

Высокомолекулярные соединения (серия А), 2021, T. 63, № 4, стр. 283-292
МОДЕЛИРОВАНИЕ ЛОКАЛЬНОЙ ДИНАМИКИ МЕЖМОЛЕКУЛЯРНЫХ ВЗАИМОДЕЙСТВИЙ В БУТАДИЕН-НИТРИЛЬНЫХ СОПОЛИМЕРАХ С КАРБОКСИЛЬНЫМИ И ГИДРОКСИЛЬНЫМИ ГРУППАМИ
М. Е. Соловьев a, *, А. Б. Раухваргер a, В. И. Иржак b
a Ярославский государственный технический университет
150023 Ярославль, Московский пр., 88, Россия
b Институт проблем химической физики Российской академии наук
142432 Московская обл., Черноголовка, пр. ак. Семенова, 1, Россия
* E-mail: soloviev56@gmail.com
Поступила в редакцию 05.01.2021
После доработки 02.03.2021
Принята к публикации 16.03.2021
Аннотация
Исследована структура и динамика межмолекулярных комплексов, образуемых карбоксильными и гидроксильными группами в бутадиен-нитрильных сополимерах. Определена энергия межмолекулярных взаимодействий при оптимизации геометрии комплексов и моделировании классической динамики с помощью квантово-химического метода функционала плотности DFT B3LYP/6-311G**. Установлено, что в зависимости от химической природы функциональных групп при соблюдении конкретных условий (температура и деформация) они могут формировать узлы физической сетки эластомера, способные воспринимать внешнюю нагрузку, либо, разрушаясь, просто изменять вязкоупругие свойства эластомера по классическому механизму.
ВВЕДЕНИЕ
Полимерные гели и студни в последнее время активно используют в самых различных областях, среди которых медицина и пищевая промышленность, также всем хорошо известно их применение в быту и технике [1, 2]. Несмотря на давнюю историю изучения полимерных гелей, до сих пор существуют несколько формальных определений [3, 4]. Общепринятым является подразделение полимерных гелей на химические, в которых узлы полимерной сетки образованы за счет ковалентных связей, и физические (термообратимые) гели. В физических гелях в роли узлов полимерной сетки могут выступать микрокристаллиты, комплиментарные последовательности регулярных участков цепей, области фазового разделения, ионные и водородные связи. В то же время, выделяется широкий класс систем, которые можно назвать одновременно и химическими и физическими гелями – это сшитые эластомеры с физической сеткой [5–7]. Известным примером таких систем являются сшитые бутадиен-нитрильные эластомеры с карбоксильными и гидроксильными группами [8–13]. Композиции на основе данных эластомеров обладают рядом ценных технических свойств, диапазон которых может широко варьироваться составом сополимера и количеством функциональных групп. В зависимости от условий синтеза или модификации сополимера функциональные карбоксильные и гидроксильные группы могут случайным образом распределяться вдоль полимерной цепи, либо располагаться только на концах цепей (бутадиен-нитрильные сополимеры с концевыми карбоксильными и концевыми гидроксильными группами) [14–17]. В последнем случае появляются дополнительные возможности молекулярного дизайна за счет направленного регулирования молекулярно-массового распределения длины цепей сетки, а также создания блок-сополимеров с другими типами полимеров, используя при этом концевые функциональные группы в качестве инструмента для регулирования архитектуры сетчатого сополимера.
Для моделирования упругих свойств бутадиен-нитрильных сополимеров (NBR) используют в основном классические феноменологические подходы, базирующиеся на представлении упругой энергии в виде функции инвариантов тензора деформации [18–21]. Вместе с тем, для бутадиен-нитрильных сополимеров характер межмолекулярных взаимодействий в силу сильного различия в полярности бутадиеновых и нитрильных звеньев отличается, что приводит к особенностям их релаксационных свойств [22–24].
Особенности физических свойств рассматриваемых эластомеров обусловлены, в первую очередь, высоким уровнем межмолекулярного взаимодействия полярных функциональных групп. Несмотря на то что с химической точки зрения природа межмолекулярных взаимодействий данных функциональных групп хорошо известна, экспериментальная оценка их параметров (термодинамических функций и динамики для полимерных систем) весьма непростая задача. Наиболее информативными методами экспериментального изучения межмолекулярных взаимодействий, а также процессов ассоциации и комплексообразования в полимерах представляются методы спектроскопии ЯМР [25–27]. С их помощью удается рассчитывать состав и термодинамические функции образования комплексов, оценивать изменение времен релаксации при образовании комплекса, коэффициенты диффузии цепей и их фрагментов. Однако данные, касающиеся оценки этих параметров для рассматриваемых в настоящей работе полимеров, практически отсутствуют.
Цель настоящей работы – исследование межмолекулярных взаимодействий функциональных групп бутадиен-нитрильных сополимеров, несущих карбоксильные и гидроксильные функциональные группы методом компьютерного моделирования.
