

Высокомолекулярные соединения (серия А), 2020, T. 62, № 4, стр. 263-270
БИОРАЗЛАГАЕМЫЕ КОМПОЗИЦИИ НА ОСНОВЕ ПОЛИЭФИРОВ ПОЛИ-(3-ГИДРОКСИБУТИРАТА) И ПОЛИЛАКТИДА, ПОЛУЧАЕМЫХ ИЗ РАСТИТЕЛЬНОГО СЫРЬЯ
Л. А. Жорина a, С. З. Роговина a, *, Э. В. Прут a, О. П. Кузнецова a, А. В. Грачев a, Н. Е. Иванушкина b, А. Л. Иорданский a, А. А. Берлин a
a Федеральный исследовательский центр химической физики им. Н.Н. Семенова Российской академии наук
119991 Москва, ул. Косыгина, 4, Россия
b Институт биохимии и физиологии микроорганизмов им. Г.К. Скрябина Российской академии наук
142290 Московская обл., Пущино, пр. Науки, 5, Россия
* E-mail: s.rogovina@mail.ru
Поступила в редакцию 29.08.2019
После доработки 27.01.2020
Принята к публикации 10.02.2020
Аннотация
Методом сдвиговых деформаций при твердофазном смешении термопластичных полиэфиров поли-(3-гидроксибутирата) и полилактида в присутствии пластификатора полиэтиленгликоля получены композиции различного состава. Измерены теплофизические параметры при первичном и вторичном нагревании. Установлено, что исходные полиэфиры являются несмешивающимися полимерами. Рассчитаны степень кристалличности и энтальпия плавления полимеров в индивидуальном состоянии и в смесях. Показано, что при вторичном нагревании происходит изменение температурных переходов и степени кристалличности данных полимеров. Исследованы механические и реологические свойства композиций. Изучение стойкости к воздействию плесневых грибов продемонстрировало, что в отличие от полилактида поли-(3-гидроксибутират) является полностью биодеградируемым полимером, в то время как биоразлагаемость композиций определяется их составом.
ВВЕДЕНИЕ
Cоздание биоразлагаемых полимерных материалов, распадающихся под действием окружающей среды на безвредные для природы вещества, весьма актуально в настоящее время [1]. Такие материалы могут быть получены не только из полимеров природного происхождения или полимеров, синтезируемых из растительного сырья, но также в результате разработки на их основе различных композиций, тем самым позволяя путем подбора компонентов и варьирования состава влиять на их свойства и биоразлагаемость.
Полиэфиры, синтезируемые из растительного сырья химическим – полилактид (ПЛА) или микробиологическим – полигидроксиалканоаты (полигидроксивалериат, поли-(3-гидроксибутират) – ПГБ) методом, представляют собой альтернативу полимерам, синтезируемым из нефтяного сырья. В связи с ростом цены барреля нефти и постепенным исчерпанием нефтяных источников, а также благодаря возможности переработки ПЛА и ПГБ на традиционном полимерном оборудовании, они становятся конкурентоспособными по отношению к крупнотоннажным синтетическим полимерам. Однако эти полиэфиры имеют ряд серьезных недостатков по сравнению с синтетическими полимерами, основными из которых остаются пока еще высокая стоимость, хрупкость, низкие барьерные характеристики, а также термическая неустойчивость при переработке, что сдерживает их широкое практическое применение.
Полилактид – линейный алифатический термопластичный полиэфир, получаемый при полимеризации молочной кислоты, которая образуется при ферментативном брожении возобновляемых продуктов природного происхождения, таких как кукуруза, сахарная свекла, картофель и другие. ПЛА обладает неплохими механическими свойствами [2] и используется для изготовления волокон, пленок, некоторых видов посуды для одноразового использования, включая емкости для холодных напитков, термоформованные лотки и контейнеры с крышками, блистерные упаковки, а также находит применение в хирургии и медицине [3]. Несмотря на то что ПЛА является продуктом, производимым из природного сырья, он подвержен биоразложению лишь в достаточно жестких условиях, а именно при компостировании в почве или под действием морской воды.
