

Высокомолекулярные соединения (серия А), 2020, T. 62, № 1, стр. 46-56
МНОГОСЛОЙНЫЕ ЧАСТИЦЫ НА ОСНОВЕ БИОПОЛИЭЛЕКТРОЛИТОВ КАК ПОТЕНЦИАЛЬНЫЕ СИСТЕМЫ ДОСТАВКИ ПЕПТИДОВ
Н. Н. Зашихина a, Д. В. Юдин b, И. И. Тарасенко a, О. М. Осипова b, Е. Г. Коржикова-Влах a, b, *
a Институт высокомолекулярных соединений Российской академии наук
199004 Санкт-Петербург, Большой пр. 31, Россия
b Санкт-Петербургский государственный университет Институт химии
198504 Санкт-Петербург, Петергоф, Университетский пр., 26, Россия
* E-mail: vlakh@mail.ru
Поступила в редакцию 07.06.2019
После доработки 10.07.2019
Принята к публикации 30.07.2019
Аннотация
Получена серия амфифильных заряженных сополимеров аминокислот и проведено изучение их связывания с катионными и анионными пептидами. Образующиеся интерполиэлектролитные комплексы дополнительно стабилизированы катионными/анионными полисахаридами за счет поверхностного наслаивания. Показано положительное влияние покрытий на скорость высвобождения пептидов, снижение цитотоксичности и повышение устойчивости полученных систем доставки пептидов в биологических средах.
ВВЕДЕНИЕ
В настоящее время широкий спектр пептидных лекарственных субстанций одобрен для клинического применения [1, 2]. Существенным недостатком пептидных лекарств является их быстрая инактивация в кровотоке при парентеральном введении и еще более быстрое разрушение в желудочно-кишечном тракте при пероральном приеме [3]. В связи с этим терапевтический эффект достигается, как правило, за счет увеличения частоты приема или повышения дозировки лекарства. Учитывая то, что большинство пептидных препаратов показано при хронических состояниях и требуют ежедневного применения во время длительного лечения, увеличение частоты приема или дозировки препарата сопряжено с очевидными недостатками. Так, например, для С-пептида, имеющего высокий потенциал для терапии осложнений сахарного диабета, период полураспада в плазме составляет около 30 мин [4]. Для поддержания необходимой терапевтической концентрации данного пептида в плазме требуется внутривенное введение препарата несколько раз в день, что в свою очередь увеличивает нагрузку как на пациента, так и на медицинский персонал. В случае применения пептидных антибиотиков группы полимиксинов их быстрое выведение из организма приводит к необходимости использования высоких доз, что неизбежно вызывает проявление гепато- и нефротоксического действия препарата [5]. Таким образом, повышение эффективности терапии за счет создания более устойчивых лекарственных форм является одной из актуальных междисциплинарных задач.
Один из хорошо известных подходов для повышения стабильности пептидов – получение их конъюгатов с полимерами, например с ПЭГ [3, 6]. Так, в экспериментах на животных было показано, что применение конъюгата С-пептид–ПЭГ позволило сократить частоту приема препарата с двух раз в день до двух раз в неделю и при этом повысить эффективность терапевтического действия [7].
Другим подходом к повышению эффективности терапии с использованием пептидных лекарств является создание полимерных систем доставки [8–10]. Наиболее изученный объект пептидной природы, используемый для получения инкапсулированных форм, – инсулин [11–13]. В литературе есть сведения об использовании полимерных частиц различной природы для создания систем доставки инсулина, в частности микросфер на основе сополимера Eudragit S-100 [11], нано- и микрочастиц хитозан–пектин [14], полимерсом на основе декстран–блок-поли(молочная кислота–со–гликолевая кислота) [15] и т.д. Эффективность загрузки инсулина в зависимости от типа частиц лежит в пределах 32–90%. Исследование высвобождения в модельных физиологических условиях (PBS, pH 7.4) показало профили быстрого высвобождения инсулина из таких частиц. Например, практически 100%-ное высвобождение инсулина из частиц Eudragit S-100 достигалось в течение 8 ч [11]. В свою очередь для высвобождения инсулина из полимерсом декстран–блок–поли(молочная кислота–со–гликолевая кислота) требовалось около 9 ч, чтобы достичь уровня 70–85% в зависимости от состава полимера [15]. Известны также работы по созданию систем доставки полимиксинов. В частности, в литературе сообщается о получении ионных комплексов альгинат–полимиксин В, загруженных в липидные наночастицы [16], создании инкапсулированных форм полимиксина Е с использованием наночастиц на основе поли(молочной кислоты–со–гликолевой кислоты) [17], поли(молочной кислоты) [18] и поли-ε-капролактона [19].
В настоящее время наночастицы, сформированные за счет самосборки амфифильных блок-сополимеров, все чаще изучаются в качестве потенциальных систем доставки лекарств различной природы [20–23]. Один из перспективных классов биосовместимых и биодеградируемых полимеров, широко используемых в качестве одного из блоков амфифильных блок-сополимеров, – полиаминокислоты. В частности, в текущей литературе описано получение амфифильных сополимеров на основе конъюгатов полиаминокислот и полибутадиена [24], ПЭГ [25, 26], полилактида [27] и других [28]. Также известны работы, где были синтезированы амфифильные сополимеры, в которых оба блока являются полиаминокислотами: поли(L-лизин)–блок–поли(L-тирозин) [29], поли(L-аргинин)–блок–поли(L-лейцин) [30], поли(L-лизин)–блок–поли(L-лейцин) [31, 32], поли(аспарагиновая кислота) или поли(глутаминовая ксилота)–блок–полифенилаланин [33, 34], полифенилаланин–блок–полисерин [35], полилизин–блок–поли(аминоизомасляная кислота) [36]. Несмотря на заметное количество работ в области синтеза блок-сополимеров аминокислот, исследования в области создания на их основе систем доставки гидрофильных веществ в целом, и пептидов в частности, практически отсутствуют.
