Lett. 1993. V. 24. № 5. P. 333.
Dobrynin A.V. // Macromolecules. 2005 V. 38. № 22. P. 9304.
Gubarev A.S., Carrillo J.M.Y., Dobrynin A.V. // Macromolecules. 2009. V. 42. № 15. P. 5851.
Carrillo J.-M.Y., Dobrynin A.V. // Macromolecules. 2011. V. 44 № 14. P. 5798.
Everaers R., Milchev A., Yamakov V. // Eur. Phys. J. E. 2002. V. 8. № 1. P. 3.
Ullner M., Woodward C.E. // Macromolecules. 2002. V. 35. № 4. P. 1437.
Ulner M. // J. Phys. Chem. B. 2003. V. 107. № 32. P. 8097.
Pavlov G.M., Okatova O.V., Ul’yanova N.N., Gavrilova I.I., Panarin E.F. // Dokl. Chem. 2014. V. 454. № 13. P. 3.
Pavlov G.M., Dommes O.A., Gosteva A.A., Okatova O.V., Gavrilova I.I., Panarin E.F. // Polymer Science A. 2018. V. 60. № 2. P. 172.
Pavlov G.M., Dommes O.A., Okatova O.V., Gavrilova I.I., Panarin E.F. // Phys. Chem. Chem. Phys. 2018. V. 20. P. 9975.
Gavrilova I.I., Panarin E.F., Nesterova N.A. // Russ. J. Appl. Chem. 2012. V. 85. № 3. P. 413.
Pavlov G.M., Perevyazko I.Y., Okatova O.V., Schubert U.S. // Methods. 2011. V. 54. № 1. P. 124.
Pavlov G.M. // Analytical Ultracentrifugation / Ed. by S. Uchiyama, F. Arisaka, W.F. Stafford, T. Laue. Japan: Springer, 2016. P. 269.
Pavlov G.M., Panarin E.F., Korneeva E.V., Kurochkin C.V., Baikov V.E., Ushakova V.N. // Makromol. Chem. 1990. V. 191. № 12. P. 2889.
Pavlov G.M., Okatova O.V., Gubarev A.S., Gavrilova I.I., Panarin E.F. // Macromolecules. 2014. V. 47. № 8. P. 2748.
Pavlov G.M., Gubarev A.S., Zaitseva I.I., Sibileva M.A. // J. Appl. Chem. 2006. V. 79. P. 1423.
Staudinger H. // Nobel Lecture (1953): Macromolecular Chemistry, in Nobel Lectures, Chemistry 1942–1962. Amsterdam: Elsevier, 1964. P. 397.
Kraemer E.O. // Ind. Eng. Chem. 1938. V. 30. № 10. P. 1200.
Dommes O.A., Okatova O.V., Kostina A.A., Gavrilova I.I., Panarrin E.F., Pavlov G.M. // Polymer Science C. 2017. V. 59. № 1. P. 125.
Pavlov G.M., Okatova O.V., Mikhailova A.V., Ulyanova N.N., Gavrilova I.I., Panarin E.F. // Macromol. Biosci. 2010. V. 10. P. 790.
Flory P.J. Principles of Polymer Chemistry. New York: Cornell Univ. Press, 1953.
Burchard W. // Makromol. Chem. 1961. V. 50. P. 20.
Stockmayer W.H., Fixman M. // J. Polym. Sci. C. 1963. V. 1. P. 137.
Gray G., Bloomfield V., Hearst J. // J. Chem. Phys. 1967. V. 46. № 4. P. 1493.
Ptitsyn O.B., Eizner Y.E. // J. Tekh. Fiz. 1959. V. 29. № 9. P. 1105.
Pavlov G.M. // Analytical Ultracentrifugation VI / Ed. by W. Borchard, A. Straatmann. Berlin: Springer, 2002. P. 149. V. 119.
Павлов Г.M., Селюнин С.Г., Шилдяева Н.A., Якопсон С.M., Эфрос Л.С. // Высокомолек. соед. А. 1985. Т. 27. № 8. С. 1823.
Pavlov G.M. // Polymer Science A. 2005. V. 47. № 10. P. 1129.
Pavlov G.M. // Eur. Phys. J. E. 2007. V. 22. P. 171.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия А)