Смешение полимеров представляет собой эффективный и экономически выгодный способ создания новых материалов с усовершенствованными свойствами. Поэтому для улучшения эластичности, гибкости и прочности ПЛА были получены и исследованы его смеси с такими синтетическими биоразлагаемыми полимерами, как поликапрoлактон, поливинилбутираль, поливинилфенол, полибутиленсукцинат и полиметилметакрилат [4–9], а также с природными биоразлагаемыми полисахаридами – целлюлозой и крахмалом [10, 11].
Вместе с тем ПГБ, стоимость которого превышает стоимость ПЛА, является полностью биоразлагаемым термопластичным хрупким кристаллическим (степень кристалличности достигает 70%) полимером с высокой температурой плавления. Медленная скорость кристаллизации препятствует его крупномасштабному применению. Однако, именно благодаря биосовместимости и отсутствию токсичности, ПГБ активно используется в биомедицине и фармацевтике, а добавление к нему ПЛА придает этим композициям еще бόльшую биоразлагаемость. Присутствие ПЛА позволяет также снизить стоимость изделий на их основе по сравнению с материалами, получаемыми только из ПГБ. Отличительной особенностью таких композиций является и то, что входящие в них компоненты представляют собой полимеры, вырабатываемые полностью из растительного сырья, а сами композиции проявляют способность к биоразложению [12, 13].
Таким образом, композиции на основе термопластичных полиэфиров ПЛА и ПГБ – альтернатива композициям синтетических полимеров, получаемых из нефти, что обусловливает перспективность их производства.
В основном смешение ПЛА с ПГБ проводят в растворе (хлороформе) [14], что требует использования большого количества растворителя, тем самым делая процесс долгим, дорогим и экономически невыгодным. В то же время, твердофазное смешение этих полиэфиров под действием сдвиговых деформаций является экологически чистым методом формирования на их основе композиционных материалов с требуемыми свойствами, достигаемыми путем варьирования состава смесей.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Объектами исследования служили полилактид марки 4043-D (“Nature Works”, США) с Тпл = 155°С, прозрачность 2.1%; поли-(3-гидроксибутират) (“Biomer”, Германия) с М = 3 × 105, ПЭГ с М = = (0.4, 0.6 и 1.0) × 103 (“SigmaR”, Германия).
Смешение проводили в смесителе “Brabender” (Германия) при 180оС в течение 10 мин.
Для дальнейших исследований на прессе “Carver” (США) прессовали пленки толщиной 0.3 мм при 180°С и давлении 10 МПа в течение 10 мин с последующим охлаждением со скоростью ∼15 град/мин.
Теплофизические характеристики и термическую стабильность исходных полимеров и их смесей изучали методами ДСК и ТГА на калориметре “Netzch”, модель DSC-204 F1 (Германия) при скорости нагревания 10 град/мин в диапазоне температуры 30–200°С. Навески образцов составляли ~10 мг. Точность измерения температуры 0.1°С. Эксперимент включал в себя первичное нагревание, охлаждение и вторичное нагревание с одной и той же скоростью.
Оценку биодеструкции композиций осуществляли двумя методами: проведением микробиологических испытаний композиций различного состава на стойкость к воздействию плесневых грибов и закапыванием в почву с рН около 7 с последующим измерением потери массы образцов через определенные промежутки времени.
Лабораторные испытания образцов на стойкость к воздействию плесневых грибов проводили в соответствии с требованиями ГОСТ 9.049-91 в условиях, обеспечивающих рост грибов только за счет питательных веществ, содержащихся в исследованных материалах.