МЕТОДЫ МОДЕЛИРОВАНИЯ
Расчет полных электронных значений энергии и термодинамических функций исследуемых соединений проводили квантово-химическим методом функционала плотности [28, 29] c гибридным обменно-корреляционным функционалом Беке, Ли, Янга и Парра [30, 31] DFT B3LYP/6-311G** с открытыми оболочками [32], используя программный комплекс “NWChem” [33]. Наличие особых точек на поверхностях потенциальной энергии контролировали общепринятыми методами на основании вибрационного анализа [34]: минимумов – по отсутствию отрицательных собственных значений матрицы Гессе в точке предполагаемого экстремума, а седловых точек – по наличию одного отрицательного собственного значения. Молекулярно-динамическое моделирование осуществляли путем решения классических уравнений движения Ньютона при вычислении градиента энергии на каждом шаге интегрирования из решения квантово-химической задачи методом DFT B3LYP/6-311G**. Интегрирование уравнений движения выполняли в соответствии с алгоритмом Verlet, шаг интегрирования по времени составлял 0.2419 × 10–15 с. Начальное распределение скорости отвечало распределению Максвелла–Больцмана, для поддержания температуры, которая в экспериментах составляла 323 К, применяли термостат [35], параметр релаксации составлял 100 шагов интегрирования по времени.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Простейшая, хотя и поверхностная, оценка различия в энергии межмолекулярных взаимодействий отдельных фрагментов цепи и функциональных групп изучаемых полимеров может быть сделана путем анализа теплоты испарения низкомолекулярных соединений различных классов, содержащих рассматриваемые типы функциональных групп. В рамках линейного приближения энтальпия испарения химического соединения ΔH, содержащего N-типов функциональных групп, представляется в виде суммы инкрементов этих групп ΔHi, умноженных на их количество ni:
Далее показаны инкременты в энтальпию испарения при стандартных условиях фрагментов цепи и функциональных групп бутадиен-нитрильных сополимеров с карбоксильными и гидроксильными группами, рассчитанные методом наименьших квадратов на основе экспериментальных данных работы [36] по выборкам для низкомолекулярных соединений на основе олефинов, нитрилов, карбоновых кислот и спиртов (кДж/моль):
Видно, что перечисленные выше типы функциональных групп занимают три верхние позиции по энергии межмолекулярных взаимодействий, тогда как фрагментам звеньев бутадиена отвечают наименьшие значения энергии. Следует отметить, что энтальпия испарения характеризует энергию, которую нужно затратить при стандартных условиях, чтобы перевести вещество из жидкого состояния в газообразное. Однако в реальном полимере разрыв межмолекулярных связей не приводит к их полному разрушению, поскольку при этом происходит замена одного вида взаимодействия другим. Более адекватную картину можно было бы получить путем вычисления инкрементов из данных по энтальпиям смешения низкомолекулярных жидкостей с различными типами функциональных групп. Только такие экспериментальные данные в необходимом количестве в литературе отсутствуют. Поэтому в настоящей работе была сделана попытка применения квантово-химических вычислений для расчета энергии межмолекулярных взаимодействий функциональных групп, формирующих узлы физической сетки.
Ниже приведены схемы структур, использовавшиеся для моделирования межмолекулярных взаимодействий, и их условные обозначения. Структуры I–III отвечают образованию узлов физической сетки в полимере в результате взаимодействий карбоксильных, гидроксильных и нитрильных групп соответственно; структуры IV–VI моделируют разрушенные физические узлы, что рассматривается как межмолекулярное взаимодействие соответствующей полярной функциональной группы с неполярным звеном бутадиена:
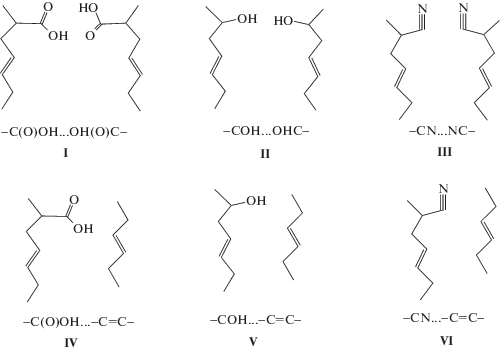
Таким образом, изменение термодинамических функций при разрушении физических узлов можно рассматривать аналогично химическим реакциям как разность термодинамических функций структур, моделирующих взаимодействие двух функциональных групп и структур, моделирующих взаимодействие функциональной группы с неполярным углеводородным фрагментом. В результате, изменение свободной энергии Гиббса при разрушении физического узла из двух карбоксильных групп составило 42.4 кДж/моль, а для гидроксильных и нитрильных групп 17.0 и 13.7 кДж/моль. Качественно этот результат согласуется с порядком расположения рассмотренных функциональных групп по значениям инкрементов в энтальпию испарения.
Изменение термодинамических функций при разрушении межмолекулярных взаимодействий функциональных групп свидетельствует о том, что образование физических узлов в полимере термодинамически выгодно. Однако остается вопрос о том, способны ли они воспринимать механическую нагрузку со стороны полимерной цепи, или же они являются настолько лабильными, что под влиянием приложенной механической нагрузки будут легко разрушаться и образовываться в новых местах, либо будут скользить вдоль полимерной цепи без изменения энергии. Были выполнены две серии расчетов: в первой серии проводилась условная оптимизация геометрии молекулярных моделей, отвечающих структурам I–III, при заданном расстоянии между концевыми атомами углерода смежного с функциональной группой бутадиенового звена; во второй серии расчетов осуществлялось молекулярно-динамическое моделирование данной структуры при фиксированном расстоянии между концевыми углеродными атомами.