В отличие от большинства публикаций, посвященных созданию систем доставки пептидов, в настоящей работе изучена возможность получения наночастиц на основе амфифильных статистических сополимеров α-аминокислот, загруженных катионными или анионными пептидами и дополнительно стабилизированных катионными/анионными полисахаридами.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы
γ-бензиловый эфир L-глутаминовой кислоты (Glu(OBzl), ε-карбоксибензил-L-лизин (Lys(Z)), D-фенилаланин (DPhe), трифосген, α-пинен, н-гексиламин, трифторуксусная кислота, трифторметансульфокислота приобретены в компании “Sigma-Aldrich” (Германия) и использовались без дополнительной очистки. C-пептид продукт компании “Bachem” (Швейцария), пептид U7 был любезно предоставлен А.Д. Вилесовым (ИВС РАН), полимиксин В был приобретен в компании “Sigma-Aldrich” (Германия). Низкомолекулярные полисахариды (гепарин, хитозан и альгинат) были приобретены в фирмах “AppliChem” (Германия) и “AlfaAesar” (США).
Все растворители для синтеза и выделения мономеров и сополимеров приобретали в компании “Вектон” (Россия) и очищали перегонкой по стандартным протоколам. Все соли для буферных растворов также были приобретены в компании “Вектон” и были аналитической степени чистоты. Буферные растворы готовили растворением соответствующих компонентов в бидистиллированной воде и дополнительно фильтровали через мембранные фильтры “Millipore” (“Merck”, Германия) с диаметром пор 0.45 мкм. Мембранные пробирки для ультрафильтрации Amicon (MWCO 3000 и 10 000) были продуктами компании “Merck” (Германия), а диализные мешки Spectra/Pore® (MWCO 1000) – компании “Spectra” (США).
Методы
Синтез мономеров и сополимеров. Мономеры N-карбоксиангидриды (КA) соответствующих α-аминокислот синтезировали как описано в работе [37] с использованием диоксана для синтеза КA Lys(Z) и ТГФ для синтеза КA Glu(OBzl) и N-КA DPhe. Очистку проводили перекристаллизацией из системы этилацетат–н-гексан. Выход КA Lys(Z) составил 75%, КA Glu(OBzl) – 80%, КA DPhe – 45%. Структуру и чистоту мономеров подтверждали методом спектроскопии ЯМР 1H (“Bruker 400 MHz Avance”) при 25°C в СDCl3: КA Lys(Z): δ 7.43–7.28 (м, 5H), 6.97 (с, 1H), 5.12 (с, 2H), 4.97 (с, 1H), 4.32–4.23 (т, J = 5.2, 1H) (с, 1H), 3.29–3.14 (м, 2H), 2.03–1.90 (м, 1H), 1.90–1.75 (м, 1H), 1.73–1.28 (м, 4H); КA Glu(OBzl): 2.05–2.39 (м, 2H), 2.63 (т, 2H), 4.39 (т, 1H), 5.17 (с, 2H), 6.40 (уш. с., 1H), 7.39 (м, 5H); КA D-Phe: 2.94–3.35 (м, 2H), 4.55 (м, 1H), 6.12 (с, 1H), 7.19–7.41 (м, 5H).
Для синтеза P(Lys(Z)–co–DPhe) и P(Glu(OBzl)–co–DPhe) методом полимеризации с раскрытием цикла в качестве инициатора использовали н-гексиламин. Мольное соотношение КA Glu(OBzl) или КA Lys(Z) к DPhe составляло 80 : 20. Соотношение мономеры : инициатор равно 100 для P(Lys(Z)–co–DPhe) и 50 для P(Glu(OBzl)–co–DPhe). Для синтеза сополимеров готовили 4%-ный раствор мономеров в ТГФ. Полимеризацию проводили при температуре 25°C в течение 24 ч. По окончании заданного времени полимер осаждали диэтиловым эфиром. Состав сополимера P(Glu(OBzl)–co–DPhe) определяли методом спектроскопии ЯМР 1Н в ДМСО-d6 по соотношению интегральной интенсивности I протонов Glu(OBzl) и DPhe в области 6.7–7.4 м.д., используя формулу
(1)
$\frac{{\left[ {{\text{Glu}}\left( {{\text{OBzl}}} \right)} \right]}}{{\left[ {{\text{DPhe}}} \right]}} = \left( {\frac{{I{{{\left( {{\text{Glu}}\left( {{\text{OBzl}}} \right)} \right)}}_{{7.25 - 7.4~\;{\text{м}}{\text{.д}}{\text{.}}}}}}}{{I{{{\left( {{\text{DPhe}}} \right)}}_{{6.7 - 7.25~\;{\text{м}}{\text{.д}}{\text{.}}}}}}}} \right){\text{\;}}$Относительную молекулярную массу сополимеров P(Lys(Z)–co–DPhe) и P(Glu(OBzl)–co–DPhe) оценивали методом эксклюзионной хроматографии, которую проводили с использованием хроматографической системы “Shimadzu LC-20 Prominence” с рефрактометрическим детектированием (Япония). Прибор был оснащен колонкой “Styragel Column HMW6E Waters” размером 7.8 мм × 300 мм, упакованной стационарной фазой с диаметром зерна 15–20 мкм (США). Анализ проводили при температуре 60°C, элюентом служил 0.1 M LiBr в ДМФА. Скорость подвижной фазы 0.3 мл/мин. Значения Mw, Mn и Đ рассчитывали с помощью программы GPC LC Solutions software Shimadzu (Япония) и калибровочной зависимости, построенной по стандартам полиметилметакрилата.
Деблокирование боковых защитных групп сополимеров проводили с использованием смеси 5%-ного раствора трифторметансульфокислоты в трифторуксусной кислоте при температуре 25°C в течение 4 ч. После удаления защитных групп, продукты осаждали избытком диэтилового эфира, осадок трехкратно промывали избытком диэтилового эфира и сушили на воздухе.