Сущность метода заключается в выдерживании образцов (пластины 40 × 60 мм), зараженных водной суспензией спор плесневых грибов в оптимальных для их роста условиях (температура 29 ± 2°С, относительная влажность воздуха более 90%). Для осуществления испытаний использовали тест-организмы из фонда Всероссийской коллекции микроорганизмов: Aspergillus brasiliensis Varga et al. 2007 ВКМ F-1119; Aspergillus terreus Thom 1918 ВКМ F-1025; Aspergillus sojae Sakaguchi et K. Yamada ex Murakami 1971 ВКМ F-2096; Chaetomium globosum Kunze 1817 ВКМ F-109; Paecilomyces variotii Bainier 1907 ВКМ F-378; Penicillium chrysogenum Thom 1910 ВКМ F-245; Penicillium aurantiogriseum Dierckx 1901 ВКМ F-265; Penicillium pinophilum Thom 1910 ВКМ F-1115; Trichoderma virens J. Sheldon 1904 ВКМ F-1117.
Концентрация спор каждого штамма в используемой смешанной суспензии равнялась 1–2 млн/см3. Испытания вели в течение 84 суток с промежуточным осмотром через 10, 15, 21, 28 и 50 суток. Оценку грибостойкости (показателя степени развития грибов) образцов проводили по изменению их окраски, формы и текстуры согласно шестибалльной шкале.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В смесителе Брабендер под действием сдвиговых деформаций при 180°С были получены смеси ПЛА с ПГБ. Поскольку эти полиэфиры являются жесткими и хрупкими полимерами, то их смешение осуществляли в присутствии пластификатора ПЭГ различной молекулярной массы – М = (0.4, 0.6 и 1.0) × 103, увеличивающего пластичность полимеров [15, 16]. Для последующих измерений были отпрессованы пленки ПГБ, ПЛА и их смесей.
На рис. 1 представлены ДСК-кривые ПЛА, ПГБ и их смеси при первичном и вторичном нагревании. Видно, что на ДСК-кривой ПЛА при первичном нагревании присутствуют четыре пика: пик стеклования, Тс = 65°С; пик холодной кристаллизации, Тх.к = 124°С; пики дублета плавления с температурами плавления Tпл1 = 159.4°C и Tпл2 = 161.8°C. При вторичном нагревании вследствие изменения размеров кристаллитов ПЛА температура переходов снижается на несколько градусов и соответствует следующим значениям: Тс = 63°С, Тх.к = 123.3°С, Tпл1 = 158.4°C и Tпл2 = = 161.7°C.
Рис. 1.
ДСК-кривые ПЛА (1, 1'), ПГБ (2, 2') и их смеси ПЛА : ПГБ (3, 3') в соотношении 80 : 20 мас. %, содержащей 5 мас. % ПЭГ400 при первичном (1–3) и вторичном (1'–3') нагревании.
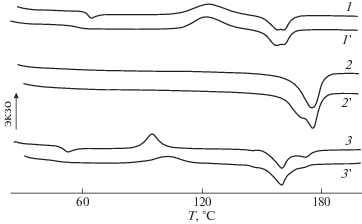
На ДСК-кривой ПГБ присутствуют пики плавления при ∼175 и 170°С при первичном и вторичном нагревании соответственно. Отсутствие пика стеклования связано с тем, что он находится ниже температуры измерений.
При анализе ДСК-кривых смеси ПЛА–ПГБ, содержащей 5% ПЭГ400, было обнаружено, что в этом случае при первичном нагревании наблюдаются пять пиков, соответствующих TсПЛА = 52°C, Тх.кПЛА = 94°С, Tпл1ПЛА = 144.4°С, Tпл2ПЛА = 159.3°С и ТплПГБ = 171°С (кривая 3). Снижение температуры переходов в ПЛА в смеси по сравнению с аналогичными переходами исходного ПЛА обусловлено присутствием как ПГБ, так и ПЭГ, влияющих на процесс кристаллизации. При вторичном нагревании (кривая 3') пик температуры стеклования ПЛА отсутствует, а величины других пиков незначительно возрастают (Тх.кПЛА = 102°С, Tпл1ПЛА = 146.4°С и Tпл2ПЛА = 159.3°С). В то же время, ТплПГБ = 170.2°С практически не меняется.
В табл. 1 представлены рассчитанные теплофизические характеристики ПЛА, ПГБ и их смесей с ПЭГ различной молекулярной массы.
Таблица 1.
