

Высокомолекулярные соединения (серия А), 2021, T. 63, № 2, стр. 110-116
ТРАНСПОРТНЫЕ СВОЙСТВА И ФИЗИОЛОГИЧЕСКАЯ АКТИВНОСТЬ КОМПЛЕКСОВ АРАБИНОГАЛАКТАНА С НЕКОТОРЫМИ АЗОТСОДЕРЖАЩИМИ СОЕДИНЕНИЯМИ
Л. А. Бадыкова a, *, Р. Х. Мударисова a, С. В. Колесов a
a Уфимский институт химии УФИЦ Российской академии наук
450054 Уфа, пр. Октября, 71, Россия
* E-mail: badykova@mail.ru
Поступила в редакцию 21.07.2020
После доработки 05.10.2020
Принята к публикации 19.10.2020
Аннотация
Исследованы диффузионно-транспортные свойства комплексов на основе арабиногалактана и его окисленных форм с азотсодержащими соединениями – 5-аминосалициловой кислотой, 4-аминосалициловой кислотой и гидразидом изоникотиновой кислоты в условиях, моделирующих биологические среды. Комплексообразование поли- и олигосахаридов с лекарственными веществами приводит к созданию пролонгированных лекарственных форм. На динамику высвобождения лекарств из комплексов оказывает влияние природа полисахаридной матрицы и молекулярная масса. Наибольшим пролонгирующим действием обладают комплексы на основе исходного арабиногалактана.
Полимеры природного происхождения широко используют в качестве потенциальных носителей лекарственных соединений [1–4]. Модификация природных полисахаридов лекарственными веществами является перспективным и рациональным способом получения полимерных систем с пролонгированным действием, т.е. систем с контролируемой скоростью высвобождения препарата [5–13]. Растительный полисахарид арабиногалактан (АГ) обладает высокой биологической активностью и может выступать как нетоксичный носитель для различных фармацевтических соединений [14–17]. Иммобилизация на макромолекуле арабиногалактана лекарственных препаратов позволяет получить производные этого полисахарида для направленного транспорта биологически активных соединений в различных системах, включая живой организм [18, 19]. Поскольку полимерная матрица во многом определяет скорость и степень высвобождения лекарственного препарата, высокомолекулярная природа АГ создает возможность регулирования фармакокинетики иммобилизованных лекарств [20, 21].
Арабиногалактаны представляют собой высокоразветвленные макромолекулы с главной цепью, состоящей из 1 → 3 связанных β-D-галактопиранозных остатков, большинство из которых несет боковые ответвления при С-6. Боковые цепи содержат 3,6-ди-О- и 6-О-замещенные остатки β-D-галактопиранозы и 3-О-замещенные остатки β-L-арабинофуранозы, а концевыми невосстанавливающими остатками являются β-D-галактопираноза, β-D-арабинофураноза и β-L-арабинопираноза [22, 23].
Для получения модифицированных соединений на основе полисахарида помимо арабиногалактана в настоящей работе были использованы его окисленные формы: высокомолекулярная (АГВМ) и низкомолекулярная (АГНМ), которые представляют собой продукты окислительной функционализации и деструкции биополимера под действием системы Н2О2 + О2 [24].
В качестве лекарственных соединений были выбраны следующие азотсодержащие соединения: 5-аминосалициловая кислота (5-АСК), обладающая противовоспалительной и противоязвенной активностью, а также 4-аминосалициловая кислота (4-АСК) и гидразид изоникотиновой кислоты (ГИНК), являющиеся противотуберкулезными препаратами.
Целью работы явилось изучение кинетики транспорта физиологически активных комплексов арабиногалактана и его окисленных форм с азотсодержащими соединениями в условиях, моделирующих биологические среды.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Использовали арабиногалактан с молекулярной массой 38.5 × 103 (ЗАО “Аметис”, Россия; ТУ 9325-008-70692 152-08). Азотсодержащие соединения 5-АСК и 4-АСК – квалификации ч.д.а., ГИНК – фармакопейной чистоты применяли без дополнительной очистки.