В качестве примера приведены молекулярные модели структуры I после условной оптимизации геометрии при различных значениях расстояний между фиксированными концевыми атомами углерода бутадиенового звена:
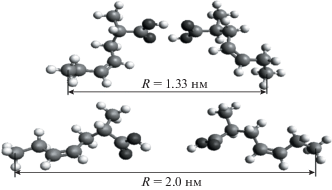
Для иллюстрации в таблице 1 представлены измеренные значения расстояний между атомами, углов и частичных зарядов на атомах для структуры I в минимуме энергии (R = 1.33 нм), т.е. для узла физической сетки, образованного карбоксильными группами, и структуры, отвечающей разрушенному узлу (R = 2.0 нм). Обозначения атомов в структуре даны ниже:
Таблица 1.
Расстояния между атомами (нм), углы (град) и частичные заряды на атомах (ед. заряда электрона по Малликену) для структуры в минимуме энергии (R = 1.33), характеризующей узел физической сетки, образованный карбоксильными группами, и структуры, отвечающей разрушению узла (R = 2.0)
| R, нм | Двугранный угол C8..O2..O3..O16, град | Угол, град | Расстояние между атомами, нм | Заряд на атоме, e | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C1..O2..H | O1..H2..O4 | O1..H2 | O3..H2 | C1..O2 | O2..O3 | O3..C2 | O1 | O2 | O3 | O4 | ||
| 1.33 | 3.0 | 110.0 | 179.5 | 0.167 | 0.099 | 0.132 | 0.267 | 0.123 | –0.432 | –0.320 | –0.432 | –0.319 |
| 2.0 | 48.3 | 110.2 | 136.0 | 0.470 | 0.097 | 0.136 | 0.566 | 0.121 | –0.339 | –0.325 | –0.341 | –0.323 |
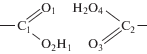
Видно, что узел формируется за счет двух водородных связей между карбоксильными группами, которые образуют практически плоскую структуру с двугранным углом между атомами C8..O2..O3..O16, близким к нулю, и практически линейным расположением атомов O1..H2..O4. Распределение зарядов на атомах кислорода при этом неоднородно, что свидетельствует о существенном перераспределении электронной плотности при образовании водородной связи. При R = 2.0 эта связь практически разрушается, так что заряды на всех четырех атомах кислорода имеют близкие значения. Вместе с тем, даже при существенном увеличении расстояния межмолекулярное взаимодействие карбоксильных групп частично сохраняется, что прослеживается по взаимному расположению атомов углерода и кислорода в карбоксильных группах.
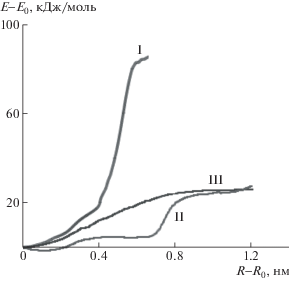
На рис. 1 показана зависимость энергии молекулярных моделей, отвечающих структурам I–III, от относительного изменения расстояния между фиксированными концевыми атомами углерода при увеличении расстояния. Так, с ростом расстояния между фиксированными концевыми атомами углерода энергия систем возрастает по кривым с насыщением, по S-образным зависимостям. В наибольшей степени рост энергии характерен для взаимодействия карбоксильных групп, для гидроксильных и нитрильных групп он выражен в существенно меньшей степени и приблизительно одинаков. Важно отметить различие в скорости нарастания энергии с увеличением расстояния для разных типов взаимодействий. Относительно карбоксильных и гидроксильных групп рост достаточно резкий, тогда как в случае нитрильных групп энергия возрастает более плавно. Это согласуется с различием в природе взаимодействующих функциональных групп. В случае нитрильных групп основной вклад в энергию дает кулоновское взаимодействие, относящееся к дальнодействующим, тогда как для гидроксильных и карбоксильных групп основной вид взаимодействий – водородные связи, которые по своей природе ближе к химическим и являются более короткодействующими. Поскольку сила является производной от энергии по расстоянию, то из S-образного характера зависимостей энергии от деформации (рис. 1) можно сделать вывод, что зависимости силы растяжения от расстояния между концевыми атомами углерода для всех трех типов функциональных групп будут проходить через максимум. Для карбоксильных и гидроксильных групп максимум будет более резким и более высоким, поскольку скорость увеличения энергии с деформацией для них существенно выше. Следовательно, и предельная нагрузка, которая способна разрушить взаимодействие, в случае карбоксильных и гидроксильных групп будет выше, чем в случае нитрильных групп. Ответ на вопрос: способна ли эта сила скомпенсировать случайные флуктуации импульса при тепловом движении при конкретной температуре, можно получить на основании молекулярно-динамического моделирования.
Рис. 1.
Зависимость энергии системы от изменения расстояния между концевыми атомами углерода относительно конформации с минимальной энергией для структур I–III.