Теплофизические характеристики ПЛА, ПГБ в их композициях различного состава при первичном (числитель) и вторичном (знаменатель) нагревании
| Состав композиции, мас. % | Тс, °С | Тпл, °С | ΔHпл, Дж/г | Тхк, °С | χкр, % |
|---|---|---|---|---|---|
| Полилактид | |||||
| ПЛА (100%) | 65.0 / 63.0 | дублет 159.4; 162.0 / | 25.6 / 27.7 | 124.0 / 123.3 | 27.3 / 29.6 |
| дублет 158.4; 161.7 | |||||
| (80 : 20) + 5% ПЭГ400 | 52.0 / – | дублет 144.4; 159.3 / | 31.3 / 36.6 | 94.0 / 102.0 | 42.0 / 49.0 |
| дублет 146.4; 159.3 | |||||
| (80 : 20) + 10% ПЭГ400 | – / – | 157.6 / 156.8 | 41.3 / 42.7 | 83.0 /дублет | 55.0 / 57.0 |
| 81.5; 127.0 | |||||
| (80 : 20) + 5% ПЭГ600 | 54.2 / – | дублет 145.6; 159.3 / | 22.8 / 32.4 | 96.0 / 106.0 | 30.0 / 43.0 |
| дублет 148.0; 159.4 | |||||
| (90 : 10) +5% ПЭГ1000 | 53.5 / – | 159.0 / – | 29.6 / – | 107.0 / – | 34.0 / – |
| Поли-(3-гидроксибутират)* | |||||
| ПГБ (100%) | – / – | 175.0 / 170.0 | 71.0 / 75.5 | – / – | 49.0 / 52.0 |
| (80 : 20) + 5% ПЭГ400 | – / – | 171.0 / 170.0 | – / – | – / – | – / – |
| (80 : 20) + 10% ПЭГ400 | – / – | 167.3 / – | – / – | – / – | – / – |
| (80 : 20) + 5% ПЭГ600 | – / – | 187.3 / – | – / – | – / – | – / – |
| (90 : 10) + 5% ПЭГ1000 | – / – | – / – | – / – | – / – | – / – |
Величины энтальпии плавления $\Delta Н_{{{\text{эксп}}}}^{0}$ ПЛА при первичном и вторичном нагревании составляют 25.6 и 27.7 Дж/г соответственно. Учитывая, что величина энтальпии плавления ПЛА при 100% кристалличности $\Delta Н_{m}^{0}$ равна 93.7 Дж/г [17], то по известному уравнению [18] были рассчитаны степени кристалличности χкр ПЛА при первичном и вторичном нагревании:
Из табл. 1 видно, что при первичном и вторичном нагревании степень кристалличности ПЛА изменяется незначительно. Известно, что величина энтальпии плавления ПГБ при 100% кристалличности $\Delta Н_{m}^{0}$ равна 146 Дж/г [19, 20], при этом измеренная энтальпия при первичном и вторичном нагревании составляет 71.0 и 75.5 Дж/г соответственно. Рассчитанные таким образом, согласно уравнению (1), степени кристалличности ПГБ при первичном и вторичном нагревании составляют 49 и 52%. При вторичном нагревании степень кристалличности как ПГБ, так и ПЛА несколько повышается, что, по-видимому, связано с изменением условий кристаллизации (табл. 1).
Так как ПГБ и ПЛА представляют собой частично кристаллические полимеры, то их степень кристалличности в смесях зависит от условий смешения. В работе [21] показано, что при добавлении ПГБ к ПЛА наблюдается рост кристалличности последнего.
Степень кристалличности ПЛА в смесях была вычислена по формуле:
где WПЛА – массовая доля ПЛА в смеси.Как и следовало ожидать, степень кристалличности ПЛА возрастает при добавлении ПГБ.
При увеличении содержания ПЭГ400 до 10 мас. % число температурных переходов снижается до трех: Тх.кПЛА = 83.2°С, TплПЛА = 157.6°С и ТплПГБ = = 167.3°С, т.е. пики температуры стеклования и один из пиков плавления ПЛА отсутствуют. Таким образом, содержание ПЭГ в смеси влияет не только на значения температурных переходов, но и на их число.
