

Высокомолекулярные соединения (серия А), 2020, T. 62, № 6, стр. 453-465
ГЕТЕРОГЕННОСТЬ ЦЕПИ В МОДЕЛЬНЫХ ПОЛИМЕРНЫХ РАСПЛАВАХ. ОРИЕНТАЦИОННАЯ АВТОКОРРЕЛЯЦИОННАЯ ФУНКЦИЯ СЕГМЕНТОВ
Е. М. Пестряев *
Уфимский государственный нефтяной технический университет
450064 Уфа, ул. Космонавтов, 1, Россия
* E-mail: physics_usptu@mail.ru
Поступила в редакцию 13.01.2020
После доработки 18.06.2020
Принята к публикации 01.07.2020
Аннотация
Динамическая гетерогенность полимерной цепи, проявляющаяся в меньших временах корреляции концевых куновских сегментов цепи по сравнению со срединными, исследована методом молекулярной динамики для свободносочлененных цепей разной длины в расплаве. Обнаружена гетерогенность анизотропии вращения, которая также меньше для концевых сегментов по сравнению со срединными. Гетерогенность анизотропии вращения количественно характеризуется остаточным внутрисегментальным диполь-дипольным взаимодействием, которое влияет на скорость поперечной магнитной релаксации различных сегментов в цепи и, в итоге, определяет форму спада свободной индукции цепи любой длины.
ВВЕДЕНИЕ
Увеличенная подвижность концевых групп цепи в расплаве, связанная с внесением ими большего свободного объема, была гипотезой, основанной на пропорциональности удельного объема полистирола обратной молекулярной массе M [1]. Полученное из этого предположения эмпирическое уравнение Фокса–Флори для температуры стеклования используется до сих пор [2–6]. Очевидно, в этом случае динамическая гетерогенность полимерной цепи в расплаве является следствием взаимодействия с окружением. Для уединенной огрубленной модели цепи динамическая гетерогенность, заключающаяся в различии времен корреляции вращательного и трансляционного движения, а также коэффициентов самодиффузии куновских сегментов, находящихся на различных расстояниях от ее концов, была выявлена в раузовской теории фантомных цепей [7–10]. В ней данное различие является следствием связанности концевых частиц (сегментов) лишь с одним соседом, а всех остальных – с двумя, в результате чего среднеквадратичное смещение концевых сегментов в некоторый характерный момент времени вдвое превышает таковое для срединных. Согласно рептационной модели, появление зацеплений в расплаве приводит к росту отношения смещений концевых и срединных сегментов, которое при стремлении длины цепи к бесконечности стремится к значению ∼5.6 [11, 12]. В то же время предполагается, что подвижность убывает постепенно от концевого куновского сегмента до первого сегмента, участвующего в создании зацепления, т.е. на длине субцепи между соседними зацеплениями NE. Два таких подвижных концевых фрагмента (или субцепи) общей длиной 2NE определяют последующее направление одномерной диффузии всей цепи по мере ее выхода из гауссово-запутанной трубки. Выбор случайного направления ограничен полусферой, заметаемой концевыми фрагментами, временно зафиксированными своими внутренними концами в точке зацепления, в связи с чем данные фрагменты выглядят “болтающимися”. Последний термин получил широкое распространение при описании характера движения концевых фрагментов цепи в химически сшитых полимерных системах, концевые сегменты которых не обладали реакционной способностью перед сшиванием [13–23]. Наличие таких болтающихся фрагментов позволило объяснить длительность механических релаксационных процессов в сшитых системах [13], хорошо описываемых в рамках рептационной модели [14].
Ниже термин “болтающийся концевой фрагмент” цепи используется в обсуждении влияния концевых фрагментов на форму спада свободной индукции ЯМР зацепленных цепей в расплавах с молекулярной массой больше критической молекулярной массы MC. Проявлением их увеличенной подвижности считается медленно релаксирующий хвост ССИ, населенность которого pE уменьшается с увеличением количества куновских сегментов в зацепленной цепи N [23–45]:
При подгонке формы ССИ его хвост описывается эмпирическим аддитивным вкладом [29, 46], который аналитически аппроксимируется в предположении раузовского движения болтающихся концевых фрагментов с сомножителем, в два–три раза увеличивающим их время корреляции по сравнению с отдельной цепью [23, 26, 27]. В многочисленных теориях ССИ полимерных расплавов всегда преследовалась цель получения выражения лишь для доминирующего вклада в ССИ от срединных заторможенных сегментов длинных цепей [23, 47–60]. Данное обстоятельство также демонстрирует признанную гетерогенность динамических свойств зацепленной цепи, которые невозможно описать в рамках одного аналитического выражения. Соответственно количественно результирующий ССИ характеризовался временем уменьшения его амплитуды e в ≈2.72 раза T2EF: очевидно, основной вклад в эту величину при больших M дают именно срединные сегменты.
Аппроксимация хвоста ССИ формулой (1) всегда была лишь качественной: из нее получались значения MC, существенно отличающиеся в меньшую сторону от полученных в реологии. Попытка увеличить величины pE исследованием поперечной магнитной релаксации в звездообразных полимерах также не привела к количественному соглашению с формулой (1) [23, 26, 27]. Тем не менее, исследования ССИ в расплавах специально синтезированных триблок-сополимеров [28, 40] и звездообразных полимеров [26, 27], в которых срединная часть цепи и ее концевые фрагменты имели разные резонирующие ядра (дейтроны и протоны), позволила обнаружить плавное уменьшение T2EF от концов болтающихся фрагментов к точке их временного закрепления или ветвления в звездообразных полимерах. Убедительный пример мультиплетности вращения закрепленных на одном конце цепочек получен при атомистическом моделировании дендримеров [61–63] и исследовании их поперечной релаксации [64, 65], где показано убывание времени корреляции при удалении от точки ветвления.
Попытка избавиться в сигнале ЯМР от хвоста ССИ добавлением к цепи полибутадиена двух концевых пердейтерированных блоков c длиной, соответствующей населенности хвоста, привела лишь к полутора-двукратному уменьшению pE в расплаве и растворах [40]. Этот результат является неожиданным, поскольку в работе убедительно продемонстрирована качественная применимость формулы (1) к исходным гомополимерам. Для его объяснения K. Saalwachter [46] предложил пересмотреть ту часть теорий полимерной динамики и упругости, которая обусловлена кооперативностью движения полимерных цепей. Однако вероятнее полученное противоречие нужно анализировать с точки зрения термодинамической неравновесности и предрасположенности к микрорасслоению смесей протонированного полимера с его пердейтерированным аналогом [66–71]. Очевидная сложность такого эксперимента, требующего специально синтезированных триблок-сополимеров с разной длиной пердейтерированных концевых блоков и их фазовых диаграмм, пока не позволила решить выявленную J.-P. Cohen-Addad [40] проблему.
В отличие от авторов работы [40] P.G. Klein с соавторами сумели избавиться от хвоста ССИ: на триблок-сополимере полибутадиена с концевыми протонированными блоками с длиной, существенно большей MC ≈ 2 × 103, и центральным пердейтерированным блоком при резонансе на дейтерии наблюдалась лишь ожидаемая начальная компонента ССИ [28]. Интересно отметить, что такая ситуация укладывается в рамки гипотезы о микрофазном расслоении, которое в рассматриваемом расплаве может привести к сегрегации срединных частей цепей, немного укорачивающей время корреляции сегментов и не вызывающей появление гетерогенности вдоль цепи, которая бы проявилась в виде хвоста ССИ.
Противоположная по смыслу, но вновь неожиданная ситуация описана в недавней работе [60], где наблюдался дейтериевый ССИ расплава высокомолекулярного поли(этилен-альт-пропилена) без медленно релаксирующего хвоста, который можно было бы отнести к концевым фрагментам цепи. Авторы предположили, что населенность данного хвоста слишком мала, чтобы быть зафиксированной в эксперименте, хотя ее теоретическое значение, согласно формуле (1), более чем десятикратно превышает уровень затухания приведенного ССИ и видимых на нем шумов.
Таким образом, краткий обзор начатых более сорока лет назад [29, 47] исследований полимерных расплавов с помощью поперечной ЯМР-релаксации показывает, что проблема полной формы ССИ весьма сложна и выходит за рамки аналитических решений для магнитной релаксации. В связи с этим в настоящей работе предпринята попытка полуколичественного описания гетерогенности свободносочлененных полимерных цепей в расплаве с помощью компьютерного моделирования – кратко она была упомянута в контексте работ [72, 73]; основной целью является развернутое обоснование такого описания на основе данных молекулярно-динамического моделирования [74–76] поперечной магнитной релаксации [77–79] полимерных расплавов в диапазоне длины цепей от раузовских до зацепленных. Однако форма ССИ однозначно определяется динамикой сегментов цепи. Информацию о ней и пытаются получить в экспериментах с помощью существующих теоретических представлений, в которых гетерогенность динамических свойств сегментов не фигурирует. Для выяснения влияния гетерогенности на форму различных компонентов ССИ необходимо вначале получить ее детальную картину в компьютерном моделировании. Иначе говоря, задача решается в два этапа, которые изложены в двух последовательных статьях.