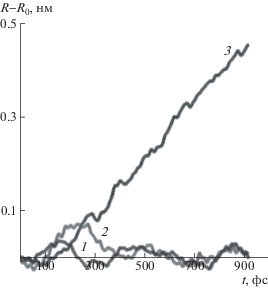
На рис. 2 приведено изменение расстояния между атомами O..H одной из пар взаимодействующих атомов в карбоксильных группах структуры I в зависимости от времени молекулярно-динамического моделирования при температуре 323 К и трех различных значениях фиксированного расстояния R между концевыми атомами углерода. Расстояние R = 1.33 нм отвечает минимуму энергии недеформированной структуры. В этом случае, соответствующем отсутствию механической нагрузки, расстояние между атомами O..H в ходе моделирования флуктуирует относительно среднего значения, которое сохраняется постоянным. Значит, при температуре 323 К и отсутствии механической нагрузки межмолекулярное взаимодействие двух карбоксильных групп не разрушается. Такая картина сохраняется и при R = 1.7 нм, хотя, как видно на рис. 1, в этих условиях на связь действует со стороны цепи уже достаточно существенная сила. При данном расстоянии между концевыми углеродными атомами расстояние между атомами O..H в начальные моменты времени несколько возрастает, но потом снова понижается и флуктуирует около значения, близкого к таковому в недеформированной системе. Отсюда можно сделать вывод, что взаимодействие двух карбоксильных групп способно передавать нагрузку на полимерную цепь, т.е. играть роль узла физической сетки. Лишь при расстоянии между фиксированными углеродными атомами R = 2.0 нм (что отвечает достижению предельного значения энергии; см. рис. 1) расстояние между атомами O..H в ходе молекулярно-динамического моделирования начинает монотонно возрастать, что соответствует разрушению физического узла.
Рис. 2.
Расстояние между атомами О..Н для структуры I в зависимости от времени молекулярно-динамического моделирования при фиксированном расстоянии между концевыми атомами углеродного звена R = 1.33 (1), 1.7 (2) и 2.0 нм (3).
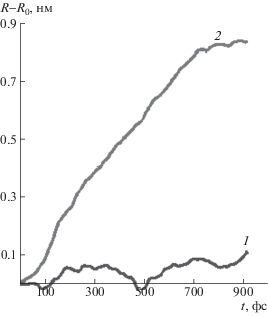
Аналогичный характер имеют зависимости расстояния между атомами O..H при молекулярно-динамическом моделировании структуры II, соответствующей физическому узлу, образованному гидроксильными группами. На рис. 3 показаны изменения расстояния между атомами О..Н для структуры II в зависимости от времени молекулярно-динамического моделирования при двух фиксированных расстояниях между концевыми углеродными атомами. Первое (R = 0.85 нм) соответствует минимуму энергии недеформированной системы, второе (R = 1.7 нм) – предельному значению энергии деформированной системы. Видно, что в недеформированной системе межмолекулярное взаимодействие гидроксильных групп образует физические узлы, разрушающиеся при деформации, превышающей предельную.
Рис. 3.
Зависимость расстояния между атомами О..Н для структуры II от времени молекулярно-динамического моделирования при фиксированном расстоянии между концевыми углеродными атомами R = 0.85 (1) и 1.7 нм (2).
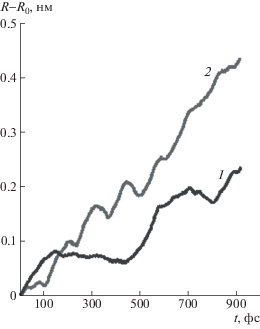
Совершенно иная картина наблюдается при моделировании структуры III, т.е. взаимодействия двух нитрильных групп. Как видно на рис. 4, даже при расстоянии между концевыми углеродными атомами бутадиеновых звеньев, отвечающем минимуму энергии недеформированной системы, расстояние между атомами N..N не флуктуирует в процессе моделирования относительно постоянной величины, а постепенно возрастает. При приложении механической нагрузки рост ускоряется. Следовательно, несмотря на то что значения энергии взаимодействия нитрильных и гидроксильных групп мало отличаются (рис. 1), межмолекулярное взаимодействие нитрильных групп не обеспечивает формирования физических узлов, способных при данных условиях выдерживать нагрузку. Так как рост энергии с увеличением расстояния в случае нитрильных групп происходит более плавно, при температуре моделирования данное взаимодействие является лабильным, что может приводить к проскальзыванию цепи по другим цепям от одной функциональной группы к другой. Это согласуется с тем, что, как продемонстрировано в работе [22], деформационные кривые сшитых бутадиен-нитрильных эластомеров, не содержащих карбоксильных и гидроксильных групп, хорошо описываются теорией “скользящих зацеплений” [37]. Следовательно, критерий, позволяющий отличить физический узел, способный подобно химическому узлу нести нагрузку, должен учитывать не только величину энергии межмолекулярного взаимодействия, но и локализацию взаимодействия в пространстве. Локализованными взаимодействиями являются, в частности, водородные связи. Обычные кулоновские взаимодействия, энергия которых с расстоянием изменяется достаточно медленно, к таким не относятся.
Рис. 4.
Зависимость расстояния между атомами N..N для структуры III от времени молекулярно-динамического моделирования при фиксированном расстоянии между концевыми углеродными атомами R = = 0.79 (1) и 1.1 нм (2).
