

Высокомолекулярные соединения (серия А), 2021, T. 63, № 4, стр. 243-248
РЕОЛОГИЧЕСКИЕ СВОЙСТВА ЖИДКОКРИСТАЛЛИЧЕСКИХ РАСТВОРОВ ПРОИЗВОДНЫХ ЦЕЛЛЮЛОЗЫ
С. А. Вшивков a, *, Е. В. Русинова a, А. С. Абу Салех a
a Уральский федеральный университет
620000 Екатеринбург, пр. Ленина, 51, Россия
* E-mail: sergey.vshivkov@urfu.ru
Поступила в редакцию 17.11.2020
После доработки 10.02.2021
Принята к публикации 05.03.2021
Аннотация
Изучены реологические свойства и жидкокристаллические переходы растворов простых эфиров целлюлозы. Концентрационные зависимости энтальпии активации вязкого течения описываются кривыми с экстремумами (максимумами и минимумами), вызванными жидкокристаллическими переходами. Эти зависимости сопоставлены с фазовыми диаграммами изученных систем.
ВВЕДЕНИЕ
Жидкие кристаллы играют огромную роль в науке и технике [1–8]. Высокая способность этих соединений к самоорганизации представляет значительный интерес для разработки новых материалов. Для макромолекул целлюлозы и ее производных характерна жесткая спиральная конформация; они способны упорядочиваться и образовывать жидкие кристаллы холестерического типа в концентрированных растворах [1, 4].
Растворы жесткоцепных полимеров имеют особую концентрационную зависимость вязкости, которая описывается кривой с резким максимумом. Впервые это было показано J. Hermans [9] для растворов ПБГ в ДМФА, J.T. Yang [10] для растворов ПБГ в м-крезоле, E. Iizuka [11] для полиалкилглутаматов. Позднее такая зависимость была обнаружена для растворов других полимеров [12–15]. Концентрация, отвечающая максимуму кривой, соответствует началу образования анизотропной фазы, что приводит к снижению вязкости за счет присутствия в системе легко ориентирующихся при течении макромолекул. C. Robinson [16] предположил, что понижение вязкости вызвано послойным течением анизотропных растворов. Минимум вязкости обусловлен завершением образования жидкокристаллической фазы по всему объему раствора. Дальнейшее повышение концентрации полимера приводит к увеличению вязкости, вызванному усилением межмолекулярного взаимодействия. Аналогичные закономерности были получены для растворов гидроксипропилцеллюлозы (ГПЦ)–полиэтиленгликоль, ГПЦ–вода, ГПЦ–диметилсульфоксид [17] и нанокомпозитов на основе гидроксипропилцеллюлозы [18]. Таким образом, увеличение вязкости с ростом концентрации полимера является типичным для однофазных (изотропных или анизотропных) растворов, в то время как вязкость уменьшается с повышением концентрации полимера в двухфазной области, где сосуществуют изотропная и жидкокристаллическая фазы.
Известно [19, 20], что вязкость связана с энтальпией активации вязкого течения ∆H уравнением
где A – постоянная величина, R – газовая постоянная, T – абсолютная температура. Из уравнения (1) следует, что появление максимума на концентрационной зависимости вязкости может быть связано с резким изменением энтальпии активации вязкого течения. Данные о концентрационной зависимости ∆H для растворов жесткоцепных полимеров в изотропной и анизотропной областях практически отсутствуют [15].Цель настоящей работы – определение концентрационной зависимости энтальпии активации вязкого течения для систем эфир целлюлозы–растворитель и сопоставление полученных данных с фазовыми диаграммами этих систем.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Эксперименты проводили со следующими образцами: ГПЦ (“Acros Organics”, Бельгия) с Mw = = 1 × 105 и молярным замещением 3.2, этилцеллюлоза (“Acros Organics”, Бельгия) с Мη = 4.7 × 104 и степенью замещения 2.6, цианоэтилцеллюлоза с Mw = 9 × 104 и степенью замещения 2.6, метилгидроксиэтилцеллюлоза (6000R) (“Hercules Culminal”, Aqulon) с Mη = 1 × 106 и степенью замещения 1.8 (–OCH3) и 0.3 (–OCH2CH2OH). Цианоэтилцеллюлозу готовили по следующей методике: хлопчатобумажный линтер обрабатывали 18%-ным раствором NaOH при 20°С в течение 2 ч, отжимали, добиваясь 3–4-кратного увеличения массы образца, и разрыхляли. Полученный продукт обрабатывали при перемешивании акрилонитрилом (20 молей на звено глюкозы) при 35°С в течение 3 ч. Реакционную массу охлаждали до 5°С, разбавляли ацетоном, нейтрализовали 15%-ным спиртовым раствором уксусной кислоты, повторно промывали водой и экстрагировали спиртом в аппарате Сокслета в течение 10 ч. Полученный сложный эфир сушили в вакууме при 50°С. Молярное замещение ГПЦ и степень замещения этилцеллюлозы и метилгидроксиэтилцеллюлозы рассчитывали по данным элементного анализа. Значение степени замещения цианоэтилцеллюлозы оценивали по азоту, определенному методом Кьельдаля. В качестве растворителей использовали диметилсульфоксид, диметилацетамид, этанол и дистиллированную воду. Чистоту растворителей оценивали по их показателям преломления [21]. Растворы готовили в течение нескольких недель при 330–350 К.
