

Высокомолекулярные соединения (серия А), 2021, T. 63, № 2, стр. 92-98
ОБРАЗОВАНИЕ ФИБРИЛЛОПОДОБНЫХ СТРУКТУР В ТОНКИХ ПЛЕНКАХ ПОЛИАНИЛИНА ПРИ РЕДОПИРОВАНИИ СМЕСЬЮ ДОДЕЦИЛБЕНЗОЛСУЛЬФОКИСЛОТЫ И N-МЕТИЛ-2-ПИРРОЛИДОНА
И. А. Лобов a, *, Н. А. Давлеткильдеев a, Д. В. Соколов a, Е. Ю. Мосур a
a Омский научный центр Сибирского отделения Российской академии наук
644024 Омск, пр. Карла Маркса, 15, Россия
* E-mail: LI__87@mail.ru
Поступила в редакцию 28.02.2020
После доработки 30.09.2020
Принята к публикации 15.10.2020
Аннотация
Исследованы особенности морфологии и свойств тонких пленок полианилина, редопированного смесью додецилбензолсульфокислоты и N-метилпирролидона. Пленки имеют развитую морфологию, образованную хаотично распределенными фибриллоподобными структурами. Показано, что формирование развитой морфологии сопровождается увеличением концентрации допанта, ростом удельной электропроводности пленки и уменьшением ширины запрещенной зоны полимера. Формирование фибриллярной структуры пленки полианилина объясняется протеканием процесса самосборки взвешенных в растворе допированных молекул полианилина.
ВВЕДЕНИЕ
В настоящее время наметилась тенденция к замене традиционных неорганических элементов в электронных устройствах на электропроводящие полимеры [1, 2]. По своим электронным характеристикам данные полимеры близки к полупроводникам, но в отличие от них физико-химические свойства полимеров можно варьировать в более широком диапазоне. Начальные характеристики полимера задаются на стадии синтеза путем контроля таких параметров, как температура, время синтеза, соотношение реагентов, наличие органических и неорганических добавок и других [1, 3, 4]. Дальнейшая модификация полимера возможна на последующих стадиях его подготовки для целевого использования.
Полианилин – один из наиболее исследуемых проводящих полимеров, основными достоинствами которого являются достаточно низкая стоимость и простота синтеза [5]. Особо следует отметить возможность сравнительно легкого управления электронными свойствами данного полимера, что можно реализовать и на стадии его синтеза, и на последующем этапе. В первом случае осуществляется синтез полимера при различных условиях полимеризации (pH, соотношение окислитель/мономер, потенциал электрода, температура синтеза и т.д.) и создание композитов с органическими или неорганическими наполнителями (оксидами металлов, углеродными материалами). Во втором случае можно провести дедопирование и повторное допирование полимера, а также последующее (ex-situ) формирование композитов полианилина путем смешивания готового полимера с наполнителями различной природы.
В качестве допантов полианилина могут выступать неорганические и органические кислоты. При синтезе полимера чаще всего используется соляная кислота как наиболее дешевый и технологически простой вариант, а комплексы полианилина с органическими кислотами формируют при повторном допировании уже готового полимера. Допирование органическими кислотами приводит к образованию более прочных комплексов полимер–кислота, которые демонстрируют повышенную термостабильность и электропроводность [6].
Продукт полимеризации анилина – электропроводящая эмералдиновая соль (ЭС-ПАНИ). Синтезированное вещество является не растворимым и не плавким, что значительно осложняет его дальнейшее использование. Дедопирование ЭС-ПАНИ в форму эмералдинового основания (ЭО-ПАНИ) влечет удаление кислоты из полимера, который становится растворимым в ряде растворителей и способным к нанесению на поверхность в виде пленок. Результатом последующего допирования служит электропроводящая форма полимера, при этом в зависимости от состава допанта и условий допирования структура и свойства ПАНИ могут быть различными [7]. Понимание механизмов, приводящих к модификации электронных свойств ПАНИ при таком допировании, дает возможность управлять характеристиками полимера для использования в различных приложениях.
