

Высокомолекулярные соединения (серия А), 2020, T. 62, № 5, стр. 392-400
О ТЕМПЕРАТУРНОЙ ЗАВИСИМОСТИ ЭНЕРГИИ АКТИВАЦИИ СТЕКЛОВАНИЯ
Д. С. Сандитов a, b, *, И. В. Разумовская c, А. А. Машанов a
a Бурятский государственный университет
670000 Улан-Удэ, ул. Смолина, 24а, Россия
b Институт физического материаловедения Сибирского отделения Российской академии наук
670047 Улан-Удэ, ул. Сахьяновой, 6, Россия
c Московский педагогический государственный университет
119435 Москва, ул. Малая Пироговская, 29/7, Россия
* E-mail: Sanditov@bsu.ru
Поступила в редакцию 03.10.2019
После доработки 28.02.2020
Принята к публикации 16.03.2020
Аннотация
Проведен расчет температурной зависимости энергии активации стеклования для аморфных органических полимеров и неорганических стекол с привлечением уравнения Вильямса–Ландела–Ферри для времени релаксации (вязкости). Полученные результаты находятся в удовлетворительном согласии с экспериментом. Обсуждается природа зависимости энергии активации стеклования от температуры.
ВВЕДЕНИЕ
Природа перехода аморфного вещества из жидкого (высокоэластического) в стеклообразное состояние до сих пор не вполне понятна. Стеклование жидкости тесно связано с повышением вязкости при охлаждении расплава, причем энергия активации стеклования, как правило, совпадает с энергией активации вязкого течения (релаксации) в области перехода жидкость–стекло.
Температурная зависимость вязкости простых маловязких жидкостей типа ацетона хорошо описывается уравнением Я.И. Френкеля [1]:
c энергией активации U = const. Здесь η0 – предэкспоненциальный множитель, R – газовая постоянная. Однако для стеклообразующих расплавов энергия активации в формуле (1) в области стеклования довольно сильно зависит от температуры: U = U(T). В связи с этим предложены эмпирические уравнения вязкости, которые в неявном виде учитывают зависимость U(T). Среди них широкое распространение получило соотношение Вильямса–Ландела–Ферри [2, 3]:(2)
$\begin{gathered} \ln {{a}_{T}} = - {{C}_{1}}\frac{{T - {{T}_{g}}}}{{T - {{T}_{g}} + {{C}_{2}}}} \\ {{a}_{T}} = \frac{{{\tau (}T{\text{)}}}}{{{\tau }({{T}_{g}})}} \cong \frac{{{\eta (}T{\text{)}}}}{{\eta ({{T}_{g}})}}, \\ \end{gathered} $Так, в работах [7, 8] был предложен вывод выражения (2) без конкретизации функций τ(Т) и η(Т):
(3)
$\ln {{a}_{T}} = - \left( {\frac{{{{A}^{2}}}}{B}} \right)\frac{{T - {{T}_{g}}}}{{T - {{T}_{g}} + \left( {\frac{A}{B}{{T}_{g}}} \right)}}$(4)
$A = - {{\left. {\frac{{\partial \ln {\eta }}}{{\partial \left( {T{\text{/}}{{T}_{g}}} \right)}}} \right|}_{{T = {{T}_{g}}}}}$(5)
$B = \frac{1}{2}{{\left. {\frac{{{{\partial }^{2}}\ln \eta }}{{\partial {{{\left( {T{\text{/}}{{T}_{g}}} \right)}}^{2}}}}} \right|}_{{T = {{T}_{g}}}}}$Было использовано разложение в ряд функции lnη(Т) вблизи Tg по малому безразмерному параметру λ = (T – Tg)/Tg (см. работу [8]).
Таким образом, постоянные уравнения (2) приобретают физический смысл – они могут быть выражены через производные А, B как
Настоящая работа посвящена оценке температурной зависимости энергии активации стеклования U(T)с привлечением данных о параметрах уравнения Вильямса–Ландела–Ферри С1 и С2. Обсуждается вариант расчета U(T) без конкретизации вида функции U(T). Поскольку основные закономерности перехода аморфного вещества из высокоэластического в стеклообразное состояние качественно одинаковы для многих стеклообразующих систем, в том числе для аморфных органических полимеров и неорганических стекол (см., например, работы [4, 9]), объектами исследования наряду с аморфными полимерами являются неорганические стекла.