Фазовое состояние раствора изучали с помощью поляризационного микроскопа OLYMPUS BX-51 и поляризационной фотоэлектрической установки [22]. Ампулу с раствором помещали в зазор между скрещенными поляризатором и анализатором и охлаждали с помощью термостатирующей рубашки. Поляризованный свет He-Ne-лазера пропускали через поляризатор и анализатор в направлении, перпендикулярном ампуле. Когда раствор был прозрачным (изотропным), интенсивность проходящего света была равна нулю. Когда система становилась мутной при изменении температуры или повышении концентрации раствора, интенсивность проходящего поляризованного света увеличивалась. Это свидетельствовало о формировании анизотропной фазы. Температуру, при которой появлялась опалесценция, принимали за температуру фазового перехода. Скорость охлаждения растворов составляла 0.2 град/мин. Вязкость растворов исследовали с использованием реометра “Rheotest RN 4.1”, снабженного коаксиальным цилиндрическим рабочим блоком.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
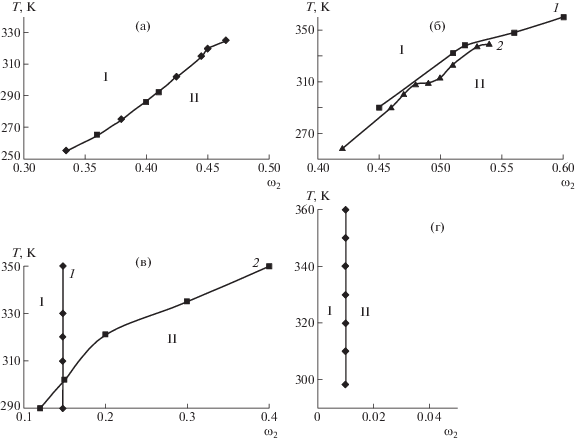
Фазовые диаграммы систем эфир целлюлозы–растворитель (рис. 1) были построены ранее и рассмотрены в работах [4, 13–15]. Пограничные кривые отделяют изотропные области от анизотропных. Влияние молекулярной массы полимера и химического строения компонентов на фазовые переходы подробно проанализировано в работе [4].
Рис. 1.
Пограничные кривые систем цианоэтилцеллюлоза–ДМАА (а), ГПЦ–ДМСО (1) и ГПЦ–этанол (2) (б), этилцеллюлоза–этанол (1) и этилцеллюлоза–ДМАА (2) (в), метилгидроксиэтилцеллюлоза–вода (г). I – область изотропных растворов, II – область анизотропных растворов. ω2 – массовая доля полимера в растворе.
На рис. 2 приведены микрофотографии растворов ГПЦ в этаноле и ДМСО. Радужная окраска свидетельствует об анизотропном фазовом состоянии растворов. Аналогичные данные получены для других изученных систем.
Рис. 2.
Микрофотографии растворов ГПЦ в скрещенных поляроидах: а – в этаноле ω2 = 0.55, б – в ДМСО ω2 = 0.50. Черные пятна отражают пузырьки воздуха. Т = 298 К.

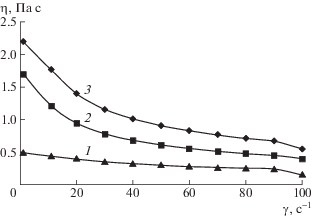
Типичные зависимости вязкости водных растворов метилгидроксиэтилцеллюлозы от скорости сдвига при разной температуре представлены на рис. 3. Кривые имеют неньютоновский характер, вызванный разрушением исходной структуры раствора полимера и ориентацией молекул в потоке, что приводит к снижению вязкости. При повышении температуры вязкость уменьшается. Такие зависимости были обнаружены для растворов всех изученных полимеров. Аналогичные результаты известны для систем Na-карбоксиметилцеллюлоза–вода [24], ГПЦ–этанол, ГПЦ–диметилсульфоксид [25], ГПЦ–этиленгликоль, этилцеллюлоза–ДМФА [26, 27], ГПЦ–м-крезол [28], цианоэтилцеллюлоза–ДМФА, цианоэтилцеллюлоза–ДМАА, ГПЦ– ДМФА [15], диацетатцеллюлоза–трифторуксусная кислота [29], цианоэтилцеллюлоза–ДМФА [30], ГПЦ–уксусная кислота [31].