При значительной доле акрилонитрила в бутадиен-нитрильном сополимере бутадиен-нитрильные группы могут образовывать блоки, что может приводить к микрофазовому разделению и росту разницы в энергии межмолекулярного взаимодействия основной цепи и полярного блока. В таком случае характер динамики взаимодействия может усложниться, и упругие свойства бутадиен-нитрильного сополимера будут более походить на свойства полимера с физической сеткой. Об этом свидетельствуют данные работы [38], в которой с помощью измерения подвижности стабильных нитроксильных радикалов–зондов показано, что с ростом содержания звеньев акрилонитрила в бутадиен-нитрильных полимерах существенно снижается локальная молекулярная подвижность, причем она характеризуется неоднородностью в распределении по энергии взаимодействий. Аналогичные эффекты могут возникать и в гидрированном бутадиен-нитрильном полимере (HNBR). При гидрировании часть двойных связей заменяется парой метиленовых звеньев. Из приведенных выше данных видно, что инкремент в энтальпию испарения у метиленовых звеньев наименьший. Соответственно, для HNBR различие в энергии межмолекулярного взаимодействия полярных и неполярных звеньев больше. Это может приводить к тому, что при той же концентрации нитрильных звеньев, что и в NBR, нитрильные группы в HNBR будут играть роль узлов физической сетки, и характер деформационных кривых для HNBR и NBR будет существенно отличаться. Такое различие наблюдали авторы работы [22], которые отметили, что при малой степени химического сшивания, когда основной вклад в упругие свойства вносит физическая сетка, деформационная кривая HNBR не описывается теорией “скользящих зацеплений”.
Чтобы проиллюстрировать возможность применения параметров межмолекулярных взаимодействий, вычисленных на основе квантово-химических расчетов, при описании физико-механических свойств бутадиен-нитрильных эластомеров с карбоксильными группами в настоящей работе использована теоретическая модель физической сетки, предложенная в работах [5, 39]. В этих работах получено равновесное распределение по числу узлов в эластомере с физической сеткой, учитывающее зависимость концентрации узлов от температуры и деформации. Следующая из данного распределения зависимость средней концентрации узлов физической сетки n от кратности растяжения и температуры представлена как
(2)
$n(\lambda ,T) = {{n}_{{{\text{ph}}}}}\frac{{x{{e}^{x}} - {{e}^{x}} + 1}}{{{{{({{e}^{x}} - 1)}}^{{}}}}}$Здесь x = $\frac{{2\Psi (\lambda ,T) - {{E}_{{{\text{ph}}}}} + TS}}{{RT}}$, nph – максимально возможная концентрация узлов физической сетки, определяемая химической структурой полимера; Eph и S – энергия и энтропия физического узла соответственно; Ψ(λ, T) – энергия деформации межузловой цепи как функция кратности растяжения λ и абсолютной температуры Т; R – универсальная газовая постоянная.
Поскольку механические испытания реальных эластомеров проводятся в неравновесных условиях, данное распределение было дополнено уравнением вязкоупругости [40], в соответствии с которым вязкоупругое напряжение в сетчатом эластомере при растяжении с постоянной скоростью $v$ имеет вид:
(3)
$\sigma = {{\sigma }_{e}}({{\lambda }_{0}} + {v}t) + \frac{{v}}{{Jg}}(1 - {{e}^{{ - \frac{{Jt}}{2}}}}),$С учетом формулы (2) для квазиравновесного напряжения можно записать выражение
(4)
${{\sigma }_{e}}(\lambda ) = 2(n(\lambda ,T) + {{n}_{{{\text{ch}}}}})\frac{{\partial \Psi (\lambda ,t)}}{{\partial \lambda }}.$Здесь nch – концентрация узлов химической сетки, которая в данном случае считается постоянной.
В работе [39] функция Ψ(λ, T) задавалась классическим соотношением теории высокоэластичности гауссовых цепей. Однако при описании процесса разрушения, необходимо учитывать конечную растяжимость цепей. Поэтому в настоящей работе для этой функции использовано выражение, полученное путем разложения в ряд до членов порядка λ4 энергии деформации цепи из N свободно-сочлененных нерастяжимых сегментов:
(5)
$\begin{gathered} \Psi (\lambda ,T) = RT\left( {\left( {1 - \frac{1}{N}} \right)\left( {{{\lambda }^{2}} + \frac{2}{\lambda } - 3} \right){\text{/}}2} \right. + \\ \left. { + \frac{3}{{5N}}\left( {1 - \frac{{47}}{{40N}}} \right)\left( {{{\lambda }^{4}} + \frac{8}{{3{{\lambda }^{2}}}} + \frac{4}{3}\lambda - 5} \right)} \right) \\ \end{gathered} $В основу построения кинетической модели разрушения эластомеров может быть положена идея термофлуктуационной теории прочности, развитая в работах С.Н. Журкова, А.И. Слуцкера и других авторов [41, 42]. Согласно уравнению Журкова, долговечность полимера τ, находящегося под постоянным напряжением σ, описывается формулой
где предэкспоненциальный множитель τ0 имеет порядок 10–13–10–12 с, Е – энергия активации разрушения, по величине близкая к энергии разрушения химических связей, γ – структурно чувствительный коэффициент.Непосредственно уравнение Журкова не может быть использовано в модели разрушения эластомеров при постоянной скорости растяжения, когда напряжение не является константой. Кроме того, даже при постоянном напряжении уравнение Журкова не применимо для описания долговечности эластомеров [43], разрушение которых контролируется релаксационными процессами. Вместе с тем, идею уравнения (6) можно применить для построения уравнения кинетики деструкции эластомерных цепей. В числителе показателя экспоненты уравнения (6) член γσ характеризует снижение энергии разрушения химических связей под действием приложенного напряжения. С учетом того, что в случае эластомера энергия деформации цепей описывается функцией Ψ(λ, T), дифференциальное уравнение кинетики деструкции цепей, несущих нагрузку, можно записать в виде
(7)
$\frac{{d\nu }}{{dt}} = - {{k}_{0}}{{e}^{{ - \,\frac{{{{E}_{{{\text{ch}}}}} - \alpha \Psi (\lambda ',T)}}{{RT}}}}}\nu .$Здесь ν – концентрация цепей, несущих нагрузку; предэкспоненциальный множитель k0 имеет смысл $\frac{1}{{{{\tau }_{0}}}}$ в уравнении (6); Ech – энергия разрыва химических связей цепей сетки; безразмерный коэффициент α характеризует степень концентрации энергии на деформируемых цепях, кратность растяжения которых λ' не совпадает с кратностью растяжения всего образца λ в силу неафинной деформации в устье растущей трещины.