РАСЧЕТ ТЕМПЕРАТУРНОЙ ЗАВИСИМОСТИ ЭНЕРГИИ АКТИВАЦИИ СТЕКЛОВАНИЯ С ПОМОЩЬЮ УРАВНЕНИЯ ВИЛЬЯМСА–ЛАНДЕЛА–ФЕРРИ
Легко убедиться в том, что уравнение (2) алгебраически выводится из соотношения Фогеля–Фульчера–Таммана (см. работу [4]):
(8)
$\eta = {{\eta }_{0}}\exp \left( {\frac{{{{B}_{{\text{0}}}}}}{{T - {{T}_{{\text{0}}}}}}} \right),$Считая предэкспоненциальные множители в формулах для вязкости (1) и (8) равными, получаем следующую зависимость энергии активации вязкого течения от температуры:
Поскольку для многих аморфных полимеров и неорганических стекол известны значения параметров уравнения Вильямса–Ландела–Ферри С1 и С2 [2–11], с помощью выражений (9) и (10) целесообразно перейти от зависимости (11) к величине U(T) как функции этих параметров:
Из данного равенства вытекает простое соотношение для расчета энергии активации стеклования Ug = U(Tg) при температуре стеклования T = Tg:
Для листового (оконного) силикатного стекла, например, С1 = 36 и Tg = 807 К [10], откуда имеем Ug = 241 кДж/моль. Методы релаксационной спектрометрии [4, 10, 11] для этого стекла приводят практически к такому же значению Ug = = 242 кДж/моль. Для полиизобутилена (Tg = 202 К, С1 = 38) и натурального каучука (Tg = 300 К, С1 = = 38) величины Ug, равные 64 и 95 кДж/моль соответственно, также согласуются с данными релаксационной спектрометрии [11].
Известно, что многие стеклующиеся системы переходят из жидкого в стеклообразное состояние практически при одной и той же вязкости ηg ~ 1012 Па с (приближенное правило постоянства вязкости при Tg). Высокотемпературный предел вязкости η0 ~ η(T → ∞) также оказывается приблизительно постоянным, η0 ~10–3.5 Па с. Поэтому энергию активации при температуре стеклования Ug = U(Tg) можно оценить по формуле (1) при T = Tg, ηg ~ η(Tg) ~ 1012 Па с и η0 ~ 10–3.5 Па с [4]:
(14)
${{U}_{g}} = \left( {\ln \frac{{{{\eta }_{g}}}}{{{{\eta }_{0}}}}} \right)R{{T}_{g}} \approx 36R{{T}_{g}},$Следовательно, слабая зависимость параметра С1 от природы стекол С1 ~ const ~ 35–40 (табл. 1) [2, 4] объясняется приближенным постоянством вязкостей ηg и η0:
Таблица 1.
Параметры уравнения Вильямса–Ландела–Ферри и энергия активации стеклования при T = Tg для аморфных органических полимеров и неорганических стекол (по данным работ [2, 4, 30])
| Аморфные вещества | Tg, К | C1, К | C2, К | Ug, кДж/моль |
|---|---|---|---|---|
| PbO–SiO2 (42.07 мол. % PbO) | 711 | 36 | 250 | 212 |
| Na2O–B2O3 (2.8 мол. % Na2O) | 544 | 29 | 130 | 131 |
| Листовое стекло | 807 | 36 | 305 | 241 |
| Полиизобутилен | 202 | 38 | 104 | 64 |
| Поливинилацетат | 305 | 38 | 104 | 96 |
| Полиуретан | 238 | 36 | 33 | 71 |
| Натуральный каучук | 300 | 38 | 54 | 95 |
Примечание. Ug раcсчитана по формуле (13).