Для определения состава сополимера навески сополимера массой 3 мг подвергали полному кислотному гидролизу с 6 M HCl, содержащей 0.0001% фенола, для чего запаянный в ампулу раствор полимера инкубировали в течение 4 суток при температуре 110°С. Растворы кислоты отгоняли на роторным испарителе, остаток растворяли в воде, которую снова отгоняли. Процедуру повторяли несколько раз до достижения раствором нейтрального значения рН. Аминокислотный анализ осуществляли с использованием хроматографической системы “LC-MS-8030” (“Shimadzu”) с тройным квадрупольным масс-спектрометрическим детектированием (Япония). Прибор был оснащен колонкой Luna C18 column, размером 2 мм × 150 мм и упакованной частицами диаметром 5 мкм. Элюентом служила смесь 0.1%-ный ацетонитрил–муравьиная кислота (5 : 95). Скорость подвижной фазы 0.3 мл/мин. Количественное определение концентраций аминокислот проводили с помощью предварительно построенной калибровки в диапазоне концентраций аминокислот 0.005–0.5 мкг/мл.
Получение наночастиц. Для получения наночастиц полимер растворяли в ДМФА и проводили диализ против воды через мембрану с отсекаемой массой 1000 в течение 48 ч. В процессе инверсии органической фазы на водную происходила самосборка амфифильных полимерных макромолекул в наночастицы. После этого водная дисперсия частиц была заморожена и лиофильно высушена. Сухие наночастицы хранили при температуре 4°С. В дальнейшем в зависимости от задачи навеску нужной массы редиспергировали в воде или буферной среде с требуемым рН под кратковременным действием ультразвука (30 с) с использованием УЗ-зонда “UP 50H Hielscher Ultrasonics” (Германия). Средний гидродинамический диаметр и индекс полидисперсности наночастиц измеряли методом динамического рассеяния света (ДРС), а ζ-потенциал – методом электрофоретического рассеяния света с использованием прибора “Malvern Zetasizer Nano-ZS” (Великобритания). Концентрация наночастиц в дисперсионной среде составляла 0.1–0.5 мг/мл. ζ-потенциал измеряли для дисперсий с концентрацией 0.1 мг/мл, приготовленных в дистиллированной воде, содержащей 10–3 моль/л NaCl. рН в диапазоне 3–12 задавался доведением до нужного значения с помощью 0.1 M HCl/NaOH.
Морфологию частиц исследовали методом просвечивающей электронной микроскопии (ПЭМ) с использованием микроскопа “Jeol JEM-2100” (Япония) при ускоряющем напряжении 160 кВ. Образцы наносили на поверхность медных сеток с углеродным напылением и контрастировали 2%-ным раствором уранил ацетата. Средний диаметр наночастиц по микрофотографиям ПЭМ определяли с использованием программы для анализа изображений ImageJ (США).
Покрытие поверхности наночастиц полисахаридами осуществляли, внося дробно водную дисперсию наночастиц с концентрацией 0.5–1.0 мг/мл в раствор полисахарида с рН 6.0 для достижения заданного соотношения наночастицы/полисахарид в пределах от 20 до 1, после чего инкубировали при температуре 25°С в течение 2 ч. Хитозан растворяли в солянокислом растворе с рН 2.0 и затем нейтрализовали до рН 5.5–6.0. Дисперсию частиц анализировали на предмет изменения гидродинамического диаметра и ζ-потенциала.
Инкапсулирование пептидов. 100 мкл раствора пептида с заданной концентрацией в диапазоне 0.1–1.0 мг вносили в дисперсию с концентрацией частиц 0.5–1.0 мг/мл сразу после ее приготовления в 0.01 М натрий-фосфатном буферном растворе, рН 7.4. Смесь инкубировали в течение 30 мин при термостатируемом орбитальном шейкере при температуре 25°С, после чего оставляли на ночь при температуре 4°С. Загрузку пептида (LC) и эффективность инкапсулирования (EE) определяли с использованием следующих уравнений:
где mi – начальная масса пептида (мг); ms – масса пептида, оставшегося в растворе (мг); mNP – масса наночастиц (мг).Массу пептида, оставшегося в растворе, определяли методом ВЭЖХ после отделения супернатанта методом ультрафильтрации через мембраны с отсекаемой массой 10000 для С-пептида и 3000 для U7 и полимиксина В.
Высвобождение пептидов. Кинетику высвобождения пептидов изучали в 0.01 М фосфатно-солевом буферном растворе (pH 7.4), содержащем 0.15 моль/л хлорида натрия при температуре 37°С. Для проведения процесса брали 1.0 мг частиц, загруженных максимальным количеством целевого пептида. Инкубирование частиц выполняли для серии систем в течение 48 ч (0.5, 1, 2, 4, 6, 8, 24 и 48 ч). Одна система соответствовала определенному времени. Анализ супернатанта на появление и накопление целевого пептида проводили троекратно. Супернатант, содержащий высвободившийся пептид, отделяли методом ультрафильтрации, как описано выше.
Количественный анализ пептидов в растворе. Количественное определение пептидов осуществляли методом ионообменной ВЭЖХ с УФ-детектированием (λ = 215 нм) и использованием в качестве стационарной фазы макропористых монолитных дисков (12 мм × 3 мм) фирмы “BIA Separations” (Словения), а именно, CIM DEAE для анализа С-пептида и CIM SO3 для анализа U7 и полимиксина В.
0.01 М фосфатный буферный раствор, pH 7.0 (элюент A) и 0.01 М фосфатный буферный раствор, pH 7.0 с добавлением 1 M NaCl (элюент Б) использовали в качестве подвижных фаз при анализе С-пептида. Скорость потока подвижной фазы составляла 0.5 мл/мин, объем петли ввода пробы – 20 мкл. Анализ проводили в условиях бинарного градиента: 0–2 мин – 100% элюент A, 2–17 мин – от 0 до 100% элюент Б, 17–20 мин – 100% элюент А. Время удерживания С-пептида 10.4 мин.