На сегодняшний день имеются работы, посвященные допированию ПАНИ кислотами в присутствии растворителей [8–10]. В этих работах порошок ЭО-ПАНИ, органическая кислота и полярный растворитель смешивали между собой в разных пропорциях. Пленки ПАНИ, осажденные из таких растворов, обладали проводимостью до 103 См/см. Авторы связывают это с образованием более делокализованного катион-радикала. Известны также работы, в которых показано, что присутствие растворителя при допировании ПАНИ не всегда положительно влияет на конечный продукт. В работе [11] было выявлено, что ЭО-ПАНИ, растворенный в N-метилпирролидоне, не может быть эффективно допирован слабой кислотой 6-циано-2-нафтолом, в то время как высушенная пленка ЭО-ПАНИ легко ею допируется. Объяснить это можно формированием водородной связи между растворителем и полимером, препятствующей процессу допирования. Таким образом, присутствие растворителя при допировании как правило улучшает электронные характеристики полимера. Однако работы в этом направлении немногочисленны и недостаточны для понимания механизма роста проводимости полимера.
Цель настоящей работы – изучить влияние растворителя в составе допирующей смеси на основе органической кислоты на изменение морфологии и электронных свойств тонких пленок ЭО-ПАНИ.
МАТЕРИАЛЫ И МЕТОДЫ
Для синтеза ПАНИ использовали окислительную полимеризацию анилина персульфатом аммония (ПСА) в водном растворе соляной кислоты HCl. Предварительно готовили два раствора с эквимольным соотношением мономера анилина и ПСА: первый раствор содержал 3 мл анилина, 25 мл 3 моль/л HCl и 40 мл изопропилового спирта; второй – 7.5 г ПСА и 15 мл 3 моль/л HCl. Полученные растворы охлаждали до 0°С в ледяной бане и оставляли там на время проведения реакции полимеризации (2 ч), для осуществления которой к раствору анилина по каплям добавляли раствор окислителя при постоянном перемешивании. В ходе экзотермической реакции происходило выпадение густого черно-зеленого осадка полимера. Осадок пропускали через воронку Бюхнера, отмывали раствором 3 моль/л HCl, ацетоном и абсолютным этиловым спиртом, а затем сушили на воздухе при комнатной температуре.
Так как, в результате описанного выше процесса сформировался полианилин в нерастворимой форме эмералдиновой соли соляной кислоты, поэтому его переводили в форму эмералдинового основания – дедопировали в водном растворе аммиака 1% в течение 24 ч. ЭО-ПАНИ растворяли в N-метил-2-пирролидоне (МП), после чего наносили методом центрифугирования в виде тонких пленок на подложки SiO2/Si, кварц и CaF2 для исследования методами атомно-силовой микроскопии, рентгеновской дифракции, УФ-вид- и ИК-спектроскопии. Повторное допирование пленок осуществляли водным раствором додецилбензолсульфокислоты (ДБСК) и ее смесью с МП (соотношение 1 : 2 по объему). Продолжительность допирования составляла 30 мин. Далее образцы отмывали дистиллированной водой и абсолютным этиловым спиртом, затем высушивали на воздухе при комнатной температуре.
Для проведения сравнительного анализа в работе использовали три типа образцов: пленки исходного ЭО-ПАНИ, редопированные пленки ЭС-ПАНИ/ДБСК : H2O и ЭС-ПАНИ/ДБСК : МП.
Морфологию образцов изучали на атомно-силовом микроскопе “MFP-3D SA” (“Asylum Research”). Применяли кантеливеры серии “Gоlden NSG01” (NT-MDT SI) c резонансной частотой 150 кГц и типичным радиусом закругления зонда 10 нм. Измерения проводили на воздухе в полуконтактном режиме сканирования.
Для определения структурных особенностей полученных пленок использовали метод рентгеновской дифракции. Измерения выполняли на дифрактометре “Bruker D8 Advance” (Cu-Kα источник излучения с λ = 1.54 Å). Диапазон измерения углов 2θ от 10° до 70°, скорость сканирования 0.05 град/сек.
Спектры инфракрасного пропускания пленок регистрировали с помощью ИК-фурье-спектрометра “VERTEX 70” (“Bruker”). Спектры оптического пропускания снимали на спектрофотометре “UV-VIS 2501PC” (“Shimadzu”). Пропускание T пересчитывали в коэффициент поглощения α в соответствии с законом Бугера–Ламберта–Бера:
где d – толщина пленки, измеряемая интерферометрическим методом до и после редопирования для каждого образца.Оптическую ширину запрещенной зоны пленок ПАНИ находили согласно теории Тауца, в соответствии с которой коэффициент поглощения в области края поглощения и ширина запрещенной зоны для прямых разрешенных переходов связаны выражением
Здесь hν – энергия фотона, B – коэффициент пропорциональности, Eg – ширина запрещенной зоны.