Настоящая статья посвящена описанию заданных параметров и вытекающих из них свойств модельного расплава в диапазоне от олигомеров до зацепленных цепей – приведено развернутое доказательство такого перехода. Для всего рассмотренного диапазона длины цепей построены ориентационные автокорреляционные функции (ОАКФ) отдельных куновских сегментов в цепи – уже на этом уровне проявляется гетерогенность динамики вдоль цепи. Дано количественное описание второго типа гетерогенности, присущей концевым фрагментам цепи – анизотропии вращения, к которой весьма чувствительна форма ССИ [80–83]. Детальное моделирование ССИ с использованием всех выявленных особенностей ОАКФ описано во второй статье.
ОСНОВНЫЕ ПАРАМЕТРЫ МОДЕЛЬНЫХ РАСПЛАВОВ
Первый шаг обсуждаемого компьютерного моделирования расплавов свободносочлененных полимерных цепей – задание структуры и взаимодействий частиц в системе можно просто определить ссылкой на давно апробированную модель Кремера–Греста [84]. Следующим самым емким по времени шагом является генерация фазовой траектории [74–76], которая представляет собой совокупность координат ri(t) (и скоростей, здесь не используемых) всех леннард-джонсовских частиц системы, записанных для последовательных k-х моментов времени t = kΔt, дискретно меняющегося с заданным шагом Δt. Время измеряется в единицах безразмерного модельного времени τ0 = σ (m/ε)1/2 = 1, где σ = m = ε = 1, σ, m, ε – диаметр и масса частиц, а также глубина леннард-джонсовского потенциала, определяющего зависимость энергии взаимодействия от расстояния между ними. Безразмерная температура модельных расплавов, измеряемая в единицах ε, во всех случаях равна единице, что при плотности 0.96 частиц на единицу объема отвечает конденсированному состоянию системы [74, 75, 84]. Генерация производилась с помощью классических алгоритмов, программная реализация которых для персональных компьютеров описана в работах [85–88].
Для достижения приемлемого отношения сигнал : шум при расчетах индивидуальных ОАКФ куновских сегментов в высокомолекулярных расплавах фазовая траектория должна быть как можно длиннее – в противном случае хвосты затухающих ОАКФ теряются в шумах. Этому требованию препятствует не только большое время генерации траектории, но и минимальная необходимая для расчетов дискретность записи на жесткий диск Δt. Чем меньше Δt, тем точнее воспроизводятся детали рассчитываемых физических функций на малых временах, что влечет за собой соответствующее увеличение объема записываемой информации [89, 90], который составляет для используемой модели сотни гигабайт на один модельный расплав. В связи с этим при расчетах ОАКФ зацепленных расплавов использовались как готовые фазовые траектории [91], так и сгенерированные дополнительно для той же модели, но содержащей уже не 32 768, а 64 000 частиц. Некоторые среднеквадратичные смещения (СКС) различных элементов цепи, приведенные для демонстрации явной трансформации динамики цепей в расплаве от раузовской к зацепленной, также были получены в работе [91].
Ниже коротко остановимся на характеристиках огрубленной модели макромолекулы, необходимых для построения автокорреляционной функции (АКФ) и ОАКФ различных ее элементов. Свободносочлененная цепь, состоящая из N + 1 леннард-джонсовских частиц, имеет N сегментов: номер первой по порядку частицы является номером сегмента, связывающего ее со следующей частицей. Частицы являются носителями массы, а упругие связи-сегменты между ними – носителями ориентации, моделирующими межспиновый вектор и используемыми для построения АКФ. Непрерывное ван-дер-ваальсово взаимодействие цепей в конденсированной фазе, а значит и зацепления, обеспечивается возникновением клеточного эффекта по всему объему системы, что происходит при достижении определенного значения плотности [91–94]. Для реализации таких свойств модельного расплава была взята количественная плотность частиц 0.96, несколько превышающая общепринятую – 0.85 [74, 84], что уменьшило производительность вычислений. Однако такая плотность частиц обеспечила относительно малую длину субцепи между соседними зацеплениями вдоль цепи
Последняя была определена с использованием базовой абстракции рептационной модели – примитивной цепи длиной LP, введенной S.F. Edwards в 1977 г. [8, 95], и алгоритма ее построения, предложенного лишь в 2004 г. [96]. В предположении гауссовой конформации исходной и примитивной цепей NE вычисляется по простой формуле [8, 10, 95, 96]
(2б)
${{N}_{{\mathbf{E}}}}{\text{/}}N = \langle R_{{{\mathbf{EE}}}}^{2}\rangle {\text{/}}\langle L_{{\mathbf{P}}}^{2}\rangle ,$Получаемая из нее величина NE оказалась чувствительна к возникновению зацеплений [91]: она растет с N для олигомерных цепей и перестает изменяться после перехода через NC, т.е. вполне может служить характеристикой возникновения сетки зацеплений в расплаве. Однако традиционно даже в компьютерном моделировании о появлении зацеплений судят по молекулярно-массовым зависимостям физических величин, которые могут быть прямо или косвенно проверены в экспериментах, например, по зависимостям для коэффициента самодиффузии, вязкости, модуля упругости и т.п. [97–107].
ПОСТРОЕНИЕ ДИНАМИЧЕСКИХ ХАРАКТЕРИСТИК МОДЕЛЬНЫХ РАСПЛАВОВ
Чтобы иметь возможность спроецировать свойства модельных расплавов на реальные, вначале покажем, что с увеличением длины цепи они демонстрируют все теоретические режимы самодиффузии рептационной модели, верифицированные как в экспериментах [108, 109], так и в моделировании [99–107]. Для этого обычно используются СКС срединных куновских сегментов цепи ց1(t), центра масс цепи ցC(t) и тех же сегментов относительно центра масс ց1С(t):
(3a)
${{g}_{1}}(t) = \left\langle {{{{\left[ {{{r}_{{\frac{{\mathbf{N}}}{2}}}}(t{\kern 1pt} '\, + t)--{{r}_{{\frac{{\mathbf{N}}}{2}}}}(t{\kern 1pt} ')} \right]}}^{2}}} \right\rangle $(3б)
${{g}_{{\mathbf{C}}}}(t) = \langle {{[{{r}_{{\mathbf{C}}}}(t{\kern 1pt} '\, + t)--{{r}_{{\mathbf{C}}}}(t{\kern 1pt} ')]}^{2}}\rangle $(3в)
${{g}_{{1C}}}(t) = \left\langle {{{{\left[ {{{r}_{{\frac{{\mathbf{N}}}{2}}}}(t{\kern 1pt} '\, + t)--{{r}_{{\frac{{\mathbf{N}}}{2}}}}(t{\kern 1pt} ')--{{r}_{{\mathbf{C}}}}(t{\kern 1pt} '\, + t) + {{r}_{{\mathbf{C}}}}(t{\kern 1pt} ')} \right]}}^{2}}} \right\rangle $Угловые скобки подразумевают усреднение по ансамблю идентичных кинетических единиц и начальному моменту t', который сканирует всю фазовую траекторию с шагом, превышающим время когерентности статистических шумов [110, 111]. Последнее определяется как минимальное t', после которого накопление сигнала приводит к уменьшению уровня шумов на длинновременном хвосте кривой. Следовательно, фазовая траектория должна быть сгенерирована и записана на интервале времени, как минимум на порядок превышающем интервал расчета СКС. Это условие просто достигается для СКС на малых временах, и весьма сложно – на максимальных, и для расчета СКС в разных временных интервалах генерируемая фазовая траектория записывалась с увеличивающимся шагом. После расчета СКС из разных интервалов “сшивались”. При сшивании иногда образовывались кривые с разрывами вследствие изъятия сильно зашумленных участков отдельных кривых на длинновременном хвосте. Такие разрывы не мешают оценке хода зависимостей в отличие от кривых, сильно “размытых” шумом; более того, при наложении СКС нескольких расплавов в интервале не зависящих от N диффузионных режимов кривые накладываются. При этом вертикальное смещение кривых относительно друг друга делает видимой погрешность их расчета, не превышающую толщину линий.
В дополнение к СКС использовались нормированные АКФ, определяемые для произвольной физической величины A (возможно, векторной) общей формулой [9, 24, 25]
(4а)
${{C}_{A}}(t) = \frac{{A(t{\kern 1pt} ') \cdot A(t{\kern 1pt} '\, + t)}}{{{{{[A(t{\kern 1pt} ')]}}^{2}}}}.{\text{\;}}$Подстановка REE(t) в выражение (4а) позволяет вычислить его вращательную АКФ, время корреляции которой τN является функцией длины цепи. Связь магнитной релаксации с тепловыми флуктуациями спин-несущих кинетических единиц молекул реализуется через нормированную ОАКФ этих единиц [77–80], получающуюся при подстановке в формулу (4а) полинома Лежандра второго порядка
(4б)
$A(t) = {{P}_{2}}[{{{\theta }}_{i}}(t)] = \frac{1}{2}[3{\text{co}}{{{\text{s}}}^{2}}{{{\theta }}_{i}}(t)--1]{\text{\;}}$Здесь θi(t) – угол i-го межспинового вектора рассматриваемой кинетической единицы по отношению к заданному направлению в пространстве, например REE или постоянному магнитному полю, как в экспериментах. В последнем случае для свободносочлененной цепи межспиновый вектор отождествляется с вектором куновского сегмента lK(t) с временем ориентационной корреляции τK [78, 79]. В компьютерном моделировании с целью дополнительной характеристики зависящего от длины цепи вращательного движения в качестве межспинового вектора использовался и сам REE, ОАКФ которого затухает со временем τEE.