Здесь необходимо отметить, что правило постоянства вязкости при Tg, согласно современным исследованиям, не всегда оправдывается (см., например, работу [12]). Тем не менее для большинства исследованных стекол значения Tg соответствуют значениям температуры, при которых вязкость находится в пределах от 1011 до 1012 Па с. Постоянство высокотемпературного предела вязкости η0 ~ const также оказывается весьма приближенным. Поэтому соотношение (14) дает лишь первое грубое приближение. Предпочтительной является оценка Ug по формуле (13).
Таким образом, для расчета U(T) в области стеклования по формуле (12) необходимо знать три величины: C1, C2 и Tg. Данные о параметрах C1, C2 и о температуре стеклования Tg для многих аморфных органических полимеров имеются в книге [2], а для неорганических стекол – в монографии [4], где описан также один из способов определения C1 и C2.
На рис. 1 приведена температурная зависимость энергии активации стеклования для листового силикатного стекла. Кривая – расчет по формуле (12), точки – экспериментальные данные. Последние получены из данных о вязкости lgη(T) с помощью уравнения (1), из которого следует
Рис. 1.
Температурная зависимость энергии активации стеклования U(Т – Tg) для листового силикатного стекла: точки – эксперимент, кривая – расчет по формуле (12).
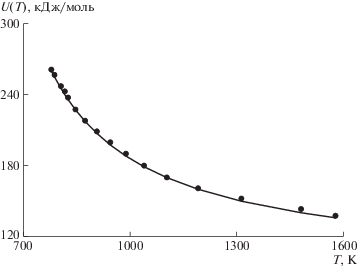
Как видно, точки ложатся на расчетную кривую, что подтверждает хорошую применимость формулы Вильямса–Ландела–Ферри. Аналогичные графики U – T построены для ряда органических аморфных полимеров (см., например, рис. 2 для поливинилацетата). В табл. 1 для некоторых стекол и полимеров приводятся значения параметров C1, C2 и Tg, входящих в соотношение (12), а также величина Ug, рассчитанная по формуле (13).
Рис. 2.
Температурная зависимость энергии активации стеклования U(Т – Tg) для поливинилацетата: точки – эксперимент, кривая – расчет по уравнению (12).
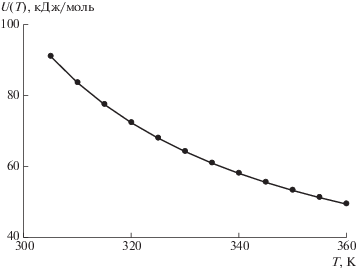
О ВОЗМОЖНОСТИ ИСПОЛЬЗОВАНИЯ РАЗЛОЖЕНИЯ ФУНКЦИИ U(T) В РЯД ТЕЙЛОРА
Представляет определенный интерес оценка зависимости энергии активации стеклования от температуры без конкретизации функции U(T). Один из таких способов расчета U(T) представлен ниже.
Разложим в ряд функцию U(T) вблизи Tg по малому приращению температуры (T – Tg) и ограничимся первыми тремя членами ряда
(18)
${{D}_{2}} = \frac{1}{2}{{\left( {\frac{{{{\partial }^{2}}U}}{{\partial {{T}^{2}}}}} \right)}_{{{{T}_{g}}}}}$Фактически параметром малости в разложении (16) по-прежнему является безразмерная величина λ = (T – Tg)/Tg, но Tg в первой и второй степени скрыто в коэффициентах D1 и D2. Рассматривая вязкость в зависимости (1) как сложную функцию температуры, включающую производные (4) и (5), и используя формулы (6) и (7), выразим D1 и D2 через параметры C1, C2 и температуру стеклования Tg (см. Приложение):
(20)
${{D}_{2}} = \frac{{{{C}_{1}}}}{{{{C}_{2}}}}R\left( {\frac{{{{T}_{g}}}}{{{{C}_{2}}}} - 1} \right)$Коэффициент D2 в С2 раз отличается от D1. По данным о С1, С2 и Tg с помощью формул (16), (19) и (20) возможно рассчитать зависимость энергии активации стеклования от температуры.