Для определения удельной проводимости пленок ЭС-ПАНИ/ДБСК при комнатной температуре применяли стандартный четырехзондовый метод измерения поверхностного электрического сопротивления. Измерение проводили на цифровом носителе удельного сопротивления 13 МП-0.5-001 с использованием измерительной головки с четырьмя линейно расположенными вольфрамовыми зондами с золотым покрытием. Поверхностное сопротивление пленок устанавливали на основе уравнения, применяемого для случая, когда толщина пленки много меньше расстояния между зондами:
Удельную электропроводность пленок определяли в соответствии с выражением
где α – поправочный коэффициент, учитывающий соотношение длины, ширины пленки и расстояния между зондами; d – толщина пленки.РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
На рис. 1 представлены типичные АСМ-изображения исследуемых пленок. На вставках показаны изображения с бóльшим увеличением, демонстрирующие гранулярную морфологию пленок, структурным элементом которых является макромолекулярная глобула (гранула) ПАНИ. На основе анализа АСМ-изображений были определены следующие параметры пленок: средний размер гранул и среднеквадратичная шероховатость поверхности. Пленка ЭО-ПАНИ (рис. 1a) имеет гранулярную морфологию со средней шероховатостью 18.3 нм и средним размером гранул 161 ± ± 33 нм. Поверхность допированной пленки ЭС-ПАНИ/ДБСК : H2O (рис. 1б) сохранила морфологию, характерную для ЭО-ПАНИ, но ее средняя шероховатость уменьшилась до 5.8 нм, а средний размер гранул увеличился до 227 ± 48 нм. Пленка ЭС-ПАНИ/ДБСК : МП (рис. 1в) имеет развитую фибриллоподобную морфологию, средняя шероховатость составляет 22.3 нм. На рис. 1в видно, что гранулы полимера сгруппированы в хаотически распределенные фибриллярные структуры толщиной 300–500 нм, при этом средний размер гранул возрос до 266 ± 40 нм. Таким образом, допирование пленок ЭС-ПАНИ смесью кислоты и растворителя дает развитую структурированную морфологию пленок. Данный эффект не удается получить при допировании раствора полимера. Пленки, высаженные из таких смесей, либо не имеют структурных особенностей, либо повторяют исходную морфологию ПАНИ (ввиду слабой растворимости) [12, 13].

Толщина исследованных пленок имела значения от 0.6 до 8.0 мкм, но после обработки допирующими смесями, как правило, она уменьшалась на 10–30%. Несмотря на то что комплекс ПАНИ–ДБСК значительно увеличивает размер гранул после допирования, уменьшение толщины пленок связывается с частичным переходом полимера в допирующую смесь.
На рис. 2 приведены типичные ИК-спектры для пленок всех трех типов. Положение основных пиков (1590, 1500, 1300, 1160 см–1) в спектрах ПАНИ соответствует литературным данным [14, 15]. Характерные полосы вблизи 1590 и 1500 см–1 связаны с деформациями C=C в хинондииминных и бензольных кольцах, пики при 1300 см–1 ассоциируются с различными колебаниями C–N. Пики вблизи 1160 и 1670 см–1 относятся к колебаниям C=N в хинондииминном кольце [16, 17].
Рис. 2.
ИК-спектры пропускания пленок ЭО-ПАНИ (а), ЭС-ПАНИ/ ДБСК : H2O (б) и ЭС-ПАНИ/ДБСК : : МП (в).
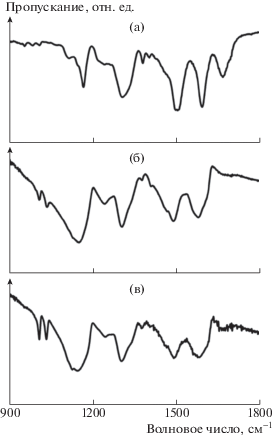
Особенностью спектров редопированных пленок ПАНИ является появление полос ⁓1030 и 1000 см–1, ассоциируемых с колебаниями O=S=O и S–O в молекулах ДБСК [18, 19]. Интенсивности пиков, связанных с ДБСК, в пленках ЭС-ПАНИ/ДБСК : : МП значительно выше, чем в ЭС-ПАНИ/ДБСК : : H2O. Такая разница свидетельствует о бóльшем содержании молекул сульфокислоты в пленках при допировании смесью кислоты и растворителя. Действительно, наличие N-метил-2-пирролидона приводит к растворению верхних слоев пленки и разрыхлению нижних. Это улучшает доступ большой молекулы ДБСК в полимерный слой. Интенсивность и положение остальных пиков не изменяются, что свидетельствует о сохранении молекулярной структуры полимера при редопировании.