УФ-спектры водных растворов снимали в кварцевых кюветах длиной 1 см на спектрофотометре “UV-VIS Specord M-40” в области 220–350 нм. Для контроля pH растворов использовали pH-метр “АНИОН 4100”. Углерод, водород и азот определяли на анализаторе марки “EUKO EA-3000”.
Спектры ЯМР 13С снимали на спектрометре “Bruker AM-300” (рабочая частота 75.47 МГц) с широкополосным подавлением по протонам и в режиме JMODXH. Применяли растворы арабиногалактана 3–5% в D2O, внутренний стандарт ТМС. Спектры записывали при температуре 25°С с задержкой между импульсами 15 с.
Окислительную функционализацию водно-пероксидных растворов АГ молекулярным кислородом проводили в стеклянном реакторе барботажного типа. Водные растворы АГ с концентрацией 10% обрабатывали перекисью водорода 10%-ной концентрации при 90°С (Н2О2 – квалификации о.с.ч.). Время реакции 6 ч, объем реакционной смеси составлял 10 мл. В результате окисления АГ выделены две фракции. Окисленную высокомолекулярную фракцию отделяли от низкомолекулярной осаждением ацетоном. Низкомолекулярную фракцию выделяли из надосадочной жидкости путем отгонки ацетона и воды с последующей сушкой продукта в вакууме. Структура выделяемых фракций АГ была подробно изучена в работе [25]. Характеристики образцов представлены в табл. 1.
Таблица 1.
Характеристики образцов арабиногалактана и его окисленных фракций
| Полимер | Состав, % | М × 10–3 | ||||
|---|---|---|---|---|---|---|
| С | Н | О | Гексозы | Уроновые кислоты | ||
| АГ | 40.18 | 7.49 | 52.33 | 71.8 | 4.70 | 38.5 |
| АГВМ | 38.75 | 7.24 | 54.01 | 46.3 | 12.0 | 22.1 |
| АГНМ | 37.64 | 6.21 | 56.15 | 20.0 | 76.0 | 4.1 |
Анализ спектра ЯМР 13С высокомолекулярной фракции показывает, что фракция состоит из структурных фрагментов не подверженных окислению моносахаридных звеньев и заметного количества фрагментов, содержащих карбоксильные группы. Синглетные сигналы при δС = 177–180 м.д., соответствующие карбоксильному атому углерода (табл. 2), указывают на наличие уроновых кислот в различных структурных фрагментах продукта частичного окисления арабиногалактана. Наиболее интенсивными являются сигналы концевых галактопирануроновых остатков.
Таблица 2.
Спектральные данные окисленных форм арабиногалактана (δС, м.д.; D2O; DSS; Т = 30°С)
| Образец | Фрагмент | Химические сдвиги δС, м.д. | |||||
|---|---|---|---|---|---|---|---|
| C(1) | C(2) | C(3) | C(4) | C(5) | C(6) | ||
| АГВМ | β-GalpA–(1→ | 105.9 | 71.9 | 73.7 | 72.2 | 75.2 | 178.9 |
| β-Galp–(1→ | 105.3 | 72.6 | 74.1 | 71.3 | 71.3 | 63.7 | |
| →3.6)-β-Galp–(1→ | 106.4 | 71.6 | 84.5 | 69.1 | 74.9 | 68.5 | |
| →6)-β-Galp–(1→ | 105.8 | 72.3 | 73.4 | 71.0 | 74.6 | 70.5 | |
| →3)-β-GalpА–(1→ | 106.0 | 72.5 | 83.7 | 73.0 | 75.1 | 178.6 | |
| -L-β-Arap–(1→ | 99.1 | 71.3 | 71.0 | 71.8 | 64.7 | – | |
| АГНМ | β-GalpА–(1→ | 105.9 | 71.8 | 73.8 | 72.1 | 75.2 | 179.5 |
| β-Galp–(1→ | 105.3 | 72.7 | 74.6 | 71.8 | 71.1 | 63.7 | |
| →3.6)-β-Galp–(1→ | 106.5 | 71.8 | 84.4 | 69.0 | 74.8 | 68.6 | |
| →6)-β-Galp–(1→ | 105.7 | 72.6 | 73.4 | 71.1 | 74.6 | 70.4 | |
| -β-Arap–(1→ | 99.4 | 71.6 | 70.8 | 73.4 | 64.7 | – | |
| →3)-Ara | 95.2 | 70.1 | 74.6 | 70.8 | 65.9 | – | |
Значительное понижение интенсивности сигналов концевых галактопиранозных групп, появление интенсивных характеристичных сигналов атомов С(5) 75.2 м.д. и С(6) 178.9 м.д. показывают, что окислению подвергаются атомы С(6) концевых β-Galp-(1 → групп. Уменьшение интенсивностей сигналов тризамещенного → 3.6)-β-Galp-(1 → звена [широких дублетных при 106.4 м.д. атома С(1) и 84.2 м.д. атома С(3) и триплетного атома 68.5 – С(6)] появление узких дублетных сигналов при δС = 106.0; 83.7 и 75.1 м.д. и синглетного сигнала 178.6 м.д. указывают на то, что при окислении АГ происходит также деструкция основной цепи полисахарида с отрывом боковых цепей и образованием → 3)-β-GalpА-(1 → галактоуроновых звеньев.