Для анализа пептида U7 в качестве подвижной фазы выбрали 0.02 М буферный раствор по Кларку–Лаббсу, рН 2.0, (элюент А) и 0.0125 М боратный буферный раствор, рН 10.5 (элюент Б). Использовали последующую программу элюирования: 0–3 мин – 100% элюент А, 3–12 мин – от 0 до 100% элюент Б, 12–15 мин – 100% элюент А. Прочие условия совпадали с таковыми для анализа С-пептида. Время удерживания пептида U7 в данных условиях 7.6 мин.
Для анализа полимиксина В в качестве элюентов брали следующие растворы: элюент А – 0.005 М натрий фосфатный буфер, pH 7; элюент Б – 2 М NaCl в элюенте А. Анализ шел в условиях бинарного градиента: 0–2 мин – 100% А, 2–7 мин – от 0 до 100% Б, 7–12 мин – 100% Б. Скорость подвижной фазы 1 мл/мин. Другие условия совпадали с таковыми для анализа С-пептида. Время элюирования полимиксина B 7.8 мин.
Концентрацию пептидов определяли по калибровочному графику, построенному предварительно с использованием стандартных растворов пептидов в диапазоне концентраций 0.05–1.00 мг/мл для С-пептида и пептида U7, и в диапазоне 0.05–4.00 мг/мл для полимиксина В.
Анализ цитотоксичности наночастиц. Анализ цитотоксичности проводили с помощью СТВ-теста. Образование флуоресцентного продукта, пропорционального количеству жизнеспособных клеток, контролировали флуориметрически (λпогл = 544, λфл = 590 нм). В каждую лунку 96-луночного планшета высевали по 8 × 103 клеток в 100 мкл культуральной среды и культивировали в течение 24 ч. Затем культуральную среду удаляли с помощью стеклянной стерильной пипетки Пастера и добавляли 200 мкл культуральной среды, содержащей наночастицы в диапазоне концентраций 0.004–1.000 мг/мл. Клетки инкубировали в CO2-инкубаторе в течение 72 ч при 37°С и по окончании инкубационного периода среду удаляли и вносили в каждую лунку по 100 мкл раствора СТВ в базальной среде (соотношение объемов 1 : 10). Клетки инкубировали еще 2 ч при 37°С и измеряли флуоресценцию раствора. Данные нормировали в процентах по отношению к контролю, т.е. лункам, содержащим клетки, инкубированные без тестируемых веществ. Для каждой концентрации анализ проводили в четырех повторностях.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Пептиды
Для изучения особенностей включения пептидов в наночастицы на основе амфифильных сополимеров α-аминокислот, содержащих заряженный гидрофильный фрагмент, были выбраны анионный линейный пептид (С-пептид) и катионные линейный (U7) и циклический (полимиксин В) пептиды (табл. 1). Как упоминалось во Введении, С-пептид имеет значение для лечения осложнений диабета. Полимиксины представляют собой группу эффективных антибиотиков, которые на сегодняшний день используются в комплексной терапии осложненных бактериальных инфекций, устойчивых к монотерапии другими антибактериальными средствами. U7 был взят как модельный линейный катионный пептид.
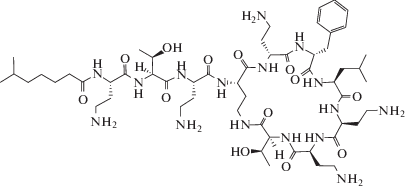
Синтез амфифильных сополимеров α-аминокислот и получение наночастиц на их основе
Амфифильная природа сополимеров достигалась за счет использования гидрофильной аминокислоты лизина или глутаминовой кислоты и гидрофобного фенилаланина. Лизин был выбран в качестве функциональной аминокислоты для связывания анионного С-пептида, в то время как для связывания катионных пептидов применяли сополимер, содержащий глутаминовую кислоту.
Синтез сополимеров осуществляли методом полимеризации с раскрытием цикла N-карбоксиангидридов соответствующих аминокислот. Для получения KA лизина и глутаминовой кислоты использовали защищенные производные аминокислот, а именно, Lys(Z) и Glu(OBzl). KA синтезировали непосредственно перед полимеризацией, как описано в Экспериментальной части. Инициирование проводили с помощью первичного амина – н-гексиламина. После окончания полимеризации полученные сополимеры имели гидрофобную природу и приобретали амфифильные свойства после удаления защитных групп. Структура сополимеров приведена ниже.

Относительную молекулярную массу и дисперсность сополимеров определяли методом ЭЖХ (табл. 2). Для P(Glu(OBzl)–co–DPhe) состав сополимеров находили по данным спектроскопии ЯМР 1H (табл. 2), тогда как для сополимеров на основе P(Lys(Z)–co–DPhe) расчет степени полимеризации по спектрам ЯМР 1H оказался невозможен вследствие уширения и наложения реперных сигналов. Состав обоих сополимеров был определен по данным аминокислотного ВЭЖХ-МС анализа после полного кислотного гидролиза образцов сополимеров до свободных аминокислот. Характеристики полученных сополимеров приведены в табл. 2. На примере сополимера P(Glu–co–DPhe) видно, что соотношение аминокислот, рассчитанное по данным ЯМР 1H и аминокислотного ВЭЖХ-анализа, было сопоставимо.
Таблица 2.