ДИФФУЗИОННЫЕ РЕЖИМЫ КУНОВСКИХ СЕГМЕНТОВ В РАМКАХ РЕПТАЦИОННОЙ МОДЕЛИ
Теоретические зависимости для построенных СКС зацепленных цепей описываются несколькими последовательными асимптотическими режимами, доминирующими в определенных интервалах времени и пространственного масштаба [8, 10, 24, 106]:
(5а)
${\text{ }}{{g}_{1}}({{\tau }_{1}} \ll t \ll {{\tau }_{{\mathbf{E}}}}) = \frac{2}{{{{{\pi }}^{{3/2}}}}}l_{{\mathbf{K}}}^{2}{{\left( {\frac{t}{{{{\tau }_{1}}}}} \right)}^{{\frac{1}{2}}}}$(5б)
$l_{{\mathbf{K}}}^{2} \ll {{g}_{1}} \ll d_{{\mathbf{T}}}^{2} = {{N}_{{\mathbf{E}}}}l_{{\mathbf{K}}}^{2},\quad {{\tau }_{{\mathbf{E}}}} = N_{{\mathbf{E}}}^{2}{{\tau }_{1}}$(6а)
${{g}_{1}}\left( {{{\tau }_{{\mathbf{E}}}} \ll t \ll {{\tau }_{{\mathbf{R}}}}} \right) = N_{{\mathbf{E}}}^{{\frac{1}{2}}}\,l_{{\mathbf{K}}}^{2}{{\left( {\frac{t}{{{{\tau }_{1}}}}} \right)}^{{\frac{1}{4}}}}$(6б)
$d_{{\text{T}}}^{2} \ll {{g}_{1}} \ll {{d}_{{\text{T}}}}{{R}_{{{\mathbf{EE}}}}},\quad {{\tau }_{{\mathbf{R}}}} \propto {{N}^{\delta }}{{\tau }_{1}}$(7а)
${{g}_{1}}({{\tau }_{{\mathbf{R}}}} \ll t \ll {{\tau }_{{\mathbf{D}}}}) = \frac{2}{{\pi }}l_{{\mathbf{K}}}^{2}{{\left( {\frac{{{{N}_{{\mathbf{E}}}}}}{{3N}} \cdot \frac{t}{{{{\tau }_{1}}}}} \right)}^{{\frac{1}{2}}}}$(7б)
${{d}_{{\mathbf{T}}}}{{R}_{{{\mathbf{EE}}}}} \ll {{g}_{1}} \ll R_{{{\mathbf{EE}}}}^{2},\quad {{\tau }_{{\mathbf{D}}}} = 3\frac{{{{N}^{3}}}}{{{{N}_{{\mathbf{E}}}}}}{{\tau }_{{\mathbf{E}}}}$(8)
${{g}_{1}}({{\tau }_{{\mathbf{D}}}} \ll t) = \frac{2}{{3{{{\pi }}^{2}}}} \cdot \frac{{{{N}_{{\mathbf{E}}}}}}{{{{N}^{2}}}}l_{{\mathbf{K}}}^{2}\left( {\frac{t}{{{{\tau }_{1}}}}} \right)$(9а)
${{g}_{{1{\mathbf{C}}}}}\left( {t \leqslant {{\tau }_{{\mathbf{E}}}}} \right) = {{g}_{{\mathbf{C}}}}\left( t \right),\quad {{g}_{{1{\mathbf{C}}}}}\left( {t > {{\tau }_{{\mathbf{R}}}}} \right) = R_{{\mathbf{G}}}^{2}$(9б)
${{g}_{{\mathbf{C}}}}({{\tau }_{{\mathbf{R}}}}) \approx 2R_{{\mathbf{G}}}^{2},\quad R_{{\mathbf{G}}}^{2} = \frac{1}{6}R_{{{\mathbf{EE}}}}^{2}$Здесь τ1 – время трансляционной корреляции куновского сегмента, которое определяет все остальные крупномасштабные времена: время зацеплений τE, раузовское время τR и время выхода из трубки τD; dТ – диаметр абстрактной силовой трубки, ограничивающей латеральное движение цепи, RG – радиус инерции цепи; δ ≈ 2 для олигомерных цепей и δ ≈ 3 для зацепленных цепей. Совокупность перечисленных СКС, представленная на рис. 1 в логарифмических координатах, позволяет определить $R_{{\text{G}}}^{2}$ как плато кривой 2, согласно выражению (9а), и τR как момент пересечения кривой 3 и координаты $2R_{{\text{G}}}^{2}$, согласно формуле (9б).
Рис. 1.
Зависимость от времени СКС (1–3) и АКФ (4–6) различных элементов цепи в расплаве с N = 127. 1, 4 – срединные куновские сегменты; 2 – они же относительно центра масс цепи; 3 – центр масс цепи; 4 – ОАКФ куновского сегмента; 5 – ОАКФ REE; 6 – АКФ REE. Стрелки отмечают значения 2R$_{{\text{G}}}^{2}$ и τR на осях; горизонтальная линия пересекает все АКФ (4–6) в точках затухания в e раз. Цветные рисунки можно посмотреть в электронной версии.
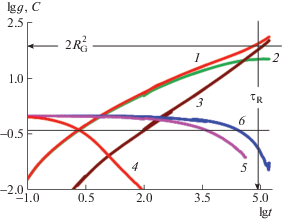
Извлечение дальнейшей информации основано на идентификации точек перегибов СКС, отделяющих последовательные режимы диффузии согласно формулам (5)–(9). Для акцентирования перегибов, ց1(t) для цепей с различными N представлены на рис. 2 в приведенных координатах, делающих участок с режимом диффузии ∼t1/2 горизонтальным. В связи с этим по прошествии времени τE, когда наблюдается переход в режим диффузии ∼t1/4, кривая проходит через максимум, положение которого определяет величину τE = 300 ± 20. Данный максимум, согласно рептационной модели, отсутствует для раузовских цепей (кривые 1–3) и совпадает для всех зацепленных цепей (кривые 4–6), т.е. качественное поведение всех кривых полностью соответствует модели.
Рис. 2.
Зависимость от времени СКС срединных куновских сегментов, приведенные к режиму ∼t1/2, для цепей с длиной N = 7 (1), 15 (2), 31 (3), 63 (4), 127 (5) и 255 (6). Стрелка τE отмечает общий для всех зацепленных цепей момент смены режима ∼t1/2 на ∼t1/4, стрелка τR – значение τR для самой длинной цепи, показывающей точный режим ∼t1/4.
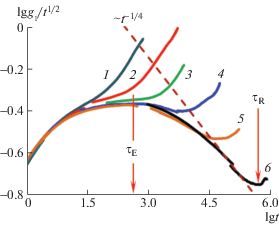
Следующая особенность, также предсказываемая моделью, – все кривые на рис. 2 заканчиваются резким подъемом, который начинается по достижении τR цепи с данным N и обусловлен переходом к режиму диффузии ∼t1/2 в суперпозиции с режимом ∼t. Последнее обстоятельство объясняется сравнительно малой величиной NE, определяющей отличие разномасштабных времен корреляции как видно в формулах (5)–(9). Тем не менее, указанный подъем, который для раузовских цепей лежит левее τE, а для зацепленных – правее, позволяет продублировать оценку значения τR в дополнение к методике, основанной на рис. 1, и уменьшить статистический разброс его зависимости от длины цепи. Кривые 4, 5 и 6 на рис. 2 сливаются от самых малых времен наблюдения до τR каждой цепи, что свидетельствует о совпадении их мелкомасштабной и среднемасштабной подвижности в течение времени пребывания в трубке. При этом лишь самая длинная цепь с N = 255 демонстрирует режим диффузии ∼t1/4, который у более коротких зацепленных цепей не проявляется вследствие суперпозиции с предыдущим режимом ∼t1/2.
Затухающие кривые на рис. 1 представляют собой различные АКФ, время корреляции которых вследствие неэкспоненциальности затухания определяется как время спада в e ≈ 2.72 раза. Быстроспадающая кривая 4 – ОАКФ куновского сегмента, с не зависящим от длины цепи временем ориентационной корреляции τК = 2.8 ± 0.3. Кривые 5 и 6 – крупномасштабные ОАКФ и АКФ REE соответственно; они приведены для сопоставления τR с их временами корреляции τEE и τN, определяемыми точками пересечения кривых с горизонтальной линией lgе.