В спектрах ЯМР 13С низкомолекулярной фракции по сравнению с высокомолекулярной наблюдали понижение интенсивностей сигналов атомов моносахаридных остатков и рост интенсивностей сигналов уроновых кислот.
Методика получения комплексов: полисахарид или олигосахарид в количестве 5.5 осново-ммоль растворяли в 20 мл воды. Соответствующую кислоту в количестве 5.5 ммоль перемешивали в 20 мл воды и доводили рН до 7.0. К раствору полисахарида при интенсивном перемешивании и комнатной температуре прикапывали раствор кислоты. Реакцию проводили в течение 3 ч. Далее продукт выделяли осаждением этиловым спиртом, переосаждали из воды в спирт, осадок отделяли и промывали 3 раза спиртом, затем диэтиловым эфиром и высушивали под вакуумом.
Кинетику высвобождения комплексов из капиллярного волокна изучали при температуре 25 и 38°С следующим образом. Капиллярное полиамидное волокно (l = 30 см, d = 0.9 мм), хорошо набухающее в водных средах, через микрошприц наполняли раствором соответствующего комплекса (50 мкл) или индивидуального лекарственного соединения и помещали в пробирку, наполненную 10 мл физиологического раствора (водный раствор NaCl 0.9%), рН 7.3–7.4. Концентрация растворов составляла 0.1 моль/л в пересчете на лекарственное соединение. Для определения количества комплекса отобранную пробу физиологического раствора анализировали на приборе “Specord M-40” при соответствующей длине волны для каждого комплекса и индивидуального лекарственного соединения.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Известно, что прием традиционных лекарственных препаратов не пролонгированного действия связан с колебанием уровней концентрации лекарственного вещества в плазме крови, сначала концентрация резко возрастает, а затем медленно падает. Терапевтически активная концентрация сохраняется примерно в течение 2–3 ч, после чего происходит значительное понижение концентрации препарата [26]. Такие перепады приводят к вредным побочным эффектам. Для оптимизации терапии и уменьшения побочных эффектов используют лекарственные формы с пролонгируемым высвобождением. Полагали, что иммобилизация выбранных физиологически активных азотсодержащих соединений в полимерные матрицы (АГ, АГВМ, АГНМ) позволит обеспечить плавный и равномерный профиль высвобождения действующего вещества, создавая необходимую концентрацию в плазме крови в течение определенного времени, тем самым повышая эффективность терапии.
Как было показано ранее [27], при взаимодействии арабиногалактана и его окисленных фракций с 5-АСК, 4-АСК и ГИНК происходит образование молекулярных комплексов состава 1 : 1, т.е. на одно дисахаридное звено арабиногалактана приходится одна молекула лекарственного вещества. Содержание азотсодержащих соединений в полученных комплексах составило 0.04–0.05 для АГ, 0.1–0.2 для АГВМ и 0.5–0.6 моль/осново-моль полимера для АГНМ. Реакция протекает по карбоксильным группам поли- и олигосахаридов и аминогруппам лекарственных соединений.
С целью оценки возможности пролонгирования действия азотсодержащих препаратов, иммобилизованных на АГ, методом УФ-спектроскопии была исследована кинетика высвобождения из капиллярного полиамидного волокна комплексов АГ и его окисленных фракций в модельные биологические среды.