Характеристики полученных амфифильных сополимеров
| Сополимер | Выход, % | ЭЖХ | ЯМР 1H | ВЭЖХ | |||||
|---|---|---|---|---|---|---|---|---|---|
| Mw × 10–3 | Ɖ | [Glu(OBzl)] : [Phe] | n | m | ωPhe, % | [Lys/Glu] : [Phe] | ωPhe, % | ||
| P(Lysn–co–DPhem) | 65 | 24.3 | 1.13 | – | – | – | – | 4.3 | 21 |
| P(Glun–co–DPhem) | 70 | 8.1 | 1.20 | 3.0 | 37 | 12 | 24 | 2.8 | 29 |
Частицы на основе синтезированных сополимеров приготавливали методом инверсии органической (диметилформамид) фазы на водную (0.01 М натрий-фосфатный буферный раствор, рН 7.4). Согласно микрофотографиям, полученным методом ПЭМ, полимерные частицы на основе сополимеров P(Lys–co–DPhe) и P(Glu–co–DPhe) имели сферическую форму и размер в сухом состоянии около 55±13 и 67±25 нм соответственно (рис. 1).
Рис. 1.
Микрофотографии ПЭМ, полученные для частиц на основе P(Lys–co–DPhe) (a) и P(Glu–co–DPhe) (б).

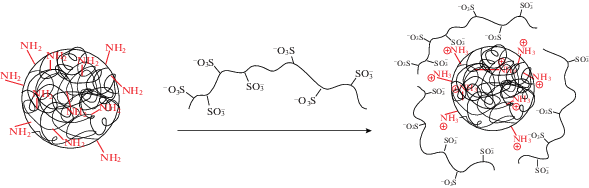
Гидродинамический диаметр DH и дисперсность Ð частиц, полученных в 0.01 М натрий-фосфатном буферном растворе, определяли методом ДРС. Характеристики полученных частиц представлены в табл. 3.
Расхождение в размерах частиц в сухом состоянии (ПЭМ) и в водной среде (ДРС) связано с сольватацией гидрофильных фрагментов и электростатическим отталкиванием одноименно заряженных групп в водной среде, что приводит к экспонированию гидрофильных фрагментов в водную фазу и, таким образом, к увеличению диаметра частиц. В свою очередь высушивание образца на поверхности сетки перед регистрацией изображения методом ПЭМ вызывает дегидратацию сольватной оболочки и колапсирование полимерных цепей, и, как следствие, уменьшение диаметра частиц [36, 38].
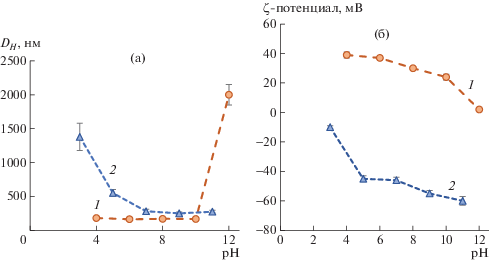
Изучение зависимости гидродинамического диаметра от рН среды, в которой проводится редиспергирование частиц, показало независимость данного параметра в диапазоне рН от 5 до 11 для анионных и от 4 до 10 для катионных частиц (рис. 2). Резкое увеличение DН наблюдалось при значениях pH ниже 4 и выше 11, что связано с протонированием γ-карбоксильных групп глутаминовой кислоты или соответственно депротонированием ε-аминогрупп лизина.
Рис. 2.
Зависимость гидродинамического диаметра (a) и ζ-потенциала (б) частиц на основе сополимеров P(Lys–co–DPhe) (1) и P(Glu–co–DPhe) (2) от pH раствора, используемого при формировании частиц.
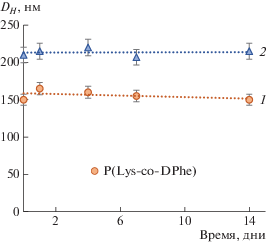
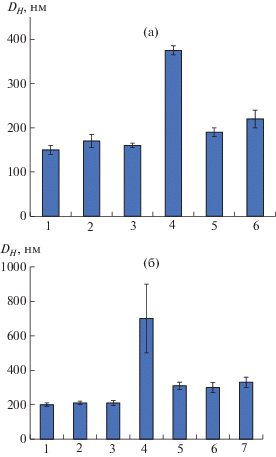
Наличие высокого абсолютного значения ζ-потенциала (более 30 мВ), наряду с относительно небольшим размером частиц позволяет предполагать высокую стабильность получаемых коллоидных систем. Действительно, дисперсии наночастиц были стабильны в модельной буферной среде как минимум в течение двух недель (рис. 3), что выражалось в сохранении гидродинамического диаметра частиц, отсутствии агрегации и седиментации частиц.
Рис. 3.
Мониторинг гидродинамического диаметра частиц методом ДРС в течение 14 дней в фосфатно-солевом буферном растворе (37°С). 1 – P(Lysn–co–DPhem), 2 – P(Glun–co–DPhem).
Инкапсулирование пептидов. Основными силами, обеспечивающими взаимодействие между выбранными сополимером и пептидом, являются ионные и гидрофобные взаимодействия. В первом случае, распределение пептида будет иметь место как во внутреннем объеме частицы, так и на ее поверхности. В то же время, гидрофобные фрагменты, локализующиеся внутри частицы, будут удерживать пептид преимущественно внутри нее. Данные по загрузке выбранных пептидов, а также характеристики загруженных частиц приведены в табл. 4. Видно, что все пептиды включались в частицы с достаточно высокой эффективностью (76–91%) и высокой общей загрузкой (380–860 мкг/мг частиц).
Таблица 4.