При вторичном нагревании смеси также наблюдаются три перехода, но при других значениях температуры: Тх.кПЛА = 81.5°С, Тх.кПЛА = 127.2°С и TплПЛА = 156.8°С, т.е. в этом случае отсутствует пик плавления ПГБ.
Степени кристалличности ПЛА в этом случае при первичном и вторичном нагреваниях оказались приблизительно одинаковыми.
Таким образом, введение ПЭГ400 приводит к изменению термодинамических свойств смеси и возрастанию кристалличности ПЛА, причем кристалличность растет с увеличением содержания пластификатора.
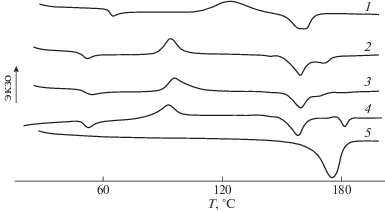
Аналогичными были результаты для смесей, содержащих ПЭГ600 и ПЭГ1000 (рис. 2; табл. 1), однако наиболее заметные изменения прослеживались у композиций, содержащих наиболее низкомолекулярный ПЭГ400.
Рис. 2.
ДСК-кривые ПЛА (1), ПГБ (5) и их смесей ПЛА : ПГБ в соотношении 80 : 20 мас. %, содержащих 5 мас. % ПЭГ с М = 0.4 × 103 (2), 0.6 × 103 (3) и 1.0 × 103 (4).
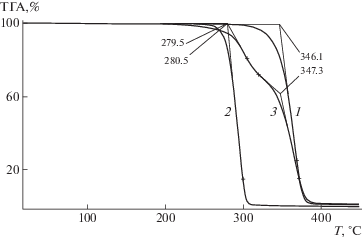
На рис. 3 приведены кривые, полученные при термогравиметрическом анализе ПЛА, ПГБ и их смеси. Начальную температуру разложения определяли по пересечению касательных на кривых потери массы. Рассчитанные таким образом значения температуры составляют ~280 и 346°С для ПГБ и ПЛА соответственно, а на кривой разложения смеси существуют два перегиба, практически совпадающие с температурами разложения ПГБ и ПЛА, что является еще одним подтверждением несовместимости этих полимеров.
Рис. 3.
ТГА-кривые ПЛА (1), ПГБ (2) и их смеси ПЛА : ПГБ в соотношении 80 : 20 мас. %, содержащей 5 мас. % ПЭГ400 (3).
Механические характеристики композиций ПЛА–ПГБ, содержащих ПЭГ различной молекулярной массы, приведены в табл. 2. Из таблицы видно, что механические характеристики ПЛА и ПГБ характерны для стеклообразных полимеров с низкой деформацией при разрыве, причем ПГБ является более жестким полимером с модулем упругости Е = 2900 МПа и разрывным удлинением εр = 1%. Аналогичные результаты представлены в работе [22], где показано, что предел прочности при растяжении σр и удлинение при разрыве εр смесей ПЛА–ПГБ уменьшаются с увеличением содержания ПГБ. При более высоком содержании ПГБ в смесях напряжение при растяжении и удлинение при разрыве характеризуются более низкими значениями, чем у исходного ПЛА.
Таблица 2.