Эффективность высвобождения оценена по накоплению комплекса в физиологическом растворе, в который была помещена полиамидная капиллярная трубка, имитирующая кровеносный сосуд. По данным УФ-спектроскопии следует, что во всех отбираемых пробах индивидуальные лекарственные соединения присутствуют исключительно в виде комплекса с полимерными матрицами. Как известно, у 5-АСК, 4-АСК и ГИНК характерный максимум поглощения обнаруживается при λ = 330, 266 и 263 нм. Комплексообразование приводит к сдвигу полосы поглощения, у соответствующих выделенных комплексов длина волны сдвигается до 315–317, 270 и 250 нм соответственно, что и наблюдается при спектроскопии растворов с высвободившимися продуктами. Следовательно, из волокна происходит диффузия именно комплекса, а не лекарственного вещества. В табл. 3 представлены результаты диффузии синтезированных соединений.
Таблица 3.
Сравнительные характеристики высвобождения комплексов
| Соединение | Время, ч | Количество выделившегося комплекса, % | Т, °С | λ, нм |
|---|---|---|---|---|
| АГ + 5-АСК | 72.0 | 70 | 25 | 317 |
| АГ + 5-АСК | 60.0 | 72 | 38 | 316 |
| АГВМ + 5-АСК | 48.0 | 87 | 25 | 315 |
| АГВМ + 5-АСК | 30.0 | 86 | 38 | 315 |
| АГНМ + 5-АСК | 24.0 | 92 | 25 | 315 |
| АГНМ + 5-АСК | 19.0 | 88 | 38 | 315 |
| 5-АСК | 4.5 | 95 | 25 | 330 |
| АГ + 4-АСК | 24.0 | 66 | 25 | 270 |
| АГВМ + 4-АСК | 24.0 | 68 | 25 | 270 |
| АГНМ + 4-АСК | 18.0 | 87 | 25 | 270 |
| 4-АСК | 2.5 | 90 | 25 | 266 |
| АГ + ГИНК | 48.0 | 60 | 25 | 250 |
| АГВМ + ГИНК | 49.0 | 68 | 25 | 250 |
| АГНМ + ГИНК | 18.0 | 86 | 25 | 250 |
| ГИНК | 2.5 | 70 | 25 | 263 |
Как видно, индивидуальные азотсодержащие соединения диффундируют из волокна на 70–95% примерно за 2.5–4.5 ч, а комплексы в количестве 60–92% обнаруживаются в модельной среде в течение 18–72 ч. В среднем наибольшим пролонгирующим действием ожидаемо обладают комплексы на основе исходного АГ и АГВМ.
Содержание лекарственного препарата в полимерном комплексе оказывает влияние на его высвобождение (на примере системы АГ+ 5-АСК). Как показано на рис. 1, наиболее оптимальным является мольный состав АГ : 5-АСК = 1 : 1, т.е. стехиометрическое соотношение. При сравнении кривых выхода комплексов с соотношениями больше и меньше стехиометрических видно, что его высвобождение происходит быстрее.
Рис. 1.
Динамика выхода комплексов АГ + 5-АСК из волокна при 25°С. Соотношение АГ : 5-АСК = 1.0 : 0.1 (1), 1.0 : 0.5 (2), 1 : 1 (3) и 1 : 3 (4). А – оптическая плотность раствора.
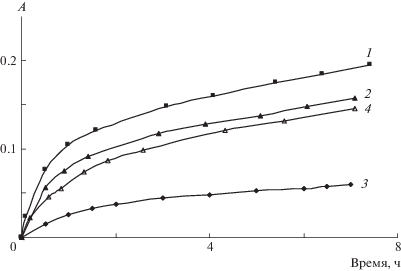
Было обнаружено, что при 38°С скорость выхода возрастает примерно на 30–40%; она также зависит и от химической природы лекарственного препарата. Так, 5-АСК и комплексы 5-АСК с полисахаридами демонстрируют более выраженный эффект пролонгирования по сравнению с 4-АСК и ГИНК.