Характеристики частиц, загруженных пептидами
| Пептид | Сополимер | Загрузка пептида, мкг/мг частиц | EE, % | ||
|---|---|---|---|---|---|
| LC1 (ионные взаимодействия) | LC2 (гидрофобн. взаимодействия) | LCmax* | |||
| С-пептид | P(Lys–co–DPhe) | 630 ± 30 | 232 ± 20 | 860 ± 20 | 86 |
| U7 | P(Glu–co–DPhe) | 105 ± 10 | 275 ± 20 | 380 ± 15 | 75 |
| Полимиксин В | P(Glu–co–DPhe) | 165 ± 10 | 260 ± 15 | 425 ± 20 | 91 |
Для оценки вклада ионных взаимодействий применяли метод анион- или катион-обменной ВЭЖХ. Использование DEAE- или SO3-содержащих макропористых монолитных сорбентов позволило за счет ионного обмена со стационарной фазой высвободить из частиц пептид, локализованный вблизи поверхности, и проанализировать его количество. При этом пептид, находящийся в более глубоких слоях частицы, и наиболее вероятно связанный за счет гидрофобных взаимодействий, не высвобождался. Для олигоаниона (С-пептид) загрузка в большей степени достигалась за счет ионных взаимодействий с поликатионным фрагментом амфифильного сополимера (LC1 > LC2) (табл. 4). Однако, как и предполагалось, некоторая часть пептидов удерживалась за счет гидрофобных взаимодействий. В отличие от С-пептида, для пептида U7 и полимиксина В доля вещества, удерживаемого за счет гидрофобных взаимодействий была выше, чем доля пептида, связанного за счет ионных взаимодействий (LC2 > LC1). Важно, что масса целевого пептида, удерживаемого в наночастице за счет гидрофобных взаимодействий, практически не зависела от типа пептида и сополимера (232–275 мкг/мг наночастиц), при том что общая загрузка отличалась существенно. Данный факт может косвенно указывать на идентичность структуры полученных наночастиц, равно как и на схожесть процессов, лежащих в основе инкапсулирования заряженных амфифильных пептидов.
Измерение гидродинамического диаметра наночастиц, дисперсности Ð и ζ-потенциала загруженных частиц показало увеличение величины DH приблизительно на 10–15%, независимость Ð и снижение абсолютного значения ζ-потенциала на 8 ± 1 мВ для U7, 15 ± 1 мВ полимиксина В и 20 ± 2 мВ для С-пептида относительно исходных характеристик частиц.
Изучение цитотоксичности полученных частиц
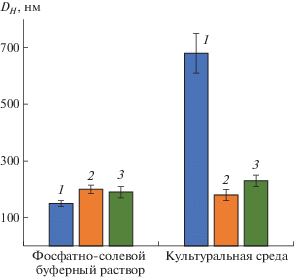
Перед тестированием полученных частиц на клеточных культурах была изучена их стабильность в культуральной среде, содержащей эмбриональную бычью сыворотку (рис. 4). Указанная культуральная среда в дальнейшем использовалась для проведения экспериментов по изучению биологических свойств полученных частиц in vitro. Установлено, что гидродинамический диаметр анионных частиц на основе P(Glu–со–DPhe) менялся незначительно. Напротив, частицы, имеющие положительный заряд поверхности, агрегировали в культуральной среде.
Рис. 4.
Изменение гидродинамического диаметра частиц в течение 24 ч в фосфатно-солевом буферном растворе и культуральной среде (37°С). 1 – P(Lys–co–DPhe), 2 – P(Glu–co–DPhe), 3 – P(Lys–co–DPhe)/Гепарин.
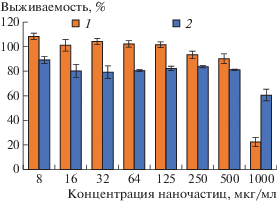
Изучение цитотоксичности наночастиц на основе P(Glu–со–DPhe) и P(Lys–co–DPhe) с использованием двух клеточных линий показало отсутствие цитотоксического эффекта во всем диапазоне тестируемых концентраций для анионных наночастиц, стабильных в кульутральной среде, и, его наличие уже при концентрациях 32–64 мкг/мл для катионных наночастиц, агрегирующих в кульутральной среде (рис. 5). В данном случае токсический эффект обусловлен как катионной природой частиц, так и их агрегацией в клеточной среде [39].

Модификация поверхности частиц
Модификация поверхности наночастиц за счет дополнительного покрытия биосовместимыми полимерами позволяет снизить цитотоксичность и/или повысить их устойчивость в биологических средах. С точки зрения размера, устойчивости к ферментативной деградации и эффективности загрузки анионного С-пептида наночастицы на основе P(Lys–co–DPhe) перспективны для парентерального введения лекарственных форм. Однако существенным ограничением наночастиц данного типа является их цитотоксичность. Для снижения цитотоксичности наночастиц на основе P(Lys–co–DPhe) была изучена возможность поверхностного наслаивания отрицательно заряженного биодеградируемого полисахарида гепарина:
Увеличение концентрации гепарина в растворе приводило к уменьшению ζ-потенциала и нелинейному изменению DH частиц (рис. 6а). При массовом соотношении наночастицы : гепарин, равном двум, наблюдалось падение ζ-потенциала практически до нуля, что приводило к увеличению гидродинамического диаметра наночастиц. При таком соотношении наночастицы : гепарин мольное соотношение звеньев [Lys] : [глюкорунат-2-сульфат/N-ацетилглюкозамин-6-сульфат] было равно 3.1. При массовом соотношении наночастицы : гепарин, равном единице, гидродинамический диаметр частиц не изменялся, что связано со сменой знака заряда и отталкиванием отрицательно заряженных частиц друг от друга (рис. 6а).
Рис. 6.
Зависимость гидродинамического диаметра и ζ-потенциала наночастиц от массового соотношения P(Lys–co–DPhe)/гепарин (a) и P(Glu–co–DPhe)/хитозан (б). а: P(Lys–co–DPhe)–С-пептид : гепарин = = 2 (1), P(Lys–co–DPhe) : гепарин = 1 (2), 2 (3), 5 (4), 10 (5), 6 – P(Lys–co–DPhe); ζ-потенциал = –10 (1), – 34 (2), 2 (3), 8 (4), 15 (5), 40 (6); б: P(Glu–co–DPhe)–U7 : хитозан = 2 (1), P(Glu–co–DPhe) : хитозан = = 1 (2), 2 (3), 3 (4), 5 (5), 10 (6), 7 – P(Glu–co–DPhe).