Коэффициенты D1 и D2, вычисленные по формулам (19) и (20) для листового силикатного стекла и полиуретана, были сопоставлены с характеристиками кривых U(T – Tg), полученных из экспериментальных данных по температурной зависимости вязкости (табл. 2). Коэффициенты кривой U(T – Tg) как полинома второй степени рассчитаны методом подгонки при фиксированных значениях С1 и С2 (17.48 и 276.75 соответственно), а также с помощью программы“Origin” (рис. 3). Во втором случае температурная зависимость энергии активации описывалась полиномом второй степени, что позволяло получить значения D1 и D2. Далее учитывали, что в широком температурном интервале может начать проявляться роль кубичного члена в разложении U в ряд Тейлора. Поэтому температурная зависимость энергии активации была интерпретирована также как полином третьей степени (рис. 4), что несколько изменило значения коэффициента D2 (табл. 2).
Таблица 2.
Коэффициенты D1 и D2, рассчитанные разными способами
| Способ расчета | D1, кДж/моль К | D2, кДж/моль К2 |
|---|---|---|
| Листовое стекло | ||
| По формулам (19) и (20) при C1 = 17.48; C2 = 276.75 | 0.278 | 10–5 |
| Подгонка U(T – Tg) при C1 = 17.48; C2 = 276.75 | 0.325 | 2.1 × 10–4 |
| Описание U(T – Tg) полиномом второй степени | 0.314 ± 0.012 | 2.400 × 10–4 ± 0.182 × 10–4 |
| Описание U(T – Tg) полиномом третьей степени* | 0.384 ± 0.013 | 5.675 × 10–4 ± 0.538 × 10–4 |
| Полиуретан | ||
| По формулам (19) и (20) при C1 = 15.60; C2 = 32.60 | 0.815 | 25 × 10–3 |
| Подгонка U(T – Tg) при C1 = 15.60; C2 = 32.60 | 0.750 | 4 × 10–3 |
| Описание U(T – Tg) полиномом второй степени | 0.851± 0.050 | 4.83 × 10–3 ± 0.60 × 10–3 |
| Описание U(T – Tg) полиномом третьей степени** | 1.022 ± 0.117 | 1.018 × 10–2 ± 0.343 × 10–2 |
Рис. 3.
Описание температурной зависимости энергии активации стеклования полиномом второй степени для листового стекла (а) и полиуретана (б): точки – эксперимент, кривые – расчет с помощью программы “Origin”.
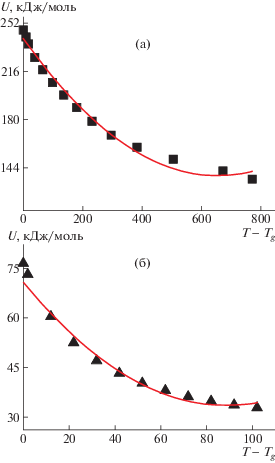
Рис. 4.
Описание температурной зависимости энергии активации стеклования полиномом третьей степени для листового стекла (а) и полиуретана (б): точки – эксперимент, кривые – расчет с помощью программы “Origin”.
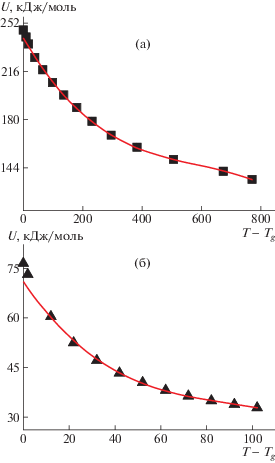
В целом в рамках рассматриваемого приближения наблюдается удовлетворительное совпадение теоретических и экспериментальных значений коэффициентов. Совпадение во всех расчетах выполняется лучше для D1, хуже – для D2 (см. табл. 2). Это объясняется тем, что расчеты проводились для широкого температурного интервала, где приближения перестают выполняться. Таким образом, температурная зависимость энергии активации в достаточно широком температурном интервале хорошо описывается полиномом второй степени.