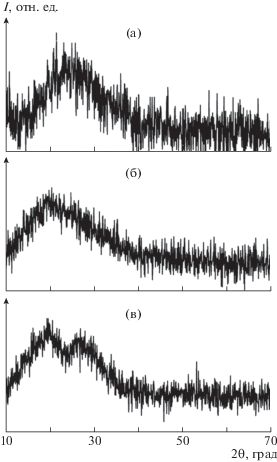
Полианилин можно назвать полукристаллическим по своей природе, он может находиться как в кристаллической фазе, где полимерные цепи параллельны и упорядочены в плотноупакованный массив, так и в аморфной фазе. На рис. 3 показаны спектры рентгеновской дифракции исследуемых пленок. Из дифрактограмм пленок ЭО-ПАНИ, ЭС-ПАНИ/ДБСК : Н2О и ЭС-ПАНИ/ДБСК : МП видно, что они имеют максимумы около 2θ = 20° и 25°, которые связывают с периодичностью параллельной и перпендикулярной упаковки полимерной цепи [20, 21]. Наличие широких пиков указывает на присутствие значительной доли аморфной фазы. Допирование полианилина ДБСК приводит к увеличению кристалличности полимера [21], что подтверждается появлением в спектрах образцов полосы при 2θ = 20°. Наличие двух разразрешаемых пиков на дифрактограмме образца ЭС-ПАНИ/ДБСК : МП свидетельствует о более высокой степени кристалличности полимера.
Рис. 3.
Спектры рентгеновской дифракции пленок ЭО-ПАНИ (а), ЭС-ПАНИ/ДБСК : H2O (б) и ЭС-ПАНИ/ДБСК : МП (в).
Оптические спектры поглощения были записаны в диапазоне длины волн от 200 до 1000 нм (рис. 4, вставки). В спектрах пленок ЭО-ПАНИ наблюдаются два характерных для этой формы полимера максимума при 330 и 625 нм. Для редопированных пленок имеются пики при 330, 425 нм и в области ~800 нм. Полоса поглощения при 330 нм свойственна для всех форм ПАНИ и обусловлена электронными π–π*-переходами в фенильных кольцах, связанных с амино- и иминогруппами. Пики поглощения при 425 нм и в области 800 нм встречаются только в электропроводящих формах ПАНИ и взаимодействуют с полярон-π*-переходами и π-полярон-переходами соответственно. Положение этих пиков не зависит от способа допирования. Ранее в работах [22, 23] было показано, что оптические спектры растворенного ПАНИ зависят от используемого растворителя. Однако при формировании пленок из этих растворов, различия в положении пиков нивелируются.
Рис. 4.
Спектры поглощения пленок ЭО-ПАНИ (а), ЭС-ПАНИ/ДБСК : H2O (б) и ЭС-ПАНИ/ДБСК : : МП (в) в координатах (αhν)2–(hν). Ширина запрещенной зоны Eg = 3.24 (а), 2.82 (б) и 2.49 (в). На вставках показаны измеренные спектры пропускания пленок.
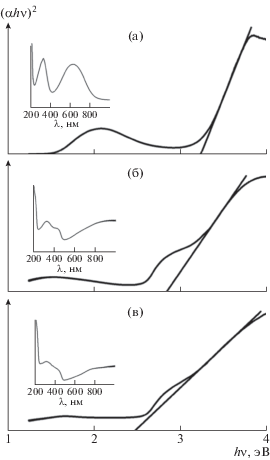
Ширина запрещенной зоны Eg образцов определялась графическим методом путем экстраполяции линейного участка зависимости (αhν)2 от hν (рис. 4) на ось абсцисс, где α – коэффициент поглощения, а hν – энергия фотона, hν = (1240/λ) эВ.
Для ЭО-ПАНИ ширина запрещенной зоны равна 3.20 эВ, что согласуется с литературными данными [24]. При допировании значение Eg уменьшается (2.82 и 2.49 эВ для ЭС-ПАНИ/ДБСК : : H2O и ЭС-ПАНИ/ДБСК : МП соответственно). Уменьшение ширины запрещенной зоны связано с образованием дополнительных электронных состояний в полимере. Вероятная причина роста числа состояний в ЭС-ПАНИ/ ДБСК : МП рассмотрена ниже.