С учетом двукратного снижения ζ-потенциала поверхности частиц после инкапсулирования C-пептида для их покрытия гепарином использовали соотношение наночастицы : гепарин, равное двум. Покрытие поверхности частиц гепарином за счет его интерполимерных взаимодействий с лизинсодержащим сополимером привело к частичному вытеснению C-пептида с поверхности. Доля C-пептида, перешедшего в раствор, составила 30 ± 3% от общего количества С-пептида, загруженного в частицы. При этом гидродинамический размер составил 220 ± 20, а ζ-потенциал снизился до –10 ± 1 мВ.
В экспериментах по изучению стабильности наночастиц в культуральной среде было подтверждено (рис. 4), что покрытие поверхности катионных частиц гепарином способствовало повышению их стабильности в данной среде. В свою очередь тестирование полученных наночастиц на культурах клеток выявило исчезновение цитотоксического эффекта (рис. 7).
Рис. 7.
Выживаемость клеток HEK-293 (эмбриональные клетки почек человека) (1) и NIH-3T3 (эмбриональные фибробласты мыши) (72 ч) (2) в присутствии наночастиц на основе P(Lys–co–DPhe), покрытых гепарином.
На примере наночастиц на основе P(Glu–co–DPhe) была изучена возможность создания многослойных систем, перспективных в качестве пероральных систем доставки пептидов. Для этого наночастицы покрывали сначала хитозаном, а затем альгинатом натрия. Такое многослойное покрытие рН-чувствительными полисахаридами может замедлять высвобождение пептида в сильно кислой среде желудка и вместе с тем интенсифицировать данный процесс в слабо кислой и слабо щелочной среде кишечника.
При покрытии наночастиц хитозаном смена знака ζ-потенциала происходила при массовом соотношении наночастицы : хитозан, равное двум, а при соотношении, равном трем, заряд поверхности был равен нулю (рис. 6б). Также, при таком массовом соотношении мольное соотношение звеньев [Glu] : [глюкозамин] было равно 3.2. Как в случае покрытия наночастиц на основе P(Lys-co-DPhe) гепарином, так и в случае покрытия наночастиц на основе P(Glu–co–DPhe) хитозаном изоэлектрическая точка достигалась при соотношении мономерных звеньев [аминокислота] : [моносахарид] ≈ 3. Таким образом, можно заключить, что на поверхности наночастиц для взаимодействия доступно около 30% звеньев заряженной аминокислоты. Кроме того, полученный результат также указывает на идентичность структуры частиц, формируемых на основе P(Lys–co–DPhe) и P(Glu–co–DPhe).
Перезарядка поверхности при покрытии наночастиц хитозаном наблюдалась при соотношении [Glu] : [глюкозамин] = 2.2 (массовое соотношение наночастицы : хитозан, равное двум). Как и в случае С-пептида, покрытие хитозаном наночастиц на основе P(Glu–co–DPhe), содержащих инкапсулированный пептид U7, способствовало частичному вытеснению поверхностно-связанного пептида. Доля пептида, перешедшего в раствор, составила 8 ± 2% от общего количества загруженного пептида, что также косвенно подтверждает преимущественное включение данного пептида по гидрофобному механизму. Гидродинамический диаметр этих систем равен 330 ± 30 нм, а ζ-потенциал 31 ± 1 мВ.
Для нанесения слоя альгината на поверхность наночастиц, покрытых хитозаном, было выбрано массовое соотношение хитозан : альгинат натрия = 1.5, что соответствовало мольному соотношению мономерных звеньев данных полисахаридов 1.4. Покрытие наночастиц альгинатом приводило к уменьшению гидродинамического диаметра наночастиц до 230 ± 20 нм, что является следствием уплотнения полимерных слоев за счет внутренней компенсации зарядов. Перезарядка поверхности наночастиц подтверждалась изменением ζ-потенциала с 46 ± 2 мВ на –41 ± 1 мВ.
Высвобождение пептидов
В табл. 5 суммированы данные по высвобождению пептидов в течение 24 ч при температуре 37°С из непокрытых и дополнительно покрытых полисахаридами наночастиц. Сравнение полученных данных позволяет заключить, что в обоих случаях дополнительное наслаивание противоположно заряженных полимеров на поверхности наночастиц способствует снижению количества высвобождаемого пептида. Так, для С-пептида данное снижение составляло 26%, а для пептида U7 – 10%.
Таблица 5.
Влияние покрытия наночастиц полисахаридами на высвобождение пептидов в фосфатно-солевом буферном растворе в течение 24 ч при 37°С
| Пептид | Наночастицы | Покрытие | Высвобождение пептида, % | |
|---|---|---|---|---|
| без покрытия | с покрытием | |||
| С-пептид | P(Lys–co–DPhe) | гепарин | 58 ± 3 | 32 ± 2 |
| U7 | P(Glu–co–DPhe) | хитозан + альгинат | 26 ± 1 | 15 ± 1 |
ЗАКЛЮЧЕНИЕ
В работе показана эффективность использования наночастиц на основе катионных и анионных сополимеров аминокислот для инкапсулирования анионных и катионных пептидов. При мольном соотношении заряженных звеньев [аминокислота] : [моносахарид] ≈ 3 электрокинетический потенциал поверхности снижается до нуля и наблюдается агрегация частиц. Несмотря на некоторое увеличение размера полимерных частиц и частичное вытеснение поверхностно связанного пептида, покрытие систем полисахаридами обеспечивает снижение цитотоксичности катионных частиц, а также количества высвобождаемого пептида. Таким образом, получение многослойных полимерных частиц может оказаться весьма перспективным для создания систем доставки пептидов.
Работа выполнена при финансовой поддержке Российского научного фонда (проект 19-73-20157).
Список литературы
Niu Z., Conejos-Sánchez I., Griffin B.T., O’Driscoll C.M., Alonso M.J. // Adv. Drug Deliv. Rev. 2016. V. 106. P. 337.
Gupta R., Mohanty S. // Colloids Surf. B. 2017. V. 154. P. 48.
