

Высокомолекулярные соединения (серия А), 2021, T. 63, № 6, стр. 422-429
ВЛИЯНИЕ НАНОРАЗМЕРНЫХ ОГРАНИЧЕНИЙ НА КРИСТАЛЛИЗАЦИЮ ПОЛИЭТИЛЕНОКСИДА В ПОРАХ ПОЛИОЛЕФИНОВ, ДЕФОРМИРОВАННЫХ ПО МЕХАНИЗМУ КРЕЙЗИНГА
А. Ю. Ярышева a, *, Н. А. Ситнов a, А. В. Бакиров b, c, Л. М. Ярышева a, М. С. Аржаков a, О. В. Аржакова a, С. Н. Чвалун b, c
a Московский государственный университет имени М.В. Ломоносова. Химический факультет
119991 Москва, Ленинские горы, Россия
b Национальный исследовательский центр “Курчатовский институт”
123182 Москва, пл. Академика Курчатова, 1, Россия
c Институт cинтетических полимерных материалов им. Н.С. Ениколопова Российской академии наук
117393 Москва, Профсоюзная ул., 70, Россия
* E-mail: alyonusha@gmail.com
Поступила в редакцию 29.04.2021
После доработки 02.07.2021
Принята к публикации 16.07.2021
Аннотация
Получены полимерные смеси полиолефинов с полиэтиленоксидом путем деформации пленок полипропилена и полиэтилена высокой плотности в водно-этанольных растворах ПЭО по механизму крейзинга. Содержание ПЭО с молекулярной массой 4 × 103 в смесях зависит от пористости полиолефиновых матриц и возрастает с увеличением степени растяжения пленок до 28% в смесях ПЭВП–ПЭО, до 32% в смесях ПП–ПЭО. Методом ДСК установлено, что кристаллизация ПЭО сопровождается его понижением температуры плавления на 4–6 К в матрице ПЭВП и на 6–7 К в ПП, а также степени кристалличности на 24–49% в ПЭВП и 44–76% в ПП по сравнению с ПЭО, закристаллизованном в “свободном” состоянии. Впервые по данным рентгеноструктурного анализа проведен расчет размеров кристаллитов ПЭО с М = 4 × 103 в порах полиолефинов, деформированных по механизму крейзинга, и показано, что макромолекулы ПЭО ориентируются перпендикулярно оси растяжения матриц ПП и ПЭВП.
ВВЕДЕНИЕ
В настоящее время широкое распространение получили исследования, направленные на изучение систем, в которых структурообразование полимеров ограничено на наноуровне. К подобным системам относятся тонкие полимерные пленки или слои, полимеры, включенные в нанопористые материалы с различной морфологией пор, совместимые и несовместимые полимерные смеси, блоксополимеры, нановолокна, нанотрубки, нанокомпозиты и многие другие системы, в том числе биологического происхождения [1–20]. Вопросы структурообразования полимеров в условиях ограниченного пространства рассматриваются как для аморфных стеклообразных полимеров, так и для систем, в которых хотя бы один из компонентов способен к кристаллизации.
Общие закономерности, наблюдаемые для полимеров в условиях ограниченного нанопространства, в первую очередь связаны с изменением молекулярной подвижности вследствие увеличения соотношения поверхности и объема. Для аморфных полимеров было обнаружено сильное понижение температуры стеклования Тс в наноразмерных дисперсиях, тонких пленках и поверхностных слоях по сравнению с соответствующей величиной блочного полимера [2, 4, 9]. Для кристаллизующихся полимеров изменение Тс определяет нижнюю границу температуры кристаллизации, и степень переохлаждения (разница между температурой кристаллизации полимера в блоке и в нанообъеме) позволяет судить о механизме кристаллизации.
В первую очередь влияние пространственных ограничений отражается на процессе зародышеобразования, поскольку критический размер зародыша кристаллизации обычно составляет несколько нанометров. По мере уменьшения объема полимерной фазы происходит изменение механизма зародышеобразования от гетерогенного, характерного для блочного полимера, к поверхностному зарождению на межфазной границе и в крайних случаях к гомогенному зародышеобразованию. Температура плавления также снижается с увеличением ограничения, но в меньшей степени. Объемные ограничения оказывают влияние и на кинетику кристаллизации полимеров. Как правило, с увеличением степени ограничения скорость кристаллизации падает, показатели степени в уравнении Аврами уменьшаются и могут достигать значений равных единице (или ниже) [1, 6–8, 10, 11, 13, 20].