Рис. 3.
Зависимость вязкости водного раствора метилгидроксиэтилцеллюлозы от скорости сдвига. T = 338 (1), 298 (2) и 288 K (3). ω2 = 0.01.
Приведенные данные использовали для расчета энтальпии активации вязкого потока. Для этой цели выбраны значения вязкости при низкой скорости сдвига (2 с–1), поскольку в работах [9, 10, 12] было показано, что концентрационная зависимость вязкости, определяемая при низкой скорости сдвига, является типичной для анизотропных растворов.
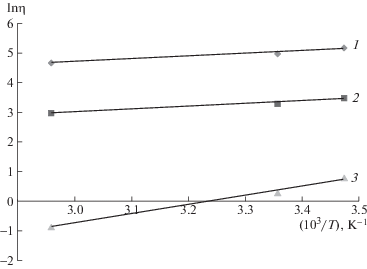
Зависимость вязкости от обратной температуры 1/T для водных растворов метилгидроксиэтилцеллюлозы при разной концентрации полимера представлена на рис. 4.
Рис. 4.
Зависимость lnη от 1/Т для системы метилгидроксиэтилцеллюлоза–вода. ω2 = 0.05 (1), 0.025 (2) и 0.01 (3).
Экспериментальные [32, 33] и рассчитанные по уравнению ∆H ≈ ∆H*/4 [19] значения энтальпии активации вязкого течения растворителей приведены в табл. 1 (∆H* – энтальпия испарения растворителя [34]).
Концентрационные зависимости энтальпии активации вязкого течения изученных систем показаны на рис. 5.
Рис. 5.
Концентрационные зависимости ∆H для систем цианоэтилцеллюлоза–ДМАА (а), цианоэтилцеллюлоза–ДМСО (б), ГПЦ–этанол (в), этилцеллюлоза–ДМАА (г), (д) этилцеллюлоза–этанол и метилгидроксиэтилцеллюлоза–вода (е).
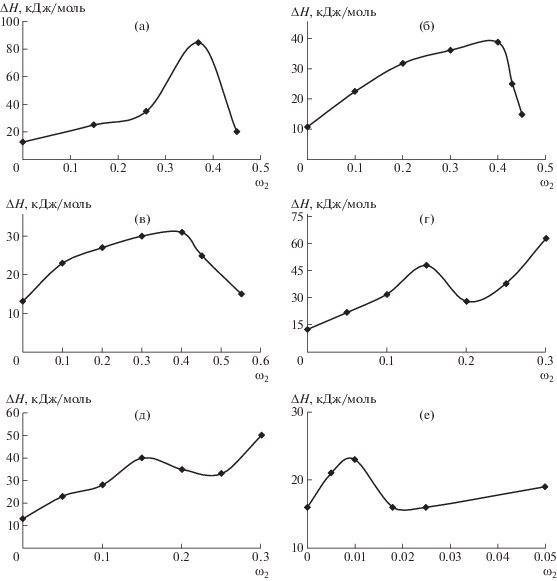
Зависимости концентрации энтальпии активации вязкого потока описаны кривыми с экстремумами (максимумами и минимумами), вызванными жидкокристаллическими переходами. Рассчитанные значения ∆Н изотропных полимерных растворов коррелируют с аналогичными данными для других полимерных систем [34]. Начальное увеличение энтальпии активации вязкого потока с повышением концентрации полимера свидетельствует о повышении взаимодействия между макромолекулами при приближении к области концентраций жидкокристаллических переходов. Область уменьшения ∆Н соответствует появлению анизотропной фазы (рис. 1). Это следует из сравнения с фазовыми диаграммами.