ОЦЕНКА ТЕМПЕРАТУРНОЙ ОБЛАСТИ ПРИМЕНИМОСТИ ТЕОРЕТИЧЕСКОГО ВЫВОДА ФОРМУЛЫ ВИЛЬЯМСА–ЛАНДЕЛА–ФЕРРИ
Получение формулы Вильямса–Ландела–Ферри в виде выражения (3) предполагало, что рассматриваются такие значения параметра малости λ = (T – Tg)/Tg, при которых модуль квадратичного члена в ряду Тейлора по крайней мере на порядок меньше, чем модуль линейного [8]. Тогда оценим значения λ для неорганического стекла и полиуретана, при которых квадратичный член на порядок меньше линейного.
На рис. 5 приведены экспериментальные зависимости ln aT от λ для неорганического стекла и полиуретана, интерпретированные как многочлен второй степени. Такие же зависимости описаны многочленом третьей степени на рис. 6. Коэффициенты полинома B1 и B2 связаны с величинами A и B, определяемыми формулами (4) и (5): B1 = –A, B2 = B. Данное разложение для двух рассматриваемых материалов имеет общие черты. Коэффициенты при λ в первой и второй степени в обоих случаях по модулю имеют одинаковый порядок величины. Коэффициент при λ3 отрицателен (как и коэффициент при λ в первой степени). Для неорганического стекла его модуль примерно тот же по порядку величины, что и первые коэффициенты ряда. Для полиуретана он на порядок меньше.
Рис. 5.
Экспериментальные зависимости ln aT от параметра λ = (T – Tg)/Tg для неорганического стекла (1) и полиуретана (2) и их описание многочленом y (x) = = B0 + B1x + B2x2: точки – эксперимент, кривые – расчет. Для стекла B0 = 0, B1 = –66.4 ± 2.8, B2 = 42.2 ± ± 3.6; для полиуретана B0 = 0, B1 = –151.6 ± 7.2, B2 = = 215.2 ± 20.7.
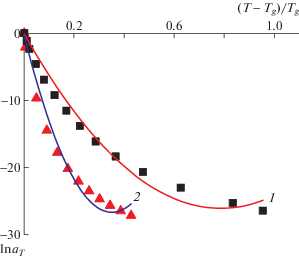
Рис. 6.
Зависимости ln aT от λ для неорганического стекла (1) и полиуретана (2) и их описание многочленом y(x) = B0 + B1x + B2x2 + B3x3: точки – эксперимент, кривые – расчет. Для стекла B0 = 0, B1 = –84.7 ± ± 2.1, B2 = 111.4 ± 7.3, B3 = –54.5 ± 5.7; для полиуретана B0 = 0, B1 = –201.7 ± 5.9, B2 = 588.8 ± 40.9, B3 = = –626.6 ± 67.7.
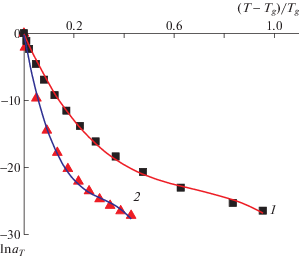
Полученные значения коэффициентов разложения показали, что представленный в настоящей работе метод вывода уравнения Вильямса–Ландела–Ферри для неорганического стекла справедлив при λ < 0.1, т.е. для температуры выше Tg примерно на 80–90 К. В этом интервале температуры должны достаточно хорошо работать выведенные соотношения между коэффициентами и сама формула Вильямса–Ландела–Ферри.
О ПРИРОДЕ ЗАВИСИМОСТИ ЭНЕРГИИ АКТИВАЦИИ СТЕКЛОВАНИЯ ОТ ТЕМПЕРАТУРЫ
Основные закономерности процесса стеклования в качественном отношении оказываются одинаковыми для подавляющего большинства аморфных веществ независимо от их природы: для неорганических стекол, аморфных органических полимеров, металлических аморфных сплавов, водных растворов, халькогенидов, что подтверждается наличием в области стеклования универсальных уравнений и правил [4, 9–11, 13–16].
Вместе с тем, для разных классов стекол при Tg замораживается подвижность различных кинетических единиц. Стеклование линейных аморфных органических полимеров является процессом физической релаксации, связанным с потерей сегментальной подвижности при температуре стеклования, а стеклование неорганических стекол с сеточной структурой относится к процессам химической релаксации и обусловлено замораживанием процесса переключения валентных связей типа кремний–кислород–кремний. Стеклование густосшитых сетчатых полимеров (например, эпоксидов) также имеет свою специфику.