Следует отметить, что наряду с общими закономерностями поведения полимеров в условиях ограниченного пространства для каждой из наносистем (тонких пленок, полимерных смесей, блоксополимеров, нанокомпозитов и других) существуют свои особенности и дополнительные факторы, влияющие на процесс кристаллизации. Большое число фундаментальных исследований по установлению влияния пространственных ограничений на структурообразование полимеров проведено на системах на основе пористых матриц с контролируемым размером пор, например трековых мембран [21], мембран на основе анодированной окиси алюминия, содержащих регулярно выстроенные цилиндрические нанопоры [1, 6, 8, 10–12, 14, 19].
В настоящей работе в качестве пористых материалов для введения высокомолекулярных соединений и исследования их структурообразования в условиях пространственных ограничений использованы полимеры, деформированные по механизму крейзинга в физически-активных жидких средах, со специфической нанопористой структурой [8, 22–24]. Крейзинг позволяет не только создавать в полимере пористую структуру, но и получать гибридные нанокомпозиты и полимер-полимерные смеси непосредственно в процессе вытяжки полимера в активных жидких средах, содержащих низко- или высокомолекулярные соединения. Преимущество крейзинга заключается в том, что изменение природы физически-активных жидких сред и условий деформирования полимера (температура, степень вытяжки) позволяют варьировать морфологию и размер пор формирующейся пористой структуры, т.е. размер “затрудняющего” кристаллизацию пространства.
В качестве вводимого компонента смеси был выбран полиэтиленоксид, который в силу доступности и узкого ММР часто служит модельным полимером для исследования кристаллизации полимеров в таких системах, как тонкие пленки, слоевые композиты, пористые темплаты и другие.
Цель данной работы – получение полимерных смесей и изучение влияния нанопористых матриц ПП и ПЭВП, деформированных по механизму крейзинга, на кристаллизацию ПЭО.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Использовали пленки ПП “Moplen” толщиной 90 мкм, ПЭВП “Stamylan” толщиной 25 мкм и полуразбавленный 20 мас. % водно-этанольный (85% этанола) раствор ПЭО (“Sigma–Aldrich”) Mw = 4 × 103. Пленки с размерами рабочей части 20 × 40 мм растягивали в растворе ПЭО со скоростью 5 мм мин–1 с помощью динамометра “Instron-1122”. После чего образцы протирали и сушили в вакууме. Объемную пористость полимера в процессе вытяжки определяли по изменению геометрических размеров образцов: W = = (Vt – V0)/Vt, где Vt – объем образца после вытяжки, V0 – исходный объем образца. Содержание ПЭО в смесях устанавливали массовым методом Δm/mt = (mt – m0)/mt × 100%, где m0 – масса исходной пленки, mt – масса смеси. Средняя ошибка измерения составила ±2%. Теоретическое содержание ПЭО в смесях вычисляли в предположении, что весь объем пор (W = ΔV/Vt) занят раствором 20% концентрации С: Δm/mt (теория) = = CWt /[(1 – Wt)ρ + CWt] × 100%, ρ – плотность 0.95 г см–3 (ПЭВП), 0.91 г см–3 (ПП).
Структуру деформированных полимеров исследовали с помощью атомно-силовой микроскопии на микроскопе “Solver BIO Olympus” (“Нанотехнология МДТ”, Россия). Сканирование проводили в контактном режиме. Полученные изображения обрабатывали в программе FemtoScan Online. Степень кристалличности ПЭО вычисляли по формуле: χ = ΔH/ΔH100% × 100%, где ΔH100% = 196.8 Дж г–1 – теплота плавления идеального кристалла ПЭО. Величину ΔH находили по данным ДСК (термоанализатор “TA 4000”, “Mettler”), масса образцов составляла 1–2 мг, скорость нагревания 10 K мин–1.
Рентгеноструктурный анализ полимерных смесей в больших углах выполняли на станции ДИКСИ Курчатовского синхротрона. Источником излучения служил поворотный магнит 1.7 Тл, дающий излучение 7.65 кэВ (1.625 А) с разрешением (dE/E) × 10–3 и потоком фотонов 109. Размер пучка на образце 0.5 × 0.3 мм, для регистрации картин дифракции использовали детектор “Dectris Pilatus 1M”.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Деформация частично кристаллических полимеров в физически-активных жидких средах по механизму межкристаллитного (делокализованного) крейзинга сопровождается формированием фибриллярно-пористой структуры с размерами пор и фибрилл в нанометровом диапазоне размерности (рис. 1). В работах [23–25] было установлено, что выбранные для работы ПЭВП с исходной row-структурой и ПП с исходной α-сферолитной структурой имеют характерные общие морфологические признаки развития деформации по механизму межкристаллитного крейзинга: раздвижение и фрагментация ламелей, перестроение ламеллярной структуры в фибриллярную с ориентацией фибрилл в направлении растяжения, формирование пор в межкристаллитном пространстве, длина и ширина которых увеличиваются с повышением степени вытяжки.