Необходимо отметить, что температура фазового перехода Tф, определенная вискозиметрическим методом, соответствует фазовой инверсии, а не появлению первой капли новой фазы. По этой причине значения Tф, полученные вискозиметрическим методом и методом точек помутнения, могут отличаться на несколько градусов. Разность в значениях Tф также может быть вызвана влиянием гидродинамического поля на фазовые переходы [13]. Результаты сопоставления концентрационной зависимости ∆H с фазовыми диаграммами при 298 К представлены в табл. 2. Обнаружено удовлетворительное совпадение значений $\omega _{2}^{*}$ и $\omega _{2}^{{**}}$, отвечающих максимуму на зависимости ∆H = f(ω2), и концентрация возникновения анизотропной фазы в растворе соответственно.
Таблица 2.
Результаты сопоставления концентрационной зависимости ∆H с фазовыми диаграммами
| Система | $\omega _{2}^{*}$ | $\omega _{2}^{{**}}$ |
|---|---|---|
| Цианоэтилцеллюлоза–ДМАА | 0.37 | 0.42 |
| ГПЦ–ДМСО | 0.40 | 0.46 |
| ГПЦ–этанол | 0.40 | 0.47 |
| Этилцеллюлоза–ДМАА | 0.15 | 0.15 |
| Метилгидроксиэтилцеллюлоза–вода | 0.10 | 0.10 |
| Этилцеллюлоза–этанол | 0.15 | 0.15 |
Таким образом, уменьшение ∆Н вызвано изменением механизма течения: в изотропной фазе происходит течение неупорядоченных макромолекул, требующее большего напряжения сдвига, в анизотропной фазе макромолекулы образуют домены, которые легко ориентируются в направлении потока. Следовательно, уменьшение значения ∆Н вызвано наличием ориентированных доменов в системе и послойным течением анизотропных растворов. Эта часть кривых соответствует сосуществованию двух фаз – изотропной и анизотропной. Появление жидкокристаллических доменов в растворах приводит к снижению ∆Н в 1.5–2.0 раза. Минимумы на кривых соответствуют формированию однофазных анизотропных систем.
Аналогичные результаты были получены ранее [35] для системы ГПЦ–ДМСО. Обнаружено резкое падение энергии активации от 50 до 30 кДж/моль при переходе от изотропных растворов к жидкокристаллическим. Авторы связывают это явление с легкой ориентацией жидкокристаллических доменов в направлении потока.
Полученные результаты согласуются с данными о концентрационной зависимости размеров рассеивающих свет частиц в растворах эфиров целлюлозы [27, 36 ] . Эти зависимости имеют максимум. Концентрации растворов с максимальным размером частиц совпадают с концентрациями перехода от изотропного раствора к анизотропному. В изотропных растворах макромолекулы и их ассоциаты не ориентированы относительно друг друга. При повышении концентрации полимера они образуют более крупные частицы в результате усиления межцепного взаимодействия. Такие частицы не имеют плотной упаковки, т.е. они содержат большое количество растворителя. Во время перехода в ЖК-состояние с дальнейшим увеличением концентрации полимера дополнительная ориентация макромолекул и доменов друг относительно друга приводит к усилению межцепного взаимодействия, что приводит к выжиманию растворителя из частиц, что проявляется в уменьшении их размера. В связи с этим снижение вязкости в данной области концентрации вызвано как более легкой ориентацией ЖК доменов, так и уменьшением их размеров.
Третья часть приведенных кривых (рис. 5г–5е) характеризует однофазные анизотропные растворы. При повышении концентрации полимера энтальпия активации вязкого потока увеличивается. Это вызвано дальнейшим усилением межцепного взаимодействия. Таким образом, концентрационная зависимость ∆Н позволяет определить концентрационные границы двухфазной области, в которой сосуществуют изотропная и анизотропная фазы.
ЗАКЛЮЧЕНИЕ
Исследованы реологические свойства и жидкокристаллические фазовые переходы систем: метилгидроксиэтилцеллюлоза–вода, гидроксипропилцеллюлоза–этанол, ГПЦ–диметилсульфоксид, цианэтилцеллюлоза–диметилацетамид, этилцеллюлоза–диметилацетамид, этилцеллюлоза–этанол. Концентрационные зависимости энтальпии активации вязкого течения ∆Н описываются кривыми с максимумами и минимумами, вызванными жидкокристаллическими фазовыми переходами. Полученные результаты согласуются с фазовыми диаграммами исследованных систем.
Список литературы
Куличихин В.Г., Голова Л.К. // Химия древесины. 1985. № 3. С. 9.
Orientation Phenomena in Polymer Solutions and Melts / Ed. by A.Ya. Malkin, S.P. Papkov. Moscow: Khimiya, 1980.
Papkov S.P., Kulichikhin V.G. Liquid Crystalline State of Polymers. Moscow: Khimiya, 1977.