Поскольку перераспределение деформации в устье трещины контролируется вязкоупругой податливостью эластомерной сетки, связь между кратностью деформации в устье трещины λ' и кратностью растяжения образца λ может быть задана с помощью простой модели вязкоупругости:
(8)
$\frac{{d\lambda '}}{{dt}} = \frac{1}{\eta }({{\sigma }_{e}}({{\lambda }_{0}} + {v}t) - {{\sigma }_{e}}(\lambda '))\lambda '$Уравнения (7), (8) с учетом определений (2), (4), (5) дают замкнутую систему, которая может быть решена численно для заданных начальных условий ${v}$(0) = ${{{v}}_{0}}$, λ'(0) = λ0. Используя полученное решение в уравнении вязкоупругости (3), можно получить деформационную кривую растяжения эластомера вплоть до разрушения. В настоящей работе при численном моделировании система уравнений (7), (8) решалась методом Рунге–Кутта с переменным шагом. Значения параметров модели, для которых проводились квантово-химические расчеты, были приняты на основании таких расчетов. В частности, это было сделано для энергии физических узлов Eph карбоксильных и нитрильных групп, приведенных выше, а также для энергии разрыва химических связей цепей сетки Ech, которая для химической связи у α-углеродного атома бутадиенового звена составила 350 кДж/моль. Величина предэкспоненты k0 в уравнении (7), равная 1.25 × 1014 с–1, была получена в результате обработки данных по молекулярно-динамическому моделированию. Для остальных параметров модели были использованы физически обоснованные значения, соответствующие выпускаемым промышленностью карбоксилированным бутадиен-нитрильным каучукам, вулканизованным серной вулканизующей группой. В том числе, концентрацию узлов химической сетки nch считали равной 70 моль/м3, а максимальную концентрацию узлов физической сетки – nph = 0.2 nch. Параметр вязкоупругости J в уравнении (3) был взят равным 0.4 с–1, что соответствует времени вязкоупругой релаксации 5 с при Т = 293 К. Зависимость времени релаксации от температуры была описана уравнением Аррениуса с энергией активации, равной величине Eph для карбоксильных групп. Температурную зависимость коэффициента вязкоупругости g вычисляли по формуле
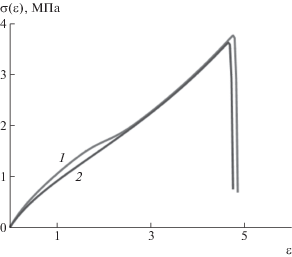
На рис. 5 приведены рассчитанные деформационные кривые для сшитого карбоксилированного и некарбоксилированного бутадиен-нитрильного каучука при Т = 293 К и скорости деформации 0.333 с–1, что отвечает растяжению стандартного образца с длиной рабочего участка 25 мм на разрывной машине со скоростью перемещения нижнего зажима 500 мм/мин. Как можно видеть, карбоксилированный каучук имеет более высокие значения напряжения при средних удлинениях, а также более высокую прочность. Это согласуется с тем, что введение карбоксильных групп в бутадиен-нитрильные эластомеры приводит в композициях на их основе к улучшению таких свойств, как сопротивление истиранию и раздиру [44]. Абсолютные значения напряжения при заданных удлинениях, прочности при растяжении и удлинении при разрыве для рассчитанных деформационных кривых находятся близко к таковым, наблюдаемым в экспериментах для ненаполненных резин на основе карбоксилированных бутадиен-нитрильных эластомеров, вулканизованных серой [23].
Рис. 5.
Рассчитанная зависимость напряжения σ от деформации ε = λ – 1 для бутадиен-нитрильного каучука, содержащего (1) и не содержащего (2) карбоксильных групп; Т = 293 К, скорость деформации 0.333 с–1.