Рис. 1.
АСМ-изображения ПЭВП (а) и ПП (б), деформированных в физически-активных жидких средах на 200%. Стрелками указано направление растяжения.
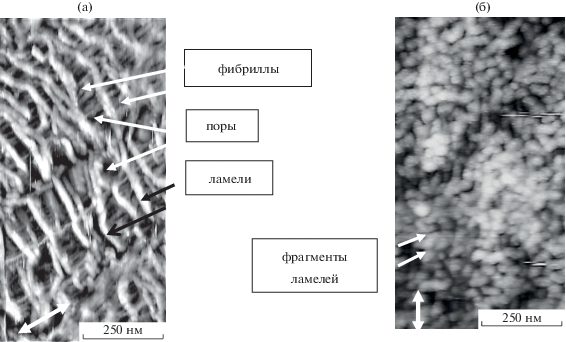
Однако, как видно на рис. 1, деформация исследуемых частично кристаллических полимеров имеет свои особенности, связанные с разной исходной надмолекулярной структурой, что приводит к разной морфологии и параметрам формирующихся пористых матриц (табл. 1). Например, после деформации по механизму крейзинга для приведенных на рисунке ПЭВП и ПП одной степени вытяжки 200% средние значения большого периода, определяемого по профилям АСМ-сечений как расстояние между вершинами ламелей, составили 75 ± 36 нм (ПЭВП) и 45 ± 17 нм (ПП) [23, 24], что позволяет оценить среднюю длину пор с учетом толщины ламелей ~60 нм для ПЭВП и 25 нм для ПП. Ширина пор, измеряемая как расстояние между вершинами фибрилл, методом АСМ была определена для ПЭВП и ПП со степенью вытяжки 300% (близость этого параметра для ПП 100, 200% степеней вытяжки и радиуса кривизны кантиллевера не позволяет однозначно его измерить). Отметим, что с увеличением степени вытяжки средняя длина пор ПЭВП и ПП и ширина пор ПЭВП увеличиваются (табл. 1), но при одинаковых степенях вытяжки размер пор в ПЭВП превышает размер пор в ПП. Выявленные особенности структуры при близкой химической природе делают матрицы полиолефинов, деформированных по механизму крейзинга, удобными модельными объектами для исследования влияния пространственных ограничений на структурообразование введенных в нанопоры кристаллизующихся веществ.
Таблица 1.
Размер пор (большой период L/ширина поры D) деформированных полиолефинов и теплофизические свойства ПЭО в смесях ПП–ПЭО, ПЭВП–ПЭО
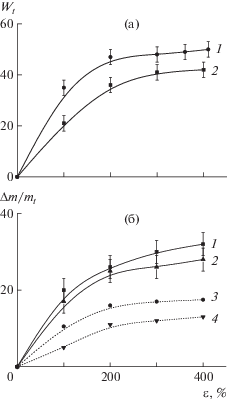
Добавление ПЭО в водно-этанольный раствор, действующий как физически-активная жидкая среда, не влияет существенно на процесс деформации ПП и ПЭВП [26–28], и сопровождается проникновением ПЭО в формирующуюся при крейзинге пористую структуру (рис. 2а), что подтверждается данными по увеличению массы пленок (рис. 2б) после удаления летучей жидкой среды. С повышением степени вытяжки содержание ПЭО увеличивается в соответствии с ростом пористости полимерной матрицы и при всех степенях вытяжки оказывается выше теоретических значений, вычисленных, как приведено выше, в предположении полного заполнения пор раствором ПЭО с концентрацией 20 мас. %.
Рис. 2.
а – Зависимость пористости W от степени вытяжки ε пленок ПП (1) и ПЭВП (2) в водно-этанольной среде (85% этанола); б – зависимость содержания ПЭО в смесях ПП–ПЭО (1) и ПЭВП–ПЭО (2), а также содержание ПЭО в смесях ПП–ПЭО (3) и ПЭВП–ПЭО (4), вычисленное в предположении, что поры ПП и ПЭВП заполняются раствором ПЭО с концентрацией 20%.