Vshivkov S.A., Rusinova E.V. // Polymer Science A. 2018. V. 60. № 1. P. 65.
Liquid Crystal Polymers / Ed. by N.A. Platé. New York: Plenum, 1993.
De Gennes P.G. The Physics of Liquid Crystals. London: Cambridge Univ. Press, 1974.
Platé N.A., Shibaev V.P. Comb-Shaped Polymers and Liquid Crystals. New York: Plenum, 1988.
Filippov A.P. // Polymer Science B. 2004. V. 46. № 3–4. P. 66.
Hermans J. // Colloid Sci. 1962. V. 17. № 7. P. 638.
Yang J.T. // J. Am. Chem. Soc. 1959. V. 81. № 15. P. 3902.
Iizuka E. //Mol. Cryst. Liq. Cryst. 1974. V. 25. № 3–4. P. 287.
Куличихин В.Г., Малкин А.Я., Папков С.П. // Высокомолек. соед. А. 1984. Т. 26. № 3. С. 499.
Vshivkov S.A. Phase Transitions and Structure of Polymer Systems in External Fields. Newcastle: Cambridge Scholars Publ., 2019.
Vshivkov S.A., Galyas A.G. // Polymer Science A. 2011. V. 53. № 11. P.1032.
Vshivkov S.A., Rusinova E.V. // Russ. J. Appl. Chem. 2011. V. 84. № 10. P. 1830.
Robinson C., Ward J.C., Beevers R.B. // Disc. Faraday Soc. 1958. V. 25. № 1. P. 29.
Kulichikhin V.G., Makarova V.V., Tolstykh M.Yu., Vasil’ev G.B. // Polymer Science A. 2010. V. 52. № 11. P. 1196.
Kulichikhin V.G., Makarova V.V., Tolstykh M.Yu., Picken S.J., Mendes E. // Polymer Science A. 2011. V. 53. № 9. P. 748.
Тагер А.А. Физикохимия полимеров. М.: Химия, 1978.
Малкин А.Я. Основы реологии. СПб.: Профессия, 2018.
Иоффе И.В. Рефрактометрические методы в химии. Л.: Химия, 1974.
Vshivkov S.A., Rusinova E.V., Kudrevatykh N.V., Galyas A.G., Alekseeva M.S., Kuznetsov D.K. // Polymer Science A. 2006. V. 48. № 10. P. 1115.
Vshivkov S.A., Rusinova E.V., Galyas A.G. // Eur. Polym. J. 2014. V. 59. № 2. P. 326.
Vshivkov S.A., Byzov A.A. // Polymer Science A. 2013. V.55. № 2. P.102.
Vshivkov S.A., Soliman T.S. // Polymer Science A. 2016. V. 58. № 3. P. 307.
Vshivkov S.A., Soliman T.S. // Polymer Science A. 2016. V. 58. № 4. P. 499.
Vshivkov S.A., Rusinova E.V. // Polymer Rheology. London: TechOpen, 2018. Ch. 1.
Asada T., Hayaahida S., Onogi S. // Repts. Progr. Polym. Phys. Jpn. 1980. V. 23. P. 145.
Deyan S., Gilli M., Sixou P. // J. Appl. Polym. Sci. 1983. V. 28. № 4. P. 1527.
Бельникевич Н.Г., Болотникова Л.С., Куценко Л.И., Панов Ю.Н., Френкель С. // Высокомолек. cоед. Б. 1985. Т. 27. № 5. С. 332.
Navard P.J., Haudin M. // J. Polym. Sci., Polym. Phys. Ed. 1986. V. 24. P. 189.
Rennsky I.A., Kamenskaya T.A., Rudnitskaya A.A. Compensation Effect in Thermodynamics of Activation of a Viscous Flow of n-alkanes. National Techn. Univ. of Ukraine “KPI”, 2010.
Maximov E.A., Pashayev B.G., Gasanov G.Sh., Gasanov N.G. // Adv. Modern Natural Sci. 2015. V. 10. P. 32.
Lide D.R. CRC Handbook of Chemistry and Physics. 2004.
Куличихин В.Г., Платонов В.А., Браверман Л.П., Рождественская Т.А., Коган Е.Г., Васильева Н.В., Волохина А.В. // Высокомолек. соед. А. 1987. Т. 29. № 12. С. 2537.
Panfilova A.A., Platonov V.A., Kulichikhin V.G., Kalmikova V.D., Papkov S.P. // Colloid J. 1975. V. 37. № 1. P. 210.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия А)