Факт существования универсальных закономерностей перехода жидкость–стекло означает, что процессы стеклования разных систем могут быть описаны общей теорией, способной предсказывать, например, характер эволюции макроскопических свойств системы.
Создание такой общей теории стеклования представляет собой исключительно сложную задачу, которая едва ли будет решена в ближайшие несколько десятилетий. Дискуссия о природе стеклования имеет столетнюю историю (см. обзоры [13–16]). Обрисованная картина ярко отражается в существовании большого количества феноменологических подходов. Кратко рассмотрим один из них.
Исходим из представления о том, что физической причиной резкого повышения энергии активации стеклования с понижением температуры является конфигурационное изменение структуры стеклообразующего расплава [15, 17, 18]. При этом микроскопическая природа конфигурационного структурного изменения не раскрывается. В развиваемой модели делокализованных атомов [19–22] формально фигурируют микроскопические понятия (“энергия делокализации атома”, “элементарный объем, необходимый для предельного смещения атома”). Однако модель в целом носит феноменологический характер и применима к различным аморфным системам.
В рамках этой модели у стеклообразующих жидкостей энергия активации стеклования (вязкого течения) в уравнении (1) представляет собой сумму двух слагаемых [14, 23]:
где U∞ – потенциал перехода кинетической единицы (атома, молекулы) в новое локальное положение, US(T) – потенциал конфигурационного изменения структуры, который является функцией температуры:(22)
${{U}_{s}}(T) = RT\left[ {\exp \left( {\frac{{\Delta {{{\varepsilon }}_{e}}}}{{RT}}} \right) - 1} \right]$Здесь ∆εe – энергия делокализации атома (его предельного смещения из локального равновесного положения). Делокализация атома обусловлена перегруппировкой соседних частиц (флуктуацией ближнего порядка) и отражает локальное конфигурационное изменение структуры.
При повышенных значениях температуры RT ≫ ∆εe потенциал конфигурационного структурного изменения US(T) = 0 и уравнение вязкости (1) с учетом выражений (21) и (22) описывает зависимость с постоянной энергией активации U = U∞:
Таким образом, величина U∞ оказывается высокотемпературным пределом энергии активации вязкого течения.
При низких значениях температуры в области стеклования энергия делокализации атома ∆εe оказывается сравнимой с энергией тепловых колебаний решетки (~3RT), и относительное число Ne/N делокализованных атомов, ответственных за текучесть выше показателя Tg, уменьшается по закону $\exp ( - \Delta {{{\varepsilon }}_{e}}{\text{/}}kT)$ [14]. Структура расплава уплотняется, и для активационного перескока атома в новое положение требуется предварительное локальное конфигурационное изменение (“разрыхление”) структуры вблизи него: потенциал конфигурационного структурного изменения US(T) резко возрастает. Этим объясняется практически экспоненциальный рост энергии активации текучести в области стеклования.
Идея разделения энергии активации текучести на две составляющие была высказана Я.И. Френкелем в книге [24]. Дальнейшее развитие она получила в работах В.Н. Филиповича и С.В. Немилова [17, 18], а также в трудах научной школы университета Шеффилда [25–27], в которых было получено двухэкспоненциальное уравнение вязкости. При более строгом подходе под U(T) следует понимать свободную энергию активации [17, 18, 28, 29], как в классических работах Эйринга.
Таким образом, конфигурационное изменение структуры, описываемое в рамках модели делокализованных атомов, оказывается ответственным за температурную зависимость энергии активации стеклования (вязкого течения) в области перехода из высокоэластического состояния в стеклообразное.
ЗАКЛЮЧЕНИЕ
Для аморфных органических полимеров и неорганических стекол рассмотрены способы расчета температурной зависимости энергии активации стеклования U(T). Данная зависимость удовлетворительно описывается вблизи значений Tg с привлечением уравнения Вильямса–Ландела–Ферри для вязкости (времени релаксации). Показано, что существует способ оценки U(T) с использованием разложения функции U(T) в ряд Тейлора вблизи температуры стеклования. В первом приближении вполне достаточно ограничиться линейным и квадратичным членами ряда. В рамках модели делокализованных атомов резкий рост энергии активации в области стеклования объясняется возрастанием потенциала конфигурационного изменения структуры при охлаждении стеклообразующего расплава.