В работе [29] была рассмотрена зависимость коэффициента разделения (отношения концентрации полимера в поре к его концентрации в растворе) от радиуса пор. При отсутствии взаимодействия между макромолекулами и стенками пор, коэффициент разделения меньше единицы вследствие потери энтропии при вхождении макромолекул в нанопоры. Однако коэффициент разделения в порах может резко возрастать, если существуют взаимодействия между макромолекулами и стенками пор [30–32]. Видимо, наблюдаемый эффект концентрирования ПЭО связан с его адсорбцией в порах деформированных полимеров, которые обладают хорошими сорбционными свойствами вследствие диспергирования на наноразмерные фибриллярные агрегаты и формирования высокоразвитой поверхности (50–100 м2 г–1) в процессе крейзинга [22–24].
Исследование теплофизических свойств полученных смесей показало наличие на термограммах двух отдельных эндотермических пиков, соответствующих плавлению полимерной матрицы и ПЭО, что свидетельствует о фазовом разделении компонентов. Значения температуры плавления ПЭВП и ПП в смесях по сравнению с исходными полимерами не изменяются. Однако в отличие от “свободной” кристаллизации в блоке, кристаллизация ПЭО в нанопорах ПП и ПЭВП приводит к изменению фундаментальных характеристик ПЭО и сопровождается понижением степени кристалличности и температуры плавления ПЭО в смесях по сравнению с ПЭО, закристаллизованным в блоке (табл. 1; рис. 3).
Рис. 3.
Термограммы плавления ПЭО с М = 4 × 103 в смесях ПП–ПЭО (а) и ПЭВП–ПЭО (б) в зависимости от степени вытяжки полимеров на 100 (1), 200 (2) и 300% (3). Штриховой линией отмечена температура плавления блочного ПЭО.
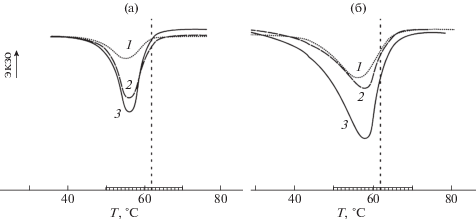
В зависимости от степени вытяжки степень кристалличности ПЭО уменьшается на 24–49% в смесях с ПЭВП и на 44–76% в смесях с ПП, размеры пор в котором заметно меньше, чем в пленках ПЭВП. Температура плавления ПЭО снижается на 4–6 К в смесях с ПЭВП и 6–7 К в смесях с ПП.
Полученные данные согласуются с результатами работ по исследованию кристаллизации полимеров в “затрудненных” условиях, когда рост ламелей хотя бы в одном из направлений ограничен нанометровыми размерами: в тонких пленках на поверхности субстратов или в тонких слоях соэкструдированных пленок, в блоксополимерах и полимерных смесях, содержащих ПЭО в виде отдельной фазы, в растворах или расплавах ПЭО, содержащих высокодисперсные частицы и в нанопористых материалах [1–20]. В указанных работах показано, что в условиях нанометровых пространственных ограничений степень кристалличности и температура плавления ПЭО уменьшаются. Степень кристалличности снижается тем больше, чем меньше размер ограничения (диаметр пор), вплоть до полной потери способности полимера к кристаллизации [1, 3, 6–8, 33, 34]. При кристаллизации полимеров в ограниченном пространстве процесс зародышеобразования превалирует над процессом роста, что приводит к уменьшению размеров кристаллитов и увеличению площади их межфазной поверхности [35]. В связи с этим плавление таких кристаллитов происходит при более низкой температуре. Очевидно, такие же причины имеет понижение температуры плавления ПЭО в смесях ПП–ПЭО и ПЭВП–ПЭО. Снижение степени кристалличности, вероятно, связано с тем, что часть ПЭО в нанопористой структуре ПП и ПЭВП оказывается в условиях, при которых ПЭО не способен закристаллизоваться из-за пространственных “затруденений”. Кроме того, адсорбция ПЭО на высокоразвитой поверхности полимеров, деформированных по механизму крейзинга, может понижать подвижность макромолекул в поверхностном слое, препятствуя их кристаллизации [34].
Как видно из таблицы, чем больше степень деформации при крейзинге, тем большего размера поры развиваются в полимерной матрице. Изменение в размерах нанопор ПП и ПЭВП, которые в данном случае выступают в качестве ограничивающего кристаллизацию ПЭО пространства, влияет на структуру ПЭО в полученных смесях. Действительно, увеличение степени вытяжки ПЭВП и ПП приводит к увеличению степени кристалличности ПЭО в смесях, хотя они так и не достигают значений ПЭО, закристаллизованного в блоке (табл. 1).