ЗАВИСИМОСТИ КРУПНОМАСШТАБНЫХ ХАРАКТЕРИСТИК ЦЕПИ ОТ ЕЕ ДЛИНЫ
Вторичные данные, полученные обработкой кривых на рис. 1 и 2, представлены на рис. 3 в логарифмических координатах. Зависимость $R_{{\text{G}}}^{2}$ от длины цепи не проявляет каких-либо особенностей во всем диапазоне исследованных N, демонстрируя неизменность структуры расплава в интервале 5 ≤ N ≤ 255 после того, как цепь начинает образовывать клубок, для которого радиус инерции практически без статистического разброса аппроксимируется уравнением
(10)
${\text{R}}_{{\mathbf{G}}}^{2} \approx \frac{1}{6}{{N}^{{1.07}}}l_{{\mathbf{K}}}^{2},\quad l_{{\mathbf{K}}}^{2} \approx 1.2$Рис. 3.
Зависимости радиуса инерции (1) и раузовского времени (2) от длины цепи. Большие светлые круги – $R_{{\text{G}}}^{2}$ субцепей разной длины внутри цепи с N = 255. Показатель степени линии тренда ∼1.07 получен без учета двух самых нижних точек. Вертикальная штрихпунктирная линия отмечает значение NC ≈ ≈ 35, перенесенное с рис. 4.
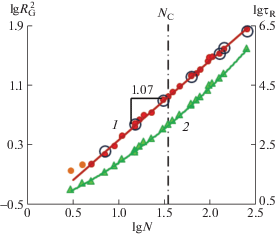
Это позволяет сделать вывод о том, что используемые модельные расплавы воспроизводят хорошо известное свойство реальных расплавов, установленное в теоретическом описании [8–10, 112, 113], моделировании [99, 114, 115] и эксперименте [116, 117]. Отличие показателя степени ∼1.07 от теоретического значения для фантомных цепей, равного единице, объясняется влиянием исключенного объема [113, 118]. Совпадение с основной зависимостью больших окружностей, отвечающих $R_{{\text{G}}}^{2}$ субцепей в составе длинной цепи, свидетельствует о достижении ими равновесной конформации, что является достаточно сложной задачей, как указано в работах [119–126]. Кривая 2 на рис. 3 для τR в этом же диапазоне длин цепей демонстрирует слабую вогнутость, по которой визуально невозможно определить ни положение, ни даже наличие NC. В связи с этим на рис. 4 показаны логарифмические зависимости трех крупномасштабных времен корреляции от длины цепи, приведенные к теоретическому скейлинговому режиму раузовской фантомной цепи τR ∝ N 2, т.е. поделенные на N 2 [7–10]. Все три кривые – τN(N), τR(N) и τЕЕ(N) имеют резкий перегиб при NC, значение которого определяется как точка пересечения трех пар асимптотических линий тренда, поскольку, согласно рептационной модели, все крупномасштабные времена корреляции должны реагировать на появление зацеплений увеличением показателя степени скейлинговых зависимостей τi ∝ N δ, в отличие от $R_{{\text{G}}}^{2}$.
Рис. 4.
Зависимость от длины цепи приведенных времен корреляции τN/N 2 (1), τR/N2 (2) и τEE/N2 (3). Числа около прямых углов – показатели степени асимптотических линий тренда, проведенных без учета точек вблизи NC ≈ 35, определяемого усредненным пересечением трех пар линий тренда.
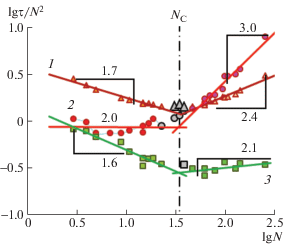
Значение NC ≈ 35 ± 2, полученное из рис. 4, на первый взгляд не согласуется с величиной NE ≈ 14.5, поскольку должно отличаться от нее вдвое как в формуле (1). Однако несогласованность легко снимается графическим анализом [91] множества экспериментальных данных по NC и NE [97, 98], которые укладываются на линейную зависимость с большим статистическим разбросом
Сравнительное небольшие значения NE и NC для модельных расплавов, с одной стороны, не позволили получить все асимптотические режимы диффузионного смещения, а, с другой – дали возможность смоделировать как докритические расплавы c N = 7, 15 и 31, так и расплавы зацепленных рептирующих цепей c N = 63, 127 и 255. Последние три зацепленные цепи на рис. 2 характеризуются полностью совпадающими приведенными СКС вплоть до момента τE, перед которым есть короткое плато, отвечающее режиму ∼t1/2, согласно формуле (5а), из которой находим
(12)
${{\tau }_{1}} = \frac{t}{{{{\pi }^{3}}}}{{\left[ {\frac{{2l_{{\mathbf{K}}}^{2}}}{{{{g}_{1}}(t)}}} \right]}^{2}} \approx 1.1 \pm 0.1{\text{\;}}$Для данной сложной зависимости цифровая обработка удобнее, чем обычная графическая обработка данных в координатах рис. 1 или рис. 2: подстановка любых пар значений ց1(t) – t дает близкие величины τ1, что было использовано для усреднения и нахождения погрешности τ1 по нескольким десяткам дискретных точек в рассматриваемом интервале. Далее подставляя значение τ1 в формулу (5б), модифицированную показателем степени из формулы (10), получаем два интервала теоретических предсказаний τE:
Сравнение с полученной из рис. 2 величиной τE = 300 ± 20, показывает, что используемый модельный расплав достаточно хорошо описывается предсказаниями рептационной модели как для фантомной цепи (формула (13а)), так и для цепи с исключенным объемом (формула (13б)).
Дополнительным подтверждением предсказательной способности применяемой модели служит совпадение показателей степени линий тренда (числа около прямых углов на рис. 4) для τR с теоретическими значениями в (6б). Расчет по соотношению (6б) дает значения τR, зависимость которых от длины цепи в пределах ошибки численного эксперимента (размер точек на рис. 3) совпадает с кривой 2. Подобная воспроизводимость предсказанного рептационной моделью длинновременного поведения аксиальных СКС срединных куновских сегментов наблюдалась для всех зацепленных цепей в работе автора [127].
Описанная согласованность характерных времен рептационной модели нарушается, если добавить значение τE, определенное на рис. 2 точкой пересечения экстраполированных диффузионных режимов ∼t1/2 и ∼t1/4, согласно формулам (5а) и (6а). Такой способ дает значение τE в 25 ± 3 раз большее, чем в формуле (13б). Подобная несогласованность была обнаружена при анализе экспериментальных данных по дисперсии спин-решеточной релаксации в рамках рептационной модели [128, 129]. А именно, при попытке сопоставить мелко- и среднемасштабную подвижность в расплаве ПБ, анализируя реологические данные в рамках рептационной модели и принципа температурно-временной суперпозиции, было установлено, что τE, полученное из перегиба СКС, в ∼42 раза выше, чем τE, рассчитанное по формуле (13а). Для ПДМС аналогичное различие составляло два десятичных порядка. Авторы [128, 129] не смогли объяснить такое расхождение погрешностями измерения, хотя использованные методы дают большой разброс значений τE вследствие диффузности переходов, наблюдаемых на логарифмических зависимостях СКС срединных сегментов цепи. Для объяснения этого рассогласования предложены новые методики обработки исходных экспериментальных зависимостей [130], включая учет аппаратурных погрешностей [131].
Продемонстрировав возможности используемой модели воспроизводить известные свойства зацепленных расплавов, в рамках существующих в периодике пока никем не понятых рассогласований, перейдем к рассмотрению редко обсуждаемых теоретических характеристик, исследование которых является целью статьи, причем некоторые из них даже в компьютерном моделировании пока могут быть оценены лишь косвенным образом.