Механические характеристики ПЛА, ПГБ и их композиций с ПЭГ
| Состав композиции ПЛА–ПГБ, мас. % | E, МПа | σр, МПа | εр, % |
|---|---|---|---|
| ПЛА (100%) | 2700 | 45.4 | 4 |
| ПГБ (100%) | 2900 | 19.5 | 1 |
| (90 : 10) + 5% ПЭГ1000 | 2500 | 37.8 | 2 |
| (85 : 15) + 5% ПЭГ1000 | 2500 | 35.7 | 3 |
| (80 : 20) + 5% ПЭГ1000 | 2500 | 31.6 | 2 |
| (75 : 25) + 5% ПЭГ1000 | 2500 | 29.7 | 1 |
| (70 : 30) + 5% ПЭГ1000 | 2450 | 28.5 | 1 |
| (80 : 20) + 5% ПЭГ600 | 2700 | 21.0 | 1 |
| (80 : 20) + 5% ПЭГ400 | 2250 | 25.3 | 2 |
| (80 : 20) + 7% ПЭГ400 | 1850 | 28.2 | 2 |
| (80 : 20) + 10% ПЭГ400 | 800 | 11.6 | 13 |
Увеличение содержания ПГБ и пластификатора ПЭГ различной молекулярной массы приводит к уменьшению модуля упругости и возрастанию предела прочности и удлинения при разрыве композиций, особенно при введении ПЭГ400. Удлинение при разрыве для всех смесей остается весьма низким за исключением композиций, содержащих 10% пластификатора ПЭГ400 (табл. 2). В этом случае, по всей видимости, происходит пластическое разрушение, и удлинение при разрыве достигает 13%.
В табл. 3 приведены величины показателей текучести расплавов композиций ПЛА–ПГБ–ПЭГ при 170°С. Видно, что для ПЛА этот показатель составляет 3.8 г/10 мин, в то время как ПГБ при тех же условиях не обладает текучестью. Так, значения показателей текучести расплавов смесей ПЛА–ПГБ (80 : 20 мас. %) изменяются при введении ПЭГ различной молекулярной массы. При добавлении ПЭГ1000 под нагрузкой 1.20 и 2.16 кг значения ПТР падают с уменьшением содержания ПЛА. В то же время, для композиций ПЛА–ПГБ (80 : 20 мас. %), содержащих 5% ПЭГ различной молекулярной массы, величина ПТР возрастает с уменьшением молекулярной массы ПЭГ, а с увеличением содержания ПЭГ400 композиция начинает течь без нагрузки.
Таблица 3.
Показатели текучести расплавов (ПТР) образцов смесей ПЛА–ПГБ–ПЭГ при 170°С
| Состав композиций, мас. % | Нагрузка, кг | ПТР, г/10 мин |
|---|---|---|
| ПЛА (100%) | 2.16 | 3.8 |
| ПГБ (100%) | 2.16 | Не течет |
| (90 : 10) + 5% ПЭГ1000 | 1.2 | 10.5 |
| (85 : 15) + 5% ПЭГ1000 | 1.2 | 8.7 |
| (80 : 20) + 5% ПЭГ1000 | 1.2 | 5.0 |
| (75 : 25) + 5% ПЭГ1000 | 2.16 | 13.2 |
| (70 : 30) + 5% ПЭГ1000 | 2.16 | 2.1 |
| (80 : 20) + 5% ПЭГ600 | 2.16 | 14.5 |
| (80 : 20) + 5% ПЭГ400 | 2.16 | 17.5 |
| (80 : 20) + 7% ПЭГ400 | Без нагрузки | 6.0 |
| (80 : 20) + 10% ПЭГ400 | Без нагрузки | 7.2 |
Испытания на грибостойкость проводили для ПЛА, ПГБ и их композиций ПЛА–ПГБ (соотношение 70 : 30 и 85 : 15 мас. %), содержащих 5% ПЭГ1000. Данные по оценке грибостойкости образцов в зависимости от времени испытаний представлены в табл. 4.
Таблица 4.