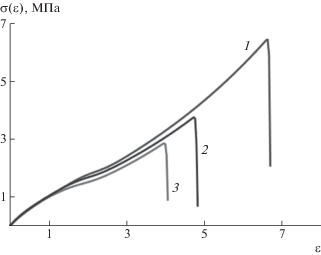
На рис. 6 и 7 представлены рассчитанные деформационные кривые карбоксилированного бутадиен-нитрильного каучука при различных значениях скорости деформации и температуры. Видно, что прочность эластомера и относительное удлинение при разрыве возрастают с увеличением скорости деформации и уменьшением температуры. Такое поведение согласуется с экспериментально наблюдаемыми зависимостями для карбоксилированных каучуков, вулканизованных серой [45], свойства которых подчиняются принципу температурно-временной суперпозиции.
Рис. 6.
Рассчитанные деформационные кривые для карбоксилированного бутадиен-нитрильного каучука при Т = 293 К и скорости деформации 0.333 (1), 0.067 (2) и 0.003 с–1 (3).
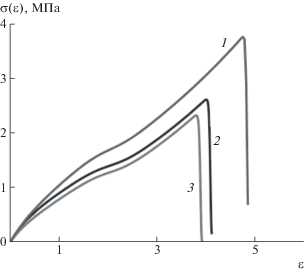
Рис. 7.
Рассчитанные деформационные кривые для карбоксилированного бутадиен-нитрильного каучука при Т = 273 (1), 293 (2) и 313 К (3). Скорость деформации 0.333 с–1.
Таким образом, результаты расчетов по простой теоретической модели качественно согласуются с экспериментальными данными, что подтверждает возможность использования результатов квантово-химических расчетов в качестве некоторых параметров модели. Следует заметить, что область применения данных параметров ограничивается видом теоретической модели. В частности, принятая модель не подходит для карбоксилированных эластомеров, вулканизуемых солями металлов. В этом случае образуются ионные поперечные связи, имеющие гетерогенный характер, и проведенные квантово-химические расчеты не применимы. Кроме того, модель вязкоупругости с одним временем релаксации справедлива только в относительно небольшом диапазоне скорости деформации и температуры. Для более широкого диапазона необходимо учитывать распределение времен релаксации.
ЗАКЛЮЧЕНИЕ
В настоящей работе с помощью квантово-химического моделирования исследованы механизм образования и локальная динамика межмолекулярных взаимодействий в бутадиен-нитрильных эластомерах с функциональными карбоксильными и гидроксильными группами. Установлено, что межмолекулярные взаимодействия карбоксильных и гидроксильных групп имеют более высокую энергию, чем взаимодействия нитрильных групп, и могут выдерживать, не разрушаясь, более высокую механическую нагрузку. Методом молекулярно-динамического моделирования показано, что поведение межмолекулярных взаимодействий карбоксильных и гидроксильных групп при воздействии механической нагрузки более соответствует понятию образования узла физической сетки, чем поведение взаимодействия нитрильных групп, которое в большей мере отвечает концепции “скользящих зацеплений”.
Проведенные квантово-химические расчеты демонстрируют, что в зависимости от химической природы функциональных групп и внешних условий (температура и деформация) межмолекулярные взаимодействия могут либо формировать узлы физической сетки эластомера, способные воспринимать внешнюю нагрузку, либо, обратимо разрушаясь, изменять вязкоупругие свойства эластомера по классическому механизму. При этом соответствующий полимер будет или не будет способен образовывать физический гель.
Для иллюстрации возможности применения квантово-химических оценок параметров межмолекулярных взаимодействий при исследовании свойств реальных полимеров предложена простая теоретическая модель, позволяющая описать упруго-прочностные свойства эластомеров с физической сеткой. Численный анализ модели, с использованием квантово-химических оценок параметров, показал ее качественное согласие с известными экспериментальными данными по упруго-прочностным свойствам карбоксилированных бутадиен-нитрильных каучуков, вулканизованных по звеньям бутадиена с помощью обычных вулканизующих групп.
Работа выполнена в рамках Госзадания ИПХФ РАН (регистрационный номер темы АААА-А19119032690060-9).
Список литературы
Gels and Other Soft Amorphous Solids / Eds by F. Horkay, J.F. Douglas, E. Del Gado. Washington: American Chemical Society, 2018.
Polymer Gels, Gels Horizons: from Science to Smart Materials / Eds by V.K. Thakur, M.K. Thakur. Singapore: Springer Nature, 2018.
Rogovina L.Z., Vasil’ev V.G., Braudo E.E. // Polymer Science C. 2008. V. 50. № 1. P. 85.
Hassan P.A., Verma G., Ganguly R. // Functional Materials / Eds by S. Banerjee, A.K. Tyagi. London: Elsevier, 2012.
Solovyov M.Y., Raukchvarger A.B., Irzhak V.I. // Progr. Coll. Polym. Sci. 1992. V. 90. P. 174.
Иржак В.И., Королев Г.В., Соловьев М.Е. // Успехи химии. 1997. Т. 66. № 2. С. 179.
Irzhak V.I., Solov’ev M.E., Irzhak T.F. // Rev. J. Chem. 2018. V. 8. № 1. P. 76.
Mandal U.K. // Polym. Int. 2000. V. 49. № 12. P. 1653.