ОАКФ ЦЕПЕЙ РАЗНОЙ ДЛИНЫ В РАСПЛАВЕ
Реальная цепь имеет несколько характерных времен вращательного движения, каждое из которых контролирует свою ОАКФ, полученную по формуле (4а) при подстановке в нее соответствующего межспинового вектора: минимальное из них – время корреляции метиленовых протонов вследствие изомеризации остова цепи τM; следующее, большее по величине на два–три порядка, время корреляции куновских сегментов τК; а максимальное, с еще большим отличием от предыдущего, у межконцевого вектора цепи τEE. Пространственная переориентация мономеров происходит вследствие всех трех типов движения, поэтому их полная ОАКФ CМ представляет собой произведение ОАКФ мономеров относительно сегмента CМK, сегмента CKE относительно REE и самого REE в пространстве CEE [8, 79–83]:
(14а)
${{C}_{{\mathbf{M}}}}\left( t \right) = {{C}_{{{\mathbf{MK}}}}}\left( {\frac{t}{{{{{\tau }}_{{\mathbf{M}}}}}}} \right){{C}_{{{\mathbf{KE}}}}}\left( {\frac{t}{{{{{\tau }}_{{\mathbf{K}}}}}}} \right){{C}_{{{\mathbf{EE}}}}}\left( {\frac{t}{{{{{\tau }}_{{{\mathbf{EE}}}}}}}} \right),{\text{\;}}$(14б)
${{C}_{{ij}}}(t) = (1--S_{{ij}}^{2})\exp \left( {--\frac{t}{{{{\tau }_{i}}}}} \right) + S_{{ij}}^{2},\quad {{\tau }_{{\mathbf{K}}}} \ll {{\tau }_{{{\mathbf{EE}}}}}.$Во-первых, отметим, что для свободносочлененной цепи внутрисегментальное движение отсутствует, поэтому опустим первый сомножитель в формуле (14а): тогда CМ(t) трансформируется в CK(t) и остается только один фактор порядка сегментов цепи по отношению к ее концам SKE > 0, поскольку для свободной цепи в расплаве фактор порядка равен нулю. Во-вторых, для определенности понятия времени корреляции все ОАКФ, как принято [80–83], аппроксимированы экспонентами, что при сильном различии времен корреляции позволяет записать произведение (14а) в виде суммы
(15)
${{C}_{{\mathbf{K}}}}(t) \approx (1--S_{{{\mathbf{KE}}}}^{2}){{C}_{{{\mathbf{KE}}}}}\left( {\frac{t}{{{{\tau }_{{\mathbf{K}}}}}}} \right) + S_{{{\mathbf{KE}}}}^{2}{{C}_{{{\mathbf{EE}}}}}\left( {\frac{t}{{{{\tau }_{{{\mathbf{EE}}}}}}}} \right).{\text{\;}}$SKE определяет плато CKE [132, 133], обусловленное анизотропией движения сегментов и определяемое формулой Куна–Грюна [46, 134–136], несущей в общем случае структурную информацию об эффективном количестве независимых кинетических единиц NEF между концами REE некоторой цепи или субцепи в ее составе:
(16)
${{C}_{{{\mathbf{KE}}}}}(\infty ) = S_{{{\mathbf{KE}}}}^{2} = {{\langle {{P}_{2}}\left[ {{{{\theta }}_{i}}(t)} \right]\rangle }^{2}} = \left\langle {{{{\left( {\frac{3}{{5 \cdot {{N}_{{{\mathbf{EF}}}}}}}} \right)}}^{2}}} \right\rangle {\text{\;}}$Первые угловые скобки подразумевают усреднение по конформациям цепи (зависимость P2 от времени) и по ансамблю рассматриваемых кинетических единиц, включая более мобильные концевые фрагменты. Поскольку θi(t) зависит от времени благодаря высокочастотным флуктуациям куновских сегментов, здесь взаимозаменяемо усреднение по конформациям различных примитивных сегментов и по времени в пределах любого из них, и требование t → ∞ в большинстве случаев легко удовлетворяется. Однако в итоге получается статистическая характеристика NEF, которая даже в пределах одной цепи вследствие распределения длины примитивных сегментов [21] характеризуется стационарным распределением и вытекающим из него распределением SКE, проявляющимся в ССИ [73]. Кроме того, во избежание недоумения при возникновении расхождений между теоретической величиной SКE и ее значениями в моделировании необходимо отметить, что формула (16) выведена для изолированной фантомной цепи [134, 135], а значит при ограничении доступного объема окружающими цепями в расплаве или растворе может дать лишь нижнюю оценку величины.
Для возникновения заметного плато на ОАКФ (15) различие во временах корреляции в формуле (14б) должно составлять несколько порядков, что бывает очень редко [137–141] и в используемой модели также не выполняется. Для выявления плато, согласно формуле (15), необходимо сделать τEE бесконечно большим, т.е. в эксперименте изучать полимерную сетку. В моделировании это делается построением ОАКФ не по отношению к направлению внешнего магнитного поля, а по отношению к REE каждой цепи. Первый вариант представлен на рис. 5а для нескольких коротких цепей с N < NC. Совпадение начальных участков всех ОАКФ указывает на идентичность мелкомасштабной подвижности, как и на рис. 2, а увеличение τEE с длиной цепи отражается в уменьшении наклона кривых на конечном наблюдаемом участке. Уже для цепей с N = 23 и 31 расхождение хвостов ОАКФ находится в пределах ширины шумовой дорожки, поэтому для более длинных цепей кривые не показаны. Необходимо отметить, что проблема отношения сигнал : шум ограничивает возможности компьютерного моделирования [90, 142, 143] так же, как и эксперимента [144–152]. Например, в работе [143] на основе атомистического моделирования расплава полипропиленоксида построены ОАКФ для цепей разной длины, демонстрирующие слияние конечных участков кривых при затухании ниже уровня 0.001 вследствие уширения шумовых дорожек.
Рис. 5.
Логарифмическое представление ОАКФ куновских сегментов относительно внешнего магнитного поля (а) и межконцевого вектора цепи (б) в расплаве цепей c числом куновских сегментов N = 3 (1), 7 (2), 15 (3), 23 (4) и 31 (5).
ОАКФ, рассчитанные по отношению к REE каждой цепи, на рис. 5б также совпадают на начальном участке и расположены в обратном порядке по отношению к рис. 5а на конечном участке: чем короче цепь, тем выше наблюдаемое на хвосте ее ОАКФ плато в согласии с формулой Куна–Грюна (16). Для трех самых длинных цепей видна лишь начальная часть плато, дальнейшему наблюдению опять мешает плохое отношение сигнал : шум.
ИНДИВИДУАЛЬНЫЕ ОАКФ КУНОВСКИХ СЕГМЕНТОВ
Следующим шагом в количественной характеристике гетерогенности цепи является получение значений τК и SKE для отдельного сегмента в зависимости от его положения в цепи, т.е. номера i, отсчитываемого симметрично с обоих концов цепи до ее середины. Для каждой пары симметричных сегментов цепи были построены ОАКФ относительно межконцевого вектора цепи. На рис. 6 показан их набор для цепи с N = 63. Индивидуальные SKE и τK сегмента вычислялись по определению [82, 83] с поправкой на недостаточное отношение сигнал : шум, позволяющее наблюдать только начало возникновения плато всех ОАКФ при t1 = 250 ≫ τK:
(17)
${{{\tau }}_{{\mathbf{K}}}} \approx \frac{{\int\limits_0^{{{t}_{1}}} {[{{C}_{{{\mathbf{KE}}}}}(t)--S_{{{\mathbf{KE}}}}^{2}]dt} }}{{1--S_{{{\mathbf{KE}}}}^{2}}},\quad S_{{{\mathbf{KE}}}}^{2} \approx {{C}_{{{\mathbf{KE}}}}}({{t}_{1}})$Рис. 6.
Полулогарифмическое представление ОАКФ куновских сегментов относительно межконцевого вектора цепи в расплаве с N = 63. Номера кривых соответствуют номерам сегментов.
Формула (17) отличается от определения заменой t → ∞ на t = t1 и отсутствием номера сегмента. Полученные результаты для цепей разной длины как олигомерных, так и зацепленных, представлены на рис. 7 в виде нормированных безразмерных зависимостей от номера сегмента i: τ'(i) = = τK (i)/τK(64) и S '(i) = SKE (i)/SKE(64). Величины τК(64) ≈ 2.8 и SKE(64) ≈ 0.037 соответствуют области плато зависимостей для самой длинной цепи из 127 сегментов. Величина SKE(64) ≈ 0.037 существенно больше ее теоретического значения ∼0.0047, даваемого формулой (16) при подстановке в нее NEF = N = 127; зато с учетом ошибки моделирования она близка к теоретическому значению для SKP = 0.041 ± 0.002, полученному при подстановке в формулу (16) NEF = NE. Здесь SKP – фактор порядка куновских сегментов по отношению к межконцевому вектору примитивного сегмента, который является следующей базовой абстракцией рептационной модели, связанной с субцепью между двумя соседними зацеплениями вдоль цепи [8–10, 73].
Рис. 7.
Зависимости относительного времени корреляции (а) и S ' (б) куновских сегментов от их номера в цепи с числом сегментов N = 7 (1), 15 (2), 31 (3), 63 (4) и 127 (5). Пояснения в тексте.
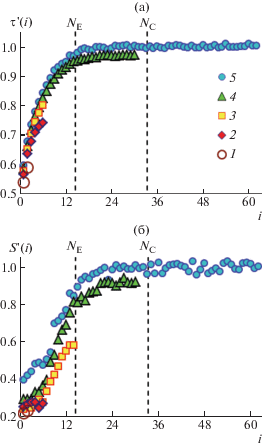
Такое несоответствие указывает на различие в зависимости структурных и динамических характеристик цепи при переходе в зацепленное состояние, как уже было видно уже по данным рис. 3. Компьютерное моделирование выявляет, что вначале происходит предусреднение диполь-дипольного взаимодействия куновского сегмента внутри примитивного сегмента до величины, определяемой SKP. Затем унаследованное примитивным сегментом остаточное диполь-дипольное взаимодействие дополнительно усредняется его ориентацией внутри цепи, оставляя ей еще меньшую величину остаточного диполь-дипольного взаимодействия, определяемую SKE. Последняя величина весьма сложно извлекаема, поскольку находится на уровне шумов для срединных сегментов и еще ниже – для концевых, как видно на рис. 6. Отсюда также вытекает, что уравнение (14а) должно содержать еще один сомножитель – ОАКФ ориентации примитивного сегмента относительно межконцевого вектора цепи, которая для упрощения выражения и вследствие недоступности даже в моделировании была опущена.