Хорошо известно, что высвобождение лекарственных соединений из химически стабильных полимерных систем (каким является полиамидное волокно) происходит по диффузионному механизму [28, 29]. Поэтому эффективную константу скорости диффузии комплексов находили путем графического анализа уравнения:
где [A]t – текущая оптическая плотность раствора в момент времени t, [A]0 – максимальная оптическая плотность раствора, kэф – эффективная константа скорости выделения комплексов.Типичные логарифмические анаморфозы кинетических кривых выделения индивидуальных веществ и комплексов представлены на рис. 2. Видно, что индивидуальные лекарственные препараты диффундируют через волокно по закону процесса первого порядка и на логарифмических анаморфозах наблюдается линейная зависимость с высоким коэффициентом корреляции R.
Рис. 2.
Логарифмические анаморфозы кинетических кривых выделения 5-АСК и комплексов АГ + 5-АСК из капиллярного волокна при 25°С: 1 – АГ + 5-АСК, 2 – АГВМ + 5-АСК, 3 – АГНМ + 5-АСК, 4 – 5-АСК. Ось ординат: lnA = ln[A]0 – ln[A]t.
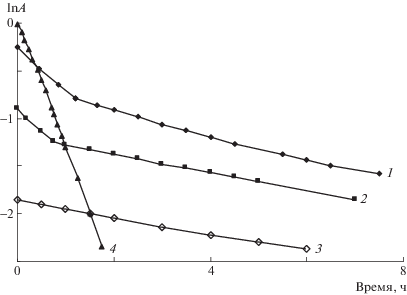
Из трансформаций кинетических кривых диффузии комплексов на основе АГ и АГВМ следует, что высвобождение комплексов в модельные среды протекает в два этапа с разной скоростью (рис. 2). Первый этап характеризуется более интенсивным увеличением концентрации комплекса в модельной системе. Далее происходит постепенное высвобождение оставшегося количества комплекса.
На начальной стадии эффективная константа скорости диффузии значительно выше по сравнению с kэф на более глубокой стадии. Определенные значения эффективных констант скорости диффузии начального участка для комплексов на основе АГ и АГВМ лежат в пределах от 1.52 × 10–3 до 3.45 × 10–3 с–1. На втором участке значения изменяются от 6.57 × 10–4 до 9.27 × 10–4 с–1.
У комплексов АГ + 5-АСК с различным содержанием кислоты начальная скорость диффузии для всех соотношений практически одинаковая (составляет ~(3.0–3.2) × 10–3 с–1), на второй стадии высвобождения при соотношении компонентов в комплексе АГ : 5-АСК равном 1 : 0.1 и 1 : 0.5 значение kэф уменьшается в ~5 раз. При избытке 5-АСК (1 : 3) скорость снижается в 2 раза.
В случае АГНМ анаморфозы кинетических кривых линейны и не содержат излома. Изломы на кинетических кривых можно связать с полидисперсностью арабиногалактана. Исходный АГ имеет, согласно работе [30], молекулярно-массовое распределение 1.6–2.3. Высокомолекулярная фракция окисленного арабиногалактана должна иметь наиболее вероятное ММР вследствие случайного механизма разрыва цепей при окислении. В обоих случаях комплексы на их основе будут содержать низкомолекулярные фракции. Видимо, на начальной стадии процесса происходит диффузия комплексов с наименьшими молекулярными массами, и поэтому наблюдается относительно высокая скорость выделения. На более глубоких стадиях процесса экспериментально проявляется диффузия более высокомолекулярных фракций комплексов.
В пользу данного предположения свидетельствуют следующие факты. Во-первых, kэф для комплекса на основе АГНМ является величиной одного порядка с kэф на начальной стадии процесса для комплексов на основе АГ и АГВМ. Во-вторых, различаются значения энергии активации исследуемых процессов. Для комплекса АГВМ + 5-АСК на начальной стадии диффузии Еакт = 51.4 кДж/моль, а на глубокой стадии Еакт = 72.8 кДж/моль. В системе АГНМ + 5-АСК Еакт = 28.6 кДж/моль. Видимо, чем меньше молекулярная масса и размеры комплексов, тем ниже энергия активации диффузии.