Список литературы
Френкель Я.И. Кинетическая теория жидкостей. М.; Л.: Изд-во АН СССР, 1945.
Ферри Дж. Вязкоупругие свойства полимеров. М.: Изд-во иностр. лит.-ры, 1963.
Williams M.L., Landel R.F., Ferry J.D. // J. Am. Chem. Soc. 1955. V.77. № 14. P. 3701.
Сандитов Д.С., Бартенов Г.М. Физические свойства неупорядоченных структур. Новосибирск: Наука, 1982.
Bestul B.A. // Glastechn. Ber. 1959. Bd. K32. S. 59.
Сандитов Д.С., Доржиев Д.Б., Балданов Ж.П. // Журн. физ. химии. 1973. Т. 47. № 12. С. 2490.
Разумовская И.В., Бартенов Г.М. // Стеклообразное состояние. Тр. V Всесоюз. совещ. по стеклообразному состоянию. Л.: Наука, 1971. С. 34.
Sanditov D.S., Razumovskaya I.V. // Polymer Science A. 2018. V. 60. № 2. P. 156.
Ростиашвили В.Г., Иржак В.И., Розенберг Б.А. Стеклование полимеров. Л.: Химия, 1987.
Бартенев Г.М., Сандитов Д.С. Релаксационные процессы в стеклообразных системах. Новосибирск: Наука, 1986.
Бартенев Г.М., Бартенева А.Г. Релаксационные свойства полимеров. М.: Химия, 1992.
Мазурин О.В. // Физ. хим. стекла. 2007. Т. 33. № 1. С. 34.
Тропин Т.В., Шмельцер Ю.В.П., Аксенов В.Л. // Успехи физ. наук. 2016. Т. 186. № 1. С. 47.
Sanditov D.S., Ojovan M.I. // Physica. B. 2017. V. 523. P. 96.
Сандитов Д.С., Ожован М.И. // Успехи физ. наук. 2019. Т. 189. № 2. С. 113.
Schmelzer J.W.P. Glasses and the Glass Transition. Weinheim: Wiley, 2011.
Филипович В.Н. // Физ. хим. стекла. 1975. Т. 1. № 3. С. 256.
Немилов С.В. // Физ. хим. стекла. 1978. Т. 4. № 2. С. 129.
Сандитов Д.С. // Журн. эксперим. теорет. физики. 2012. Т. 142. Вып. 1 (7). С. 123.
Сандитов Д.С. // Журн. эксперим. теорет. физики. 2016. Т. 150. Вып. 3 (9). С. 501.
Sanditov D.S., Mashanov A.A. // Polymer Science A. 2019. V. 61. № 2. P. 119.
Сандитов Д.С., Бадмаев С.С. // Физ. хим. стекла. 2015. Т. 40. № 5. С. 621.
Сандитов Д.С. // Докл. PАН. 2013. Т. 451. № 6. С. 650.
Френкель Я.И. // Совещание по вязкости жидкостей и коллоидных растворов. М.; Л.: Изд-во АН СССР, 1944. Т. 2. С. 24.
Ojovan M.I., Lee W.E. // J. Non-Cryst. Solids. 2010. V. 356. S. 2534.
Ojovan M.I., Travis K.P., Hand R.J. // J. Phys. Condens. Matter. 2007. V. 19. S. 415107.
Ojovan M.I. // J. Non-Cryst. Solids. 2013. V. 382. S. 79.
Мюллер Р.Л. // Журн. прикл. химии. 1955. Т. 28. № 10. С. 1077.
Глесстон С., Лейдлер К., Эйринг Г. // Теория абсолютных скоростей реакций. М.: Изд-во иностр. лит-ры, 1948.
MDL® SciGlass-7.8. Institute of Theoretical Chemistry, Shrewsbury, MA, 2012. www.sciglass.info
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия А)