В итоге описанной выше нормировки данных для цепи с N = 127 плато на обеих панелях рис. 7 имеет единичное значение и в то же время вследствие его доминирующей протяженности на графике характеризует средние по всем сегментам цепи τК и SKE. При этом τК для концевых сегментов меньше, чем для срединных в ∼ 2 раза, в то время как SKE – в ∼2.5 раза: указанные изменения происходят на концевых участках цепи протяженностью ∼1.5NE ≈ 22 куновских сегмента. В данном интервале, т.е. до выхода на плато, нормированные кривые для цепи c N = 127 аппроксимируются полиномами
(18а)
$\tau '(i\, < \,22)\, \approx \,{{10}^{{--5}}}{{i}^{3}}\,--\,0.0032{{i}^{2}}\, + \,0.0646i\, + \,0.5573~$(18б)
$\begin{gathered} S'(i < 22) \approx {{10}^{{--5}}}{{i}^{4}}--0.0006{{i}^{3}} + \\ + \,0.0117{{i}^{2}}--0.0416i + 0.4523~ \\ \end{gathered} $Рисунок 7 является наглядной демонстрацией двойной гетерогенности даже олигомерных цепей, вдоль которых время корреляции сегмента и динамический фактор порядка растут на протяжении всей цепи от концов к середине. Другими словами, олигомерные цепи гетерогенны по всей длине, в то время как зацепленные – только вдоль концевых болтающихся фрагментов длиной ∼1.5NE. В экспериментах по измерению селективной спин-решеточной релаксации на углероде-13 в углеводородных цепях, закрепленных на одном конце, двойная гетерогенность может проявиться в виде минимума на зависимости времени релаксации от номера метилена в цепочке [153, 154]. Гетерогенность олигомерных цепей по всей длине к тому же осложнена конкретной химической природой мономерного звена, которая определяет молекулярно-массовую зависимость T2EF в олигомерных расплавах [25, 27, 155–159].
Аналогично обнаруженной гетерогенности, в атомистическом моделировании полипропиленоксида было выявлено четырехкратное уменьшение времени корреляции концевых мономеров по сравнению со срединными, причем насыщение отношения времен корреляции происходило для цепей длиной более 20 мономеров, как и на рис. 7а [160]. Последнее указывает на масштабную инвариантность формулы Куна–Грюна, в которой основную роль играет количество независимых кинетических единиц в рассматриваемой цепи, а не их структура или масштаб.
Теоретическое обоснование обоих видов гетерогенности также давно известно – это возрастание мультиплетности вращения метилена по мере приближения к концу углеводородной цепочки. Однако оно редко используется вследствие громоздкости аналитических выражений [161–164], хотя гетерогенность локальной упорядоченности вдоль болтающихся углеводородных цепочек в экспериментах начала изучаться давно [153, 154, 165, 166]. Эти исследования продолжаются до сих пор [167–169], поскольку дают уникальную информацию о локальной подвижности и упорядоченности функциональных групп в биологических молекулах [170, 171]. Применимость теоретических выражений ограничивает также то, что они выведены для изолированной закрепленной на одном конце цепочки, пространственное вращение которой вокруг точки закрепления не ограничено окружающими цепями, хотя очевидно, что последние накладывают существенное ограничение на требуемый для этого свободный объем. Данное ограничение проявилось, например, в атомистическом моделировании расплава полипропиленоксида, где была обнаружена некоторая дифференциация подвижности сегментов на разном расстоянии от концов цепи и показано, что такая гетерогенность заметно искажает предсказываемую раузовской теорией динамику слабо зацепленных цепей [143].
ЗАКЛЮЧЕНИЕ
С помощью компьютерного моделирования исследованы структурные и динамические свойства полимерных расплавов в диапазоне длины цепей от раузовских до рептирующих. Переход от олигомерных к зацепленным расплавам продемонстрирован с помощью общепринятых характеристик – среднеквадратичного смещения и различных автокорреляционных функций срединных куновских сегментов цепи, которые при переходе через критическую длину цепи претерпевали все предсказанные рептационной моделью изменения. Вычислены индивидуальные ориентационные автокорреляционные функции всех сегментов цепи, что позволило количественно охарактеризовать два вида гетерогенности вдоль нее: динамическую гетерогенность и гетерогенность вращательной анизотропии. Первая описывается временем ориентационной корреляции, а вторая – остаточным магнитным диполь-дипольным взаимодействием. Обе величины зависят от номера сегмента в цепи, в совокупности определяют характер поперечной магнитной релаксации и лежат в основе анализа компонентов спада свободной индукции.
Список литературы
Fox T.G, Jr., Flory P.J. // J. Appl. Phys. 1950. V. 21. № 6. P. 581.
Danusso F., Levi M., Gianotti G., and Turri S. // Polymer. 1993. V. 34. № 17. P. 3687.
Sperling L.H.// Introduction to Physical Polymer Science. Hoboken: Wiley Interscience, 2006.
Miwa Y., Urakawa O., Nobukawa S., Kutsumizu S. // Polymer. 2015. V. 59. № 2. P. 194.
Startsev O.V., Lebedev M.P. // Polymer Science A. 2018. V. 60. № 6. P. 911.
Boyer R.F. // Macromolecules. 1974. V. 7. № 1. P. 142.
Rouse P.E. // J. Chem. Phys. 1953. V. 21. № 7. P. 1272.
Doi M., Edwards S.F. The Theory of Polymer Dynamics. Oxford: Clarendon Press, 1986.
Готлиб Ю.Я., Даринский А.А., Светлов А.А. Физическая кинетика макромолекул. Л.: Химия, 1986.
Гросберг А.А., Хохлов А.Р. Статистическая физика макромолекул. М.: Наука, 1989.
Ebert U., Schäfer L., Baumgartner A. // J. Stat. Phys. 1998. V. 90. № 5–6. P. 1325.
Baumgartner A., Ebert U., Schäfer L. // J. Stat. Phys. 1998. V. 90. № 5–6. P. 1375.
Ferry J.D. Viscoelastic Properties of Polymers. New York: Wiley, 1970.
Curro J.G., Pincus P. // Macromolecules. 1983. V. 16. № 4. P. 559.
McLoughlin K., Szeto K., Cohen C. // Macromolecules. 1996. V. 29. № 16. P. 5475.
Erman B., Queslelt J.-P., Monnerie L. // Polymer. 1998. V. 29.№ 10. P. 1823.
Gurtovenko A.A., Gotlib Yu.Ya. // J. Chem. Phys. 2001. V. 115. № 14. P. 6785.
Callaghan P.T., Samulski E.T. // Macromolecules. 2003. V. 36. № 3. P. 724.
Luo H., Kluppel M., Schneider H. // Macromolecules. 2004. V. 37. № 21. P. 8000.
Yu W., Du M., Zhang D., Lin Y., Zheng Q. // Macromolecules. 2013. V. 46. № 18. P. 7341.
Jensen M.K., Khaliullin R., Schieber J.D. // Rheol. Acta. 2012. V. 51. № 1. P. 21.
Basterra-Beroiz B., Rommel R., Kayser F., Valentín J.L., Westermann S., Heinrich G. // Macromolecules. 2018. V. 51. № 5. P. 2076.
Ries M.E., Brereton M.G., Klein P.G., Daunis P. // Polym. Gel Network. 1997. V. 5. № 3. P. 285.
Kimmich R., Fatkullin N. // Adv. Polym. Sci. 2004. V. 170. P. 1.
Kimmich R., Schnur G., Kopf M. // Progr. NMR Spectr. 1988. V. 20. № 4. P. 385.
Adams C.H., Brereton M.G., Hutching L.R., Klein P.G., McLeish T.C.B., Richard R.W., Ries M.E. // Macromolecules. 2000. V. 33. № 19. P. 7101.
Klein P.G., Ries M.E. // Progr. Nucl. Magn. Reson. Spec. 2003. V. 42. № 1–2. P. 31.
Klein P.G., Adams C.H., Brereton M.G., Ries M.E., Nicholson T.M., Hutchings L.R., Richards R.W. // Macromolecules. 1998. V. 31. № 25. P. 8871.
Федотов В.Д., Чернов В.М., Хазанович Т.Н. //Высокомолек. cоед. А. 1978. Т. 20. № 4. С. 919.
Чернов В.М., Краснопольский Г.С. // Журн. эксперим. и теорет. физики. 2008. Т. 134. № 2(8). С. 354.
Kornfield J.A., Chung G.-C. // Macromolecules. 1992. V. 25. № 17. P. 4442.
Kopf M., Schnur G., Kimmich R. // Macromolecules. 1988. V. 21. № 11. P. 3340.
Kimmich R., Kopf M., Callaghan P. // J. Polym. Sci., Polym. Phys. 1991. V. 29. № 10. P. 1025.