Противотуберкулезная активность арабиногалактана и его окисленных фракций с 4-аминосалициловой кислотой и гидразидом изоникотиновой кислоты была определена в опытах in vitro с использованием культур микобактерий Mycobacterium tuberculosis человеческого типа методом серийных разведений с использованием плотной яичной среды Левенштейна–Йенсена, к которой (перед свертыванием) были добавлены исследуемые соединения. Суспензия культур была приготовлена по бактериальному стандарту мутности 500 млн микробных тел в миллилитре (5 единиц). Эффект оценивался по количеству выросших колоний в пробирках (табл. 4).
Таблица 4.
Туберкулостатическая активность арабиногалактана и его окисленных фракций с 4-АСК и ГИНК
| Соединение | Доза, мкг/мл | Количество колоний |
|---|---|---|
| АГ | 5 | 0–20 |
| АГВМ | 5 | 20–100 |
| АГНМ | 5 | 20–100 |
| АГ + 4-АСК | 5 | 0 |
| АГВМ + 4-АСК | 5 | 0–20 |
| АГНМ + 4-АСК | 5 | 0 |
| 4-АСК | 5 | 0–20 |
| АГ + ГИНК | 1 | 0–20 |
| АГВМ + ГИНК | 1 | 0–20 |
| АГНМ + ГИНК | 1 | 0–20 |
| ГИНК | 1 | 0–20 |
| Контроль | – | <100 |
В результате испытаний установлено, что арабиногалактан обладает противотуберкулезной активностью на уровне свободного препарата, окисленные фракции лишь частично угнетают рост микроорганизмов. Комплексы АГ + 4-АСК и АГНМ + 4-АСК полностью подавляют рост культур микобактерий, даже нечувствительных к свободному 4-АСК. Комплексы на основе АГ и его окисленных фракций с ГИНК обладают активностью на уровне свободного препарата.
Проверка биологической активности исследуемых комплексов с 5-АСК продемонстрировала высокую противоязвенную и противовоспалительную активность полученных образцов [31].
Таким образом, экспериментальные данные показывают, что комплексы АГ и его окисленных форм с 5-АСК, 4-АСК и ГИНК позволяют значительно пролонгировать действие лекарственных препаратов, создавая и поддерживая оптимальную концентрацию терапевтических средств.
Установлено, что динамика диффузии комплексов АГ определяется в основном характеристиками полисахаридной матрицы – ее молекулярной массой.
Авторы выражают благодарность Х.К. Аминеву и Г.С. Хамидуллиной за помощь в исследовании биологической активности полученных соединений.
Анализы выполнены на оборудовании Центра коллективного пользования “Химия” Уфимского института химии и Регионального центра коллективного пользования уникальным оборудованием “Агидель” Уфимского федерального исследовательского центра РАН.
Работа подготовлена в рамках выполнения программы Фундаментальных научных исследований государственных академий на 2013–2020 гг. (Гос. задание № АААА-А20-120012090024-5).
Список литературы
Arifkhodzhaev A.O. // Chem. Natural Compounds. 2000. V. 36. № 3. P. 229.
Zashikhina N.N., Yudin D.V., Tarasenko I.I., Osipova O.M., Korzhikova-Vlakh E.G. // Polymer Science A. 2020. V. 62. № 1. P. 43.
Efimova A.A., Trosheva K.S., Krasnikov E.A., Krivtsov G.G., Yaroslavov A.A. // Polymer Science A. 2019. V. 61. № 6. P. 737.
Shilova S.V., Tret’yakova A.Y., Barabanov V.P. // Polymer Science A. 2019. V. 61. № 1. P. 39.
Ovodov Yu.S. // Russ. J. Bioorganic Chem. 1998. V. 24. № 7. P. 423.
Shtilman M.I. // Polymer Science A. 2010. V. 52. № 9. P. 884.
Martinho N., Damge Ch., Pinto Reis C. // J. Biomater. Nanobiotechnol. 2011. V. 2. № 5. P. 510.
Philippova O.E., Korchagina E.V. // Polymer Science A. 2012. V. 54. № 7. P. 552.
Бочков П.О., Колыванов Г.Б., Литвин А.А., Жердев В.П., Шевченко Р.В. // Фармакокинетика и фармакодинамика. 2016. № 1. С. 3.