С одной стороны, нарушение морфологической однородности пленки ЭС-ПАНИ/ДБСК : МП должно отразиться на ее электропроводности отрицательно. С другой стороны, наличие структурных особенностей в полианилине повышает электропроводность [13]. Так, проводимость вдоль отдельной структурной единицы полимера может быть на несколько порядков выше, чем в слое [25]. В дополнении к этому, более высокое содержание ДБСК как источника носителей заряда и рост степени кристалличности полимера должно увеличить электропроводность пленки. Удельная электрическая проводимость пленок ЭС-ПАНИ/ДБСК : H2O и ЭС-ПАНИ/ДБСК : МП, измеренная четырехзондовым методом, составляет в среднем 50 и 80 См/см соответственно. Значит, несмотря на морфологическую неоднородность пленки, рост содержания кислоты и наличие фибриллоподобной структуры приводит к увеличению проводимости пленки ЭС-ПАНИ/ДБСК : МП.
Процесс легирования пленки ЭО-ПАНИ водным раствором ДБСК можно описать следующим образом. ПАНИ в форме эмералдинового основания является гидрофобным [26]. Макромолекулы полимера плотно упакованы и могут содержать небольшое количество связанной воды. ПАНИ не растворяется в воде, поэтому вода в допирующей смеси не разрыхляет пленку, а действует как транспортная среда для молекул кислоты. Протонирование пленки включает собственно химическую реакцию и диффузионные процессы [27]. В ходе реакции возникает связь протона с атомом азота на иминном сегменте ЭО-ПАНИ, а в результате диффузии протон и противоион кислоты проникают в пленку. Однако проникновение довольно большой молекулы ДБСК затруднено как внутрь не разрыхленной молекулы ПАНИ, так и вглубь плотно упакованной полимерной пленки. В таком случае изменение морфологии пленки возможно только за счет увеличения размера макромолекул полимера при образовании комплексов с кислотой и последующего их смещения из исходного положения вследствие возникшего механического напряжения. Это предположение подтверждается данными, полученными методом АСМ.
При допировании пленки смесью ДБСК и МП одновременно протекают два процесса: растворение ЭО-ПАНИ и образование электропроводящих комплексов ДБСК-ПАНИ. Растворение подразумевает набухание пленки вследствие проникновения растворителя в макромолекулы полимера с последующей сольватацией. Растворитель, разрыхляя макромолекулы ПАНИ, способствует лучшему проникновению в них молекул ДБСК. Рост количества молекул кислоты в составе комплекса ПАНИ–ДБСК увеличивает концентрацию носителей заряда и число электронных состояний в полимере в целом, что подтверждается полуторакратным возрастанием удельной проводимости и значительным сужением запрещенной зоны ПАНИ в пленке ЭС-ПАНИ/ДБСК : МП. Более высокая концентрация молекул кислоты в пленке ЭС-ПАНИ/ДБСК : МП, чем в ЭС-ПАНИ/ДБСК : H2O также подтверждается данными ИК-спектроскопии.
Процесс допирования ДБСК снижает способность ПАНИ к растворению, однако это не происходит мгновенно. Постепенное изменение цвета пленки от синего до зеленого (цвет связан с уровнем допирования) в процессе допирования можно наблюдать визуально в течение нескольких минут. Для снижения скорости допирования важно использовать достаточно большую молекулу кислоты, такую как ДБСК в данном эксперименте. Макромолекулы верхних слоев пленки, успевшие раствориться до допирования, отделяются от нее и переходят в раствор, где и происходит их допирование. С ростом концентрации допированных макромолекул ПАНИ в приповерхностной области раствора они начинают взаимодействовать между собой и собираться в фибриллоподобные структуры. Центрами роста таких структур могут выступать как отдельные макромолекулы ПАНИ в растворе с последующим их осаждением на поверхность пленки, так и допированный нерастворенный верхний слой пленки. Остается неясным вопрос: почему формируются волокна, а не другие надмолекулярные структуры, например глобулы. Здесь необходимо проведение дополнительных исследований.