Bumbaca B., Li Z., Shah D.K. // Drug Metab. Pharmacokinet. 2019. V. 34. P. 42.
Faber O.K., Hagen C., Binder C. // J. Clin. Invest. 1978, V. 62. P. 197.
Wanger A., Chavez V., Huang R.S.P., Wahed A., Actor J.K., Dasgupta A. // Microbiol. Molec. Diagnosis Pathology. Amsterdam: Elsevier, 2017.
Zhang X., Wang H., Ma Z., Wu B. // Expert Opin. Drug Metab. Toxicol. 2014. V. 10. P. 1691.
Jolivalt C.G., Rodriguez M., Wahren J., Calcutt N.A. // Diabetes, Obes. Metab. 2016. V. 3. P. 147.
Nemeth C.L., Lykins W.R., Tran H., Elsayed M.E.H., Desai T.A. // Pharm. Res. 2019. V. 36. P. 89.
Kim M.R., Feng T., Zhang Q., Chan H.Y.E., Chau Y. // Polymers. 2019. V. 11. P. 288.
Hetényi G., Griesser J., Moser M., Demarne F., Jannin V., Bernkop-Schnürch A. // Int. J. Pharm. 2017. V. 523. P. 357.
Agrawal G.R., Wakte P., Shelke S. // Prog. Biomater. 2017. V. 6. P. 125.
Bloch K., Vanichkin A., Gil-Ad I., Vardi P., Weizman A. // J. Tissue Eng. Regen. Med. 2017. V. 11. P. 3263.
Saravanan S.S., Malathi M.S., Selvasubramanian S.S., Balasubramanian B.S., Pandiyan P.V. // Int. J. Biol. Macromol. 2017. V. 95. P. 1190.
Maciel V.B.V., Yoshida C.M.P., Pereira S.M.S.S., Goycoolea F.M., Franco T.T. // Molecules. 2017. V. 22. P. 1707.
Alibolandi M., Alabdollah F., Sadeghi F., Mohammadi M., Abnous K., Ramezani M., Hadizadeh F. // J. Control. Release. 2016. V. 227. P. 58.
Severino P., Chaud M.V., Shimojo A., Antonini D., Lancelloti M., Santana M.H.A., Souto E.B. // Colloids Surfaces B. 2015. V. 129. P. 191.
Shah S.R., Henslee A.M., Spicer P.P., Yokota S., Petrichenko S., Allahabadi S., Bennett G.N., Wong M.E., Kasper F.K., Mikos A.G., // Pharm. Res. 2014. V. 31. P. 3379.
Tang C., Zhang E., Li Y., Yang L. // Eur. J. Pharm. Sci. 2017. V. 102. P. 63.
Wang M., Zhou C., Chen J., Xiao Y., Du J. // Bioconjug. Chem. 2015. V. 26. P. 725.
Zhao L., Li N., Wang K., Shi C., Zhang L., Luan Y. // Biomaterials. 2014. V. 35. P. 1284.
Kita-Tokarczyk K., Grumelard J., Haefele T., Meier W. // Polymer. 2005. V. 46. P. 3540.
Pearson S., Vitucci D., Khine Y.Y., Dag A., Lu H., Save M., Billon L., Stenzel M.H. // Eur. Polym. J. 2015. V. 69. P. 616.
Gao H., Tan Y., Guan, Q., Cai T., Liang G., Wu Q. // RSC Adv. 2015. V. 5. P. 49376.
Sigel R., Łosik M., Schlaad H. // Langmuir. 2007. V. 23. P. 7196.
Gao H., Li G., Hu Z., Xiao Z., Liang G., Wu Q. // Polymer. 2014. V. 55. P. 4593.
Osada K., Christie R.J., Kataoka K., J.R. // Soc. Interface. 2009. V. 6. Suppl 3. P. 325.
Chen C., Chen L., Chen H., Li H., Yuan M. // Polym. Bull. 2013. V. 70. P. 1739.
Johnson R.P., John J.V., Kim I. // Eur. Polym. J. 2013. V. 49. P. 2925.
Huang Y.-C., Yang Y.-S., Lai T.-Y., Jan J.-S. // Polymer. 2012. V. 53. P. 913.
Holowka E.P., Pochan D.J., Deming T.J., // J. Am. Chem. Soc. 2005. V. 127. P. 12423.
Liu Z., Zhou F., Wu J., Yao Y., Guo Y., Liao X., Gao F., Qian Y. // J. Electroanal. Chem. 2018. V. 823. P. 253.
Zashikhina N.N., Volokitina M.V., Korzhikov-Vlakh V.A., Tarasenko I.I., Lavrentieva A., Scheper T., Rühl E., Orlova R.V., Tennikova T.B., Korzhikova-Vlakh E.G. // Eur. J. Pharm. Sci. 2017. V. 109. P. 1.
Tang J., Yao J., Shi J., Xiao Q., Zhou J., Chen F. // Pharmazie. 2012. V. 67. P. 756.
Vlakh E., Ananyan A., Zashikhina N., Hubina A., Pogodaev A., Volokitina M., Sharoyko V., Tennikova T. // Polymers. 2016. V. 8. P. 212.
Zhao Z., Wang Y., Han J., Wang K., Yang D., Yang Y., Du Q., Song Y., Yin X. // Int. J. Nanomedicine. 2014. V. 9. P. 5849.
Tarasenko I., Zashikhina N., Guryanov I., Volokitina M., Biond B., Fiorucci S., Formaggi F., Tennikova T., Korzhikova-Vlakh E. // RSC Adv. 2018. V. 8. P. 34603.
Wilder R., Mobashery S. // J. Org. Chem. 1992. V. 57. P. 2755.
Marsden H.R., Gabrielli L., Kros A. // Polym. Chem. 2010. V. 1. P. 1512.
Guo Q., Zhang T., An J., Wu Z., Zhao Y., Dai X., Zhang X., Li C. // Biomacromolecules. 2015. V. 16. P. 3345.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия А)