Weber H.W., Kimmich R. // Macromolecules. 1993. V. 26. № 10. P. 2597.
Ries M.E., Brereton M.G.,Cruickshank J.M., Klein P.G., Ward I.M. // Macromolecules. 1995. V. 28. № 9. P. 3282.
Callaghan P.T. // Polymer. 1998. V. 29. № 11. P. 1951.
Cohen-Addad J.P. and Domard M., Boileau S. // J. Chem. Phys. 1981. V. 75. № 15. P. 4107.
Guillermo A., Cohen-Addad J.P., Bytchenkoff D. // J. Chem. Phys. 2000. V. 113. № 12. P. 5098.
Guillermo A., Cohen-Addad J.P. // J. Chem. Phys. 2002. V. 116. № 7. P. 3141.
Schille E., Cohen-Addad J.P., Guillermo A. // Macromolecules. 2004. V. 37. № 6. P. 2144.
Chavez F.V., Saalwachter K. // Macromolecules. 2011. V. 44. № 6. P. 1549.
Litvinov V.M., Ries M.E., Baughman T.W., Henke A., Matloka P.P. // Macromolecules. 2013. V. 46. № 2. P. 541.
Fischer E., Kimmich R., Fatkullin N. // J. Chem. Phys. 1997. V. 106. № 23. P. 9883.
Simon G., Schneider H., Hauler K.-G. // Progr. Colloid Polym. Sci. 1988. V. 78. № 9. P. 30.
Simon G., Baumann K., Gronski W. // Macromolecules.1992. V. 25. № 14. P. 3624.
Saalwachter K. // Prog. Nucl. Magn. Reson. Spectr. 2007. V. 51. № 1. P. 1.
Cohen-Addad J.P. // J. Chem. Phys. 1974. V. 60. № 6. P. 2440.
Cohen-Addad J.P. // J. Phys. 1982. V. 43. № 10. P. 1509.
Cohen-Addad J.P., Dupeyre R. // Polymer. 1983. V. 24. № 4. P. 400.
Cohen-Addad J.P. // Polymer. 1983. V. 24. № 9. P. 1128.
Brereton M.G. // Macromolecules. 1989. V. 22. № 9. P. 3667.
Brereton M.G. // Macromolecules. 1990. V. 23. № 4. P. 1119.
Brereton M.G. // J. Chem. Phys. 1991. V. 94. № 3. P. 2136.
Schneider H., Hiller W. // J. Polym. Sci., Polym. Phys. 1990. V. 28. № 7. P. 1001.
Кулагина Т.П., Маникин П.С., Карнаух Г.Е., Смирнов Л.П. // Хим. физика. 2011. Т. 30. № 8. С. 68.
Кулагина Т.П., Варакина В.А., Кузина А.Н. // Хим. физика. 2014. Т. 33. № 5. С. 76.
Fenchenko K.V. // Polymer Science A. 2002. V. 44. № 6. P. 657.
Fenchenko K.V. // J. Non-Cryst. Solids. 2012. V. 358. № 3. P. 474.
Sturniolo S., Pieruccini M. // J. Magn. Res. 2012. V. 223. № 11. P. 138.
Fatkullin N.F., Körber T., Rössler E.A. // Polymer. 2018. V. 142. № 4. P. 310.
Gotlib Y.Y., Markelov D.A. // Polymer Science A. 2007. V. 49. № 10. P. 1137.
Markelov D.A., Dolgushev, Gotlib Yu.Ya., Blumen A. // J. Chem. Phys. 2014. V. 140. № 24. P. 244904.
Markelov D.A., Gotlib Y.Y., Darinskii A.A., Lyulin A.V., Lyulin S.V. // Polymer Science A. 2009. V. 51. № 3. P. 331.
Markelov D.A., Shishkin A.N., Matveev V.V., Penkova A.V., Lahderanta E., Chizhik V.I. // Macromolecules. 2016. V. 49. № 23. P. 9247.
Pinto L.F., Correa J., Martin-Pastor M., Riguera R., Fernandez-Megia E. // J. Am. Chem. Soc. 2013. V. 135. № 5. P. 1972.
Buckingham A.D., Hentschel G.E. // J. Polym. Sci. A-2. 1980. V. 18. № 4. P. 853.
Bates F.S., Wignall G.D., Koehler W.C. // Phys. Rev. Lett. 1985. V. 55. № 22. P. 2425.
Bates F.S., Wignall G.D., Dierker S.B. // Macromolecules. 1986. V. 19. № 7. P. 1938.
Bates F.S., Wiltzius P. // J. Chem. Phys. 1989. V. 91. № 5. P. 3258.
White R.P., Lipson J.E.G., Higgins J.S. // Macromolecules. 2010. V. 43. № 9. P. 4287.
Bates C.M., Bates F.S. // Macromolecules. 2017. V. 50. № 1. P. 3.
Пестряев Е.М. // Изв. Уфимского науч. центра РАН. Физика. 2014. Т. 4. № 3. С. 32.
Pestryaev E.M. // Polymer Science A. 2018. V. 60. № 4. P. 530.
Frenkel D., Smit B. // Understanding Molecular Simulation. New York: Acad. Press, 2002.
Балабаев Н.К. // Методы компьютерного моделирования для исследования полимеров и биополимеров / Отв. ред. В.А. Иванов, А.Л. Рабинович, А.Р. Хохлов. М.: Книжный дом “ЛИБРОКОМ”, 2009.
Норман Г.Э., Стегайлов В.В. // Матем. моделирование. 2012. Т. 24. № 6. С. 3.
Anderson P.W., Weiss P.R. // Rev. Modern Phys. 1953. V. 25. № 1. P. 269.
Абрагам А. // Ядерный магнетизм / Пер. с англ. под ред. Г.В. Скроцкого. М.: Изд-во иностр. лит., 1963.
Сликтер Ч. // Основы теории магнитного резонанса / Пер. с англ. под ред. Г.В. Скроцкого. М.: Мир, 1981.
Woessner D.E. // J. Chem. Phys. 1962. V. 36. № 1. P. 1.
Latanowicz L. // Conc. Magn. Reson. 2012. V. 40A. № 2. P. 66.
Halle B. // J. Chem. Phys. 2009. V. 131. № 22. P. 224507.
Meirovich E., Shapiro Yu.E., Polimeno A., Freed J.H. // Prog. Nucl. Magn. Reson. Spectr. 2010. V. 56. № 4. P. 360.
Kremer K. // NIC Series. 2004. V. 23. P. 141. www.fz-juelich.de/nic-series
Пестряев Е.М. // Матем. моделирование. 2003. Т. 15. № 12. С. 118.
Пестряев Е.М. // Матем. моделирование. 2007. Т. 19. № 4. С. 62.
Pestryaev E.M. // Polymer Science A. 2013. V. 55. № 5. P. 336.
Пестряев Е.М. // Матем. моделирование. 2014. Т. 26. № 1. С. 69.
Ramirez J., Sukumaran S.K., Vorselaars B., Likhtman A.E. // J. Chem. Phys. 2010. V. 133. № 15. P. 154103.
Likhtman A.E., Sukumaran S.K., Ramirez J. // Macromolecules. 2007. V. 40. № 18. P. 6748.
Pestryaev E.M. // J. Phys., Conf. Ser. 2011. V. 324. № 1. P. 012031.
Brazhkin V.V., Fomin Yu.D., Lyapin A.G., Ryzhov V.N., Trachenko K. // Phys. Rev. E. 2012. V. 85. № 3. P. 031203.
Бражкин В.В., Ляпин А.Г., Рыжов В.Н., Траченко К., Фомин Ю.Д., Циок Е.Н. // Успехи физ. наук. 2012. V. 182. № 11. P. 1137.
Пестряев Е.М. // Журн. физ. химии. 2018. Т. 92. № 7. С. 1086.
Edwards S.F. // Brit. Polym. J. 1977. V. 9. № 2. P. 140.
Everaers R., Sukumaran S.K., Grest G.S., Svaneborg C., Sivasubramanian A., Kremer K. // Science. 2004. V. 303. № 5659. P. 823.
Fetters L.J., Lohse D.J., Milner S.T., Graessley W.W. // Macromolecules. 1999. V. 32. № 20. P. 6847.
Fetters L.J., Lohse L.J., Richter D., Witten T.A., Zirkel A. // Macromolecules. 1994. V. 27. № 17. P. 4639.
Kremer K., Grest G. // J. Chem. Phys. 1990. V. 92. № 8. P. 5057.
Paul W., Binder K., Heermann D.W., Kremer K. // J. Chem. Phys. 1991. V. 95. № 10. P. 7726.
Paul W., Smith G.D. // Rep. Prog. Phys. 2002. V. 67. № 7. P. 1117.
Wittmer J., Paul W., Binder K. // Macromolecules. 1992. V. 25. № 26. P. 7211.
Smith S.W., Hall C.K., Freeman B.D. // J. Chem. Phys. 1996. V. 104. № 14. P. 5616.