Prochon M., Przepiorkowska A., Zaborski M. // J. Appl. Polym. Sci. 2007. V. 106. № 6. P. 3674.
Ibarra L., Rodrıguez A., Mora-Barrantes I. // Polym. Int. 2009. V. 58. P. 218.
Wang J., Jia H., Tang Y., Ji D., Sun Y., Gong X., Ding L. // J. Mater. Sci. 2013. V. 48. P. 1571.
Laskowska A., Zaborski M., Boiteux G., Gain O., Marzec A., Maniukiewicz W. // eXPRESS Polym. Lett. 2014. V. 8. № 6. P. 374.
Gaca1 M., Zaborski M. // Polimery. 2016. V. 61. № 1. P. 31.
Gusev Yu.K., Glukhovskoi V.S., Yudin V.P., Shvetsova E.A. // Int. Polym. Sci. Technol. 2010. V. 37. № 2. P. T.13.
Brzić S.J., Jelisavac L.N., Galović J.R., Simić D.M., Petković J.L. // Hem. Ind. 2014. V. 68. № 4. P. 435.
Brzić S., Ušćumlić G., Milojković A., Rodić V., Bogosavljević M. // Scientific Techn. Rev. 2015. V. 65. № 4. P. 28.
Brzić S.J., Ušćumlić G.S., Dimić M.V., Tomić M., Rodić V.Ž., Fidanovski B.Z. // Hem. Ind. 2016. V. 70. № 5. P. 547.
Thomas S., Sinturel C., Thomas R. Micro and Nanostructured Epoxy / Rubber Blends. Weinheim: Wiley-VCH, 2014. P. 320.
Jerrams S.J., Harvey S.J. // Transactions on Modelling and Simulation. 1999. V. 21. P. 533.
Wang Q., Yang F., Liu Y., Yang Q., Chen J., Guan H. // J. Thermoplast. Comp. Mater. 2012. V. 25. P. 305.
Hassan M.A., Abouel-Kasem A., El-Sharief M.A. // J. Eng. Sci., Assiut Univ. 2010. V. 38. № 1. P. 119.
Syed I.H., Stratmann P., Hempel G., Klüppel M., Saalwächter K. // Macromolecules. 2016. V. 49. P. 9004.
Ibarra L. // J. Appl. Polym. Sci. 1999. V. 73. P. 927.
Fritzsche J., Das A., Jurk R., Stöckelhuber K.W., Heinrich G., Klüppel M. // eXPRESS Polym. Lett. 2008. V. 2. № 5. P. 373.
Pastor A., Mart’ınez-Viviente E. // Coordinat. Chem. Rev. 2008. V. 252. P. 2314.
Cohen Y., Avram L., Frish L. // Angew. Chem. Int. Ed. 2005. V. 44. P. 520.
Saalwächter K. // Polymer Science A. 2012. V. 2. P. 185.
Hohenberg P., Kohn W. // Phys. Rev. 1964. V. 136. № 3B. P. B864.
Kohn W., Sham L. // Phys. Rev. 1965. V. 140. № 4A. P. A1133.
Becke A.D. // J. Chem. Phys. 1993. V. 98. P. 5648.
Miehlich B.A., Savin H.S., Preuss H. // Chem. Phys. Lett. 1989. V. 157. № 3. P. 200.
Mueller M. Fundamentals of Quantum Chemistry. Molecular Spectroscopy and Modern Electronic Structure Computation. New York: Kluwer Acad. Publ., 2002.
Valiev M., Bylaska E.J., Govind N., Kowalski K., Straatsma T.P., Van Dam H.J.J., Wang D., Nieplocha J., Apra E., Windus T.L., de Jong W.A. // Comput. Phys. Commun. 2010. V. 181. P. 1477.
Levine I.N. Phisical Chemistry. New York: McGraw-Hill, 2009.
Bussi G., Donadio D., Parrinello M. // J. Chem. Phys. 2007. V. 126. P. 014101.
Mackay D., Shiu W.Y., Ma K.-C., Lee S.C. Handbook of Physical-Chemical Properties and Environmental Fate for Organic Chemicals. Boca Raton: CRC Press, 2006.
Rubinstein M., Panyukov S. // Macromolecules. 2002. V. 35. P. 6670.
Livanova N., Karpova S. // Chem. Chem. Technol. 2011. V. 5. № 4. P. 423.
Raukhvarger A.B., Solovyov M.Y., Irzhak V.I. // Chem. Phys. Lett. 1989. V. 155. № 4–5. P. 455.
Соловьев М.Е., Раухваргер А.Б., Капустин А.А. // Каучук и резина. 2002. № 4. С. 3.
Журков С.Н., Нарзуллаев Б.Н. // Журн. техн. физики. 1953. Т. 23. № 10. С. 1677.
Регель В.Р., Слуцкер А.И., Томашевский Э.Е. Кинетическая природа прочности твердых тел. М.: Наука, 1974.
Бартенев Г.М. Прочность и механизм разрушения полимеров. М.: Химия, 1984.
Klingender R.C. Handbook of Specialty Elastomers. Boca Raton: CRC Press, 2008.
Halpin J.C., Bueche F. // J. Polym. Sci. A. 1965. V. 3. P. 3935.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия А)