ЗАКЛЮЧЕНИЕ
В работе впервые показана возможность трансформации исходно неструктурированной тонкой пленки ЭО-ПАНИ в высокопроводящую пленку с развитой фибриллоподобной морфологией, путем воздействия на нее смесью органической кислоты и растворителя. Тонкие пленки дедопированного ПАНИ редопировали смесями ДБСК : H2O и ДБСК : МП. При использовании первой смеси изменений в морфологии пленок не наблюдалось. При редопировании смесью ДБСК : МП пленки ПАНИ приобретают развитую структурированную морфологию, образованную хаотически распределенными фибриллярными структурами толщиной 300–500 нм. Показано, что изменение морфологии пленки сопровождается увеличением содержания допанта и ростом удельной электропроводности до 80 См/см. Предложен возможный механизм редопирования пленок ПАНИ смесью ДБСК : МП. Суть его заключается в том, что растворитель способствует проникновению кислоты как в макромолекулы полимера, так и в объем пленки. Использование достаточно большой молекулы кислоты позволяет снизить скорость допирования, сдвигая баланс в сторону преобладания процесса растворения ПАНИ. Формирование фибриллярной структуры пленки возможно за счет процесса самосборки взвешенных в растворе допированных молекул ПАНИ.
Работа выполнена по госзаданию Омского научного центра Сибирского отделения РАН в соответствии с Программой фундаментальных научных исследований государственных академий наук на 2013–2020 гг. по направлению II.9, проект № II.9.2.1 (номер АААА-А17-117041210227-8).
Список литературы
Xing J., Liao M., Zhang C., Yin M., Li D., Song Y. // Phys. Chem. Chem. Phys. 2017. V. 19. P. 14030.
Iqbal S., Ahmad S. // J. Ind. Eng. Chem. 2018. V. 60. P. 53.
Noskov Y., Mikhaylov S., Coddeville P., Wojkiewicz J.-L., Pud A. // Synthetic Metals. 2016. V. 217. P. 266.
Wang H., Yi S., Pu X., Yu C. // ACS Appl. Mater. Int. 2015. V. 7. P. 9589.
Tanguy N.R., Thompson M., Yan N. // Sensors Actuat. B. 2018. V. 257. P. 1044.
Jelmy E.J., Ramakrishnan S., Rangarajan M., Kothurkar N.K. // Bull. Mater. Sci. 2013. V. 36. № 1. P. 37.
Horta-Romarís L., González-Rodríguez M.V., Lasagabástera A., Rivadulla F., Abad M.-J. // Synth. Met. 2018. V. 243. P. 44.
Ghosh S. // Chem. Phys. Lett. 1994. V. 226. P. 344.
MacDiarmid A.G., Epstein A.J. // Synth. Met. 1994. V. 65. № 2–3. P. 103.
Izumi C.M.S., Rodrigues D.C., Temperini M.L.A. // Synth. Met. 2010. V. 160. P. 2552.
Das D., Datta A., Contractor A.Q. // J. Phys. Chem. B. 2014. V. 118. № 45. P. 12993.
Taş R., Gülen M., Can M., Sönmezoğlu S. // Synthetic Metals. 2016. V. 212. P. 75.
Krukiewicz K., Katunin A. // Synthetic Metals. 2016. V. 214. P. 45.
Trchová M., Morávková Z., Šeděnková I., Stejskal J. // Chemical Papers. 2012. V. 66. № 5. P. 415.
Trchová M., Stejskal J. // Pure Appl. Chem. 2011. V. 83. № 10. P. 1803.
Lin Y.-W., Wu T.-M. // Comp. Sci. Technol. 2009. V. 69. P. 2559.
Ibrahim M., Koglin E. // Acta Chim. Slov. 2005. V. 52. P. 159.
Karaoğlan N., Bindal C. // Eng. Sci. Technol. Int. J. 2018. V. 21. P. 1152.
Kabomo T.M., Scurrell M.S. // Polym. Adv. Technol. 2016. V. 27. P. 1195.
Ayad M., Zaghlol S. // Chem. Eng. J. 2012. V. 204–206. P. 79.
Ashokan S., Ponnuswamy V., Jayamurugan P., Chandrasekaran J., Subba Rao Y.V. // Superlatt. Microstructures. 2015. V. 85. P. 282.
Dubey R., Dutta D., Yadav V.K., Shami T.C., Rao K.U.B. // Synthetic Metals. 2010. V. 160. № 15–16. P. 1627.
Majidi M.R., Kane-Maguire L.A.P., Wallace G.G.G. // Polymer. 1996. V. 37. № 2. P. 359.
Chutia P., Kumar A. // Physica. B. 2014. V. 436. P. 200.
Tao Y., Li J., Xie A., Li S., Chen P., Ni L., Shen Y. // Chem. Commun. 2014. V. 50. № 84. P. 12757.
Junaid A.S., Shaochun T., Xiangkang M. // Scientific Reports. 2017. V. 7. P. 4403.
Wan M., Yang J. // J. Appl. Polym. Sci. 1995. V. 55. № 3. P. 399.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия А)