Baschnagel J., Paul W., Tries V., Binder K. // Macromolecules. 1998. V. 31. № 12. P. 3856.
Ramirez-Hernandez A., Detcheverry F.A., Peters B.L., Chappa V.C., Schweizer K.S., Muller M., de Pablo J.J. // Macromolecules. 2013. V. 46. № 15. P. 6287.
Kreer T., Baschnagel J., Muller M., Binder K. // Macromolecules. 2001. V. 34. № 4. P. 1105.
Ramos J., Vega J.F., Martínez-Salazar J. // Soft Matter. 2016. V. 12. № 17. P. 3929.
Smith B.A., Samulski E.T., Yu L.P., Winnik M.A. // Phys. Rev. Lett. 1984. V. 52. № 1. P. 45.
Elfadl A.A., Kahlau R., Herrmann A., Novikov V.N., Rossler E.A. // Macromolecules. 2010. V. 43. № 7. P. 3340.
Farris A.C.K., Li Y.W., Eisenbach M. // Comput. Phys. Comm. 2019. V. 235. № 2. P. 297.
Yi S.D., Kim B.J. // Comput. Phys. Comm. 2012. V. 183. № 8. P. 1574.
Lifshitz I.M., Grosberg A.Y., Khokhlov A.R. // Rev. Modern Phys. 1978. V. 50. № 3. P. 683.
Лифшиц И.М., Гросберг А.Ю., Хохлов А.Р. // Успехи физ. наук. 1979. Т. 127. № 3. С. 353.
McCrackin F.L., Mazur J., Guttman C.M. // Macromolecules. 1973. V. 6. № 6. P. 859.
Mazur J., Guttman C.M., McCrackin F.L. // Macromolecules. 1973. V. 6. № 6. P. 872.
Mendichi R., Šoltés L., Schieroni A.G. // Biomacromolecules. 2003. V. 4. № 6. P. 1805.
Smilgies D.M., Folta-Stogniew E. // J. Appl. Crystallogr. 2015. V. 48. № 5. P. 1604.
Panja D., Barkema G.T. // J. Chem. Phys. 2009. V. 131. № 15. P. 154903.
Auhl R., Everaers R., Grest G., Kremer K., Plimpton S.J. // J. Chem. Phys. 2003. V. 119. № 24. P. 12718.
Perez M., Lame O., Leonforte F., Barrat J.-L. // J. Chem. Phys. 2008. V. 128. № 23. P. 234904.
Subramanian G. // J. Chem. Phys. 2010. V. 133. № 16. P. 164902.
Carbone P., Karimi-Varzaneh H.A., Muller-Plathe F. // Faraday Disc. 2010. V. 144. P. 25.
Sliozberg Y.R., Andzelm J.W. // Chem. Phys. Lett. 2012. V. 523. № 1. P. 139.
Zhang G., Moreira L.A., Stuehn T., Daoulas K.Ch., Kremer K. // ACS Macro Lett. 2014. V. 3. № 2. P. 198.
De Nicola A., Kawakatsu T., Milano G. // J. Chem. Theory Comput. 2014. V. 10. № 12. P. 5651.
Moreira L.A., Zhang G., Muller F., Stuehn T., Kremer K. // Macromol. Theory Simul. 2015. V. 24. № 5. P. 419.
Pestryaev E.M. // Polymer Science A. 2019. V. 61. № 3. P. 392.
Kresse B., Hofmann M., Privalov A.F., Fatkullin N., Fujara F., Rossler E.A. // Macromolecules. 2015. V. 48. № 13. P. 4491.
Hofmann M., Fatkullin N., Rössler E.A. // Macromolecules. 2017. V. 50. № 4. P. 1755.
Mordvinkin A., Saalwachter K. // J. Chem. Phys. 2017. V. 146. № 9. P. 094902; Erratum: J. Chem. Phys. 2018.V. 148. № 8. P. 089901.
Hempel G. // Solid State Nucl. Magn. Res. 2017. V. 82. № 4–5. P. 29.
Lipari G., Szabo A. // J. Am. Chem. Soc. 1982. V. 104. № 17. P. 4546.
Lipari G., Szabo A. // J. Am. Chem. Soc. 1982. V. 104. № 17. P. 4559.
Kuhn W.V., Grun F. // Kolloid-Zeitschrift. 1942. V. 101. № 3. P. 248.
Волькенштейн М.В. // Конфигурационная статистика полимерных цепей. М.; Л.: Изд-во АН СССР, 1959.
Готлиб Ю.Я., Лифшиц М.И., Шевелев В.А., Лишанский И.С., Баланина И.В. // Высокомолек. соед. А. 1976. Т. 18. № 10. С. 2299.
Sukumaran S.K., Grest G.S., Kremer K., Everaers R. // J. Polym. Sci., Polym. Phys. 2005. V. 43. № 8. P. 917.
Snabhag S., Larson R.G. // Macromolecules. 2007. V. 40. № 8. P. 2897.
Hoy R.S., Fotenopoulou K., Kroger M. // Phys. Rev. E. 2009. V. 80. № 3. P. 031803.
Karayiannis N.C., Kroger M. // Int. J. Mol. Sci. 2009. V. 10. № 11. P. 5054.
Karatrantos A., Clarke N., Composto R.J., Winey K.I. // Soft Matter. 2013. V. 9. № 14. P. 3877.
Xi L. // Molec. Simul. 2019. V. 45. № 14–15. P. 1242.
Bormuth A., Hofmann M., Henritzi M., Vogel M., Rossler E.A. // Macromolecules. 2013. V. 46. № 19. P. 7805.
Borgia G.C., Brown R.J.S., Fantazzini P. // J. Magn. Res. 1998. V. 132. № 1. P. 65.
Borgia G.C., Brown R.J.S., Fantazzini P. // J. Magn. Res. 2000. V. 147. № 2. P. 273.
Song Y.-Q., Venkataramanan L., Hurlimann M.D., Flaum M., Frulla P., Straley C.// J. Magn. Res. 2002. V. 154. № 2. P. 261.
Weglarz W.P., Haranczyk H. // J. Phys. D. 2000. V. 33. № 15. P. 1909.
Koay C.G., Basser P.J. // J. Magn. Res. 2006. V. 179. № 2. P. 317.
Ghosh S., Keener K.M., Pan Y. // J. Magn. Res. 2008. V. 191. № 2. P. 266.
Earle K.A., Mainali L., Sahu I.D., Schneider D.J. // Appl. Magn. Reson. 2010. V. 37. № 1. P. 865.
Testamanti M.N., Rezaee R.//J. Petrol. Sci. Eng. 2019. V. 174. № 3. P. 177.
Hou X., Su G., Lu B., Wang X., Nie S. // Spectr. Lett. 2019. V. 52. № 9. P. 541.
Levine Y.K., Birdsall N.J.M., Lee A.G., Metcalfe J.C., Partington P., Roberts G.C.K. // J. Chem. Phys. 1974. V. 60. № 7. P. 2890.
Davis J.H., Jeffrey K.R., Bloom M. // J. Magn. Res. 1978. V. 29. № 2. P. 191.
McCall D.W., Douglass D.C., Anderson E.W. // J. Polym. Sci. 1962. V. 59. № 16. P. 301.
Folland R., Charlesby A. // Polymer. 1979. V. 20. № 2. P. 207.
Kimmich R., Bachus R. // Colloid Polym. Sci. 1982. V. 260. № 10. P. 911.
Kimmich R. // Polymer. 1984. V. 25. № 2. P. 187.
Brereton M.G., Ward, Boden N., Wright P. // Macromolecules. 1991. V. 24. № 8. P. 2068.
Bormuth A., Henritzi P., Vogel M. // Macromolecules. 2010. V. 43. № 8. P. 8985.
Wallach L. // J. Chem. Phys. 1967. V. 47. № 12. P. 5258.
Levine Y.K., Partington P., Roberts G.C.K. // Molec. Phys. 1973. V. 25. № 3. P. 497.
Wittebort L., Szabo L. // J. Chem. Phys. 1978. V. 69. № 4. P. 1722.
Brown M.F. // J. Chem. Phys. 1984. V. 80. № 6. P. 2808.
Davis J.H., Jeffrey K.R. // Chem. Phys. Lipids. 1977. V. 20. № 2. P. 87.
Jeffrey K.R., Wong T.C., Tulloch A.P. // Molec. Phys. 1984. V. 52. № 2. P. 289.
Douliez J.-P., Leonard A., Dufourc E. // Biophys. J. 1995. V. 68. № 5. P. 1727.
Vogel A., Katzka C.P., Waldmann H., Arnold K., Brown M.F., Huster D. // J. Am. Chem. Soc. 2005. V. 127. № 5. P. 12263.
Stahlberg S., Lange S., Dobner B., Huster D. // Langmuir. 2016. V. 32. № 8. P. 2023.
Luginbuhl P., Wuthrich K. // Progr. Nucl. Magn. Res. Spec. 2002. V. 40. № 3. P. 199.
Meirovitch E., Liang Z., Freed J.H. // J. Phys. Chem. B. 2015. V. 119. № 7. P. 2857.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия А)