Оценка грибостойкости образцов в зависимости от времени испытаний
| Время испытания, сутки | Интенсивность развития грибов, баллы | |||
|---|---|---|---|---|
| ПЛА | ПГБ | ПЛА : ПГБ | ||
| (70 : 30 мас. %) + 5% ПЭГ1000 | (85 : 15 мас. %) + 5% ПЭГ1000 | |||
| 10 | 0 | 2 | 1 | 1 |
| 15 | 0 | 2 | 1 | 1 |
| 21 | 0 | 2 | 2 | 1 |
| 28 | 0 | 5 | 2 | 1 |
| 50 | 0 | 5 | 3 | 1–2 |
| 84 | 0–1 | 5 | 3–4 | 2 |
Как видно из приведенных в таблице данных, ПЛА за время испытаний практически не подвергается деструкции под действием грибов. Только на 84 сутки испытаний на одном из трех образцов было обнаружено незначительное развитие грибного мицелия (1 балл). ПГБ в ходе испытаний показал максимальную интенсивность развития плесневых грибов, соответствующую 5 баллам, подтвердив свой статус биоразлагаемого материала (табл. 4). Уже через 10 суток под микроскопом хорошо различим грибной мицелий, покрывающий всю поверхность материала, а через 21 сутки начинается образование спор, внешний вид которых типичен для грибов Aspergillus brasiliensis, а на 28–50 сутки испытаний появляется также гриб вида Trichoderma virens. Максимальное распространение на 84 сутки получил гриб вида Paecilomyces variotii.
Для композиции ПЛА–ПГБ (70 : 30 мас. % + + 5% ПЭГ1000) незначительно развитый мицелий был выявлен на 10 сутки испытаний. Характер роста грибов позволяет предположить неравномерное распределение компонентов композиции в образцах. Грибы, формирующие спороношение, также как и для исходного ПГБ, представлены видами Aspergillus brasiliensis, Trichoderma virens и Paecilomyces variotii.
Снижение концентрации ПГБ до 15% приводит к снижению интенсивности развития грибов с 4 до 2 баллов (табл. 4). Несмотря на то, что отдельные грибные гифы были обнаружены уже на 10 сутки испытаний, интенсивность развития грибов невелика, и только на 84 сутки мицелий стал покрывать ~50% поверхности образцов.
За весь срок испытаний ни у одного из образцов не было отмечено изменение формы и текстуры, а потеря массы, составляющая 2.4%, наблюдалась только у ПГБ.
Для определения способности изучаемых композиций к биоразложению при экспонировании в почве образцы закапывали в грунт с рН 7.0–7.5 и выдерживали в течение нескольких месяцев.
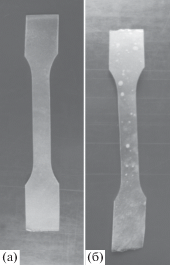
На рис. 4 приведены фотографии исходного ПГБ до и после экспонирования в течение 3 месяцев. Фотографии смеси ПЛА–ПГБ до и после экспонирования в течение 3 месяцев представлены на рис. 5. Как видно, после экспонирования образцов в течение 3 месяцев на пленках ПГБ и пленках композиций ПЛА : ПГБ = (80 : 20 мас. %) + + 10% ПЭГ400 появляются пятна, свидетельствующие о протекании деструкции пленок. Необходимо отметить, что на пленках ПЛА никаких изменений отмечено не было.
Рис. 4.
Фотографии пленок исходного ПГБ до (а) и после (б) экспонирования в почве в течение 3 месяцев.
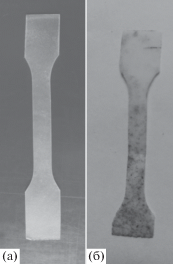
Рис. 5.
Фотографии пленок композиции ПЛА : ПГБ в соотношении (80 : 20 мас. %) + 10% ПЭГ400 до (а) и после (б) экспонирования в почве в течение 3 месяцев.
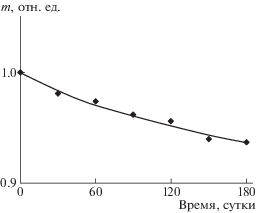
На рис. 6 приведена кривая потери массы композиционной пленки ПЛА : ПГБ = (80 : 20 мас. %) + + 10% ПЭГ400 при нахождении в почве в течение 6 месяцев, из которой видно, что потеря массы после экспонирования составила 6.3%.
ЗАКЛЮЧЕНИЕ
Изучение методом ДСК термофизических свойств композиций при первичном и вторичном нагревании показало, что ПЛА и ПГБ являются несмешивающимися полимерами. Проведен расчет степени кристалличности и энтальпии плавления индивидуальных ПЛА и ПГБ, а также в смесях. Показано, что вторичное нагревание приводит к изменению температурных переходов и степени кристалличности исследуемых полимеров. На основании данных ТГА определены начальные значения температуры разложения полиэфиров.