Le-Deygen I.M., Skuredina A.A., Kudryashova E.V. // Russ. J. Bioorganic Chem. 2017. V. 43. № 5. P. 487.
Khakamov T.Sh., Feoktistov D.V., Badykova L.A., Kornilaev P.G., Shavaleev R.R., Mudarisova R.Kh. // Russ. J. Applied Chem. 2013. V. 86. № 9. P. 1417.
Селютина О.Ю., Поляков Н.Э., Метелева Е.С., Душкин А.В. // Химия в интересах устойчивого развития. 2015. Т. 23. № 5. С. 549.
Coviello T., Matricardi P., Marianecci C., Alhaique F. // J. Controll. Release. 2007. V. 119. № 1. P. 5.
Медведева Е.Н., Бабкин В.А., Остроухова Л.А. // Химия раст. сырья. 2003. № 1. С. 27.
Хвостов М.В., Толстикова Т.Г., Борисов С.А., Душкин А.В. // Биоорган. химия. 2019. Т. 45. № 6. С. 563.
Grischenko L.A., Parshina L.N., Larina L.I., Novikova L.N., Trofimov B.A. // Carbohydr. Polym. 2015. V. 115. P. 294.
Ganenko T.V., Tantsyrev A.P., Khutsishvili S.S., Vakulskaya T.I., Sukhov B.G., Trofimov B.A., Sapozhnikov A.N., Fadeeva T.V. // Russ. J. General Chem. 2015. V. 85. № 2. P. 477.
Tolstikova T.G., Khvostov M.V., Bryzgalov A.O., Dushkin A.V., Tolstikov G.A. // Dokl. Biological Sci. 2010. V. 433. №. 1. P. 247.
Mudarisova R.Kh., Badykova L.A., Novoselov I.V. // Russ. J. General Chem. 2018. V. 88. № 12. P. 2572.
Khvostov M.V., Borisov S.A., Tolstikova T.G., Dushkin A.V., Tsyrenova B.D., Chistyachenko Yu.S., Polyakov N.E., Dultseva G.G., Onischuk A.A., An’kov S.V. // Eur. J. Drug Metabolism Pharmacokinetics. 2017. V. 42. № 3. P. 431.
Mudarisova R.Kh., Badykova L.A., Azamatova G.A., Aznabaev M.T., Islamova R.M. // Russ. J. Applied Chem. 2013. V. 86. № 4. P. 606.
Karacsonyi S., Kovacik V., Alfoldi J., Kubackova M. // Carbohydr. Res. 1984. V. 134. № 2. P. 265.
Антонова Г.Ф., Усов А.И. // Биоорган. химия. 1984. Т. 10. № 12. С. 1664.
Borisov I.M., Shirokova E.N., Babkin V.A., Tolstikov G.A., Monakov Yu.B. // Dokl. Chem. 2002. V. 383. № 4–6. P. 117.
Borisov I.M., Zimin Yu.S., Monakov Yu.B., Shirokova E.N., Mudarisova R.Kh., Muslukhov R.R., Medvedeva S.A., Tolstikov G.A. // Russ. Chem. Bulletin. 2004. № 2. P. 318.
Farmer K.C. // Clin. Ther. 1999. V. 21. № 6. P. 1074.
Mudarisova R.Kh., Badykova L.A. // Polymer Science A. 2012. V. 54. № 2. P. 106.
Ainaoui A., Vergnaud J.M. // Comput. Theor. Polym. Sci. 2000. V. 10. № 5. P. 383.
Martinelli A., D’Ilario L., Francolini I., Piozzi A. // Int. J. Pharm. 2011. V. 407. № 1–2. P. 197.
Медведева Е.Н., Федорова Т.Е., Ванина А.С., Рохин А.В., Еськова Л.А., Бабкин В.А. // Химия раст. сырья. 2006. № 1. С. 25.
Mudarisova R.Kh., Shirokova E.N., Badykova L.A., Bori-sov I.M., Tolstikova T.G., Sorokina I.V., Dolgikh M.P., Monakov Yu.B. // Pharm. Chem. J. 2005. V. 39. № 8. P. 418.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия А)