При изучении механических свойств установлено, что при добавлении к ПЛА ПГБ и пластификатора ПЭГ различной молекулярной массы происходит уменьшение модуля упругости и возрастание предела прочности и удлинения при разрыве.
При изучении грибостойкости ПЛА, ПГБ и их композиций показано, что ПГБ в отличие от ПЛА является полностью биоразлагаемым полимером. Биоразлагаемость композиций на их основе зависит от состава и возрастает с увеличением содержания ПГБ. Данные материалы могут быть использованы при изготовлении различных изделий, в частности блистеров для упаковки лекарств и одноразовой посуды (тарелок, ложек, вилок) и т.д.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (№ 18-29-05017).
Список литературы
Mohanty A.K., Misra M., Hinrichsen G. // Macromol. Mater. Eng. 2000. V. 276–277. Iss. 1. P. 1.
Carrasco F., Cailloux J., Sánchez-Jiménez P.E., Maspoch M.L. // Polym. Degrad. Stab. 2014. V. 104. P. 40.
Auras R., Harte B., Selke S. // Macromol. Biosci. 2004. V. 4. P. 835.
Wang L., Gross R. // Polym. Degrad. Stab. 1998. V. 59. P. 161.
Ferri J.M., Fenollar O., Jorda-Vilaplana A., GarcÃ-a-Sanoguera D., Balart R. // Polym. Int. 2016. V. 65. P. 453.
Khurma J., Rohindra D., Devi R. // J. Nat. Sci. 2005. V. 23. P. 22.
Zhang L., Goh S., Lee S. // Polymer. 1998. V. 39. P. 4841.
Yokohara T., Yamaguchi M. // Eur. Polym. J. 2008. V. 44. P. 677.
Li S., Woo E.M. // Polym. Int. 2008. V. 57. P. 1242.
Rogovina S.Z., Aleksanyan K.V., Kosarev K.V., Ivanushkina N.E., Prut E.V., Berlin A.A. // Polymer Science B. 2016. V. 58. № 1. P. 38.
Rogovina S.Z., Aleksanyan K.V., Loginova A.A., Ivanushkina N.E., Vladimirov L.E., Prut E.V., Berlin A.A. // Starch. 2018. V. 70. DOI: . 201700268.https://doi.org/10.1002/star
Arrieta M.P., Fortunati E., Dominici F., Rayón E., López J., Kenny J.M. // Polym. Degrad. Stab. 2014. V. 107. P 139.
González-Ausejo J., Sánchez-Safont E., Lagarón J.M., Balart R., Cabedo L., Gámez-Pérez J. // J. Appl. Polym. Sci. 2017. V. 134. DOI: . 44806.https://doi.org/10.1002/app
Iordanskii A., Karpova S., Olkhov A., Borovikov P., Kildeeva N., Liu Y. // Eur. Polym. J. 2019. V. 117. P. 208.
Arrieta M.P., Samper M.D., López J., Jiménez A. // J. Polym. Environ. 2014. V. 22. P. 460.
Courgneau C., Domenek S., Guinault A., Avérous L., Ducruet V. // J. Polym. Environ. 2011. V. 19. P. 362.
Suksut B., Deeprasertkul C. // J. Polym. Environ. 2011. V. 19. P. 288.
Handbook of Thermal Analysis and Calorimetry / Ed. by S.Z.D Cheng. Amsterdam: Elsevier, 2002. V. 3.
Azizi Samir M.A.S., Alloin F., Sanchez A. // Polymer. 2004. V. 45. P. 4149.
Kiziltas A., Gardner D.J., Han Y., Yang H. // Thermochim. Acta. 2011. V. 519. P. 38.
Ni C., Luo R., Xu K., Chen G.-Q. // J. Appl. Polym. Sci. 2009. V. 111. P. 1720.
Zhang M., Thomas N.L. // Adv. Polym. Techn. 2011. V. 30. P. 67.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия А)