Разница в значениях температуры плавления ПЭО в смесях по сравнению с исходным гомополимером с увеличением степени вытяжки пленок также уменьшается (рис. 3). Например, для смесей ПП–ПЭО, полученных деформированием ПП в растворе ПЭО на 100%, степень кристалличности ПЭО составляет всего 18%, а температура плавления по сравнению с гомополимером падает на семь градусов. Для смеси, полученной деформированием ПП в том же растворе ПЭО на 300%, степень кристалличности ПЭО возрастает до 50%, а температура плавления по сравнению с гомополимером ниже на шесть градусов. Аналогичные эффекты прослеживаются для смесей ПЭВП–ПЭО (табл. 1; рис. 3).
Сравнение теплофизических свойств смесей на основе ПП, ПЭВП и анализ АСМ-изображений (см. рис. 1) позволяет предположить, что более выраженное понижение температуры плавления и степени кристалличности ПЭО в смесях ПП–ПЭО по сравнению со смесями на основе ПЭВП связано с размерами пор ПП, которые, как было показано выше, в 2–3 раза меньше размеров пор ПЭВП при одинаковых степенях растяжения. Таким образом, процесс кристаллизации ПЭО, действительно, определяется пространственными ограничениями нанопористой полимерной матрицы.
Исследование полимер-полимерных смесей, полученных деформированием полиолефинов в растворах ПЭО на 200%, методом рентгеновского рассеяния в больших углах позволило оценить размеры области когерентного рассеяния кристаллитов ПЭО, которая составила 15 нм (в экваториальном направлении) и 21 нм (в меридиональном) в матрице ПЭВП, 10 нм (в экваториальном) и 18 нм (в меридиональном) в матрице ПП.
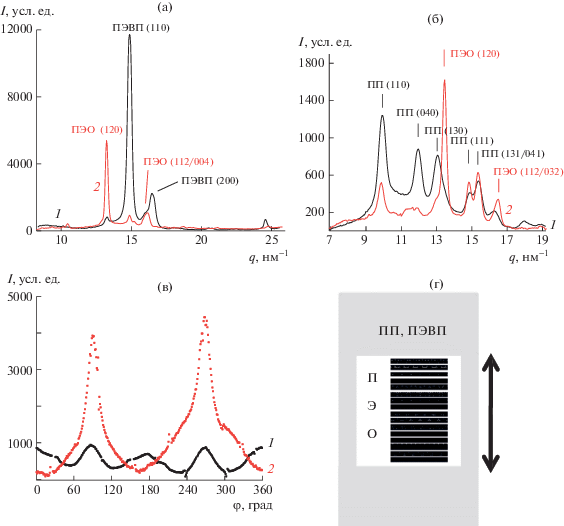
Анализ картин рентгеновского рассеяния образцами ПП–ПЭО и ПЭВП–ПЭО позволил определить ориентацию макромолекул ПЭО относительно направления вытяжки (рис. 4). На кривых рассеяния наряду с рефлексами ПЭВП (110, 200), ПП (110, 040, 130, 111 и 131/041) присутствуют рефлексы ПЭО (120) и перекрывающиеся рефлексы (112/004), соответствующие межплоскостным расстояниям 4.63 и 3.86/3.97 Å, характерным для моноклинной структуры [36]. Видно, что и в матрице ПП и в ПЭВП пик ПЭО (120) в меридиональном направлении значительно выше, чем в экваториальном (рис. 4а, 4б). Азимутальные распределения интенсивности рефлекса ПЭО (120) в смесях ПП–ПЭО и ПЭВП–ПЭО показали наличие максимумов под углом 90° к экватору (на меридиане) (рис. 4в). Локальные максимумы при 0° и 180о азимутального угла возникают из-за наложения рефлексов ПЭО (120) и ПП (130). Таким образом, в смесях ПП–ПЭО и ПЭВП–ПЭО оси макромолекул ПЭО перпендикулярны оси растяжения или незначительно отклоняются от этого направления, что позволяет сделать вывод о преимущественной ориентации макромолекул ПЭО в направлении перпендикулярном оси растяжения полиолефинов или плоскостной ориентации ламелей ПЭО по отношению к фибриллам, как показано на схеме рис. 4г.
Рис. 4.
Кривые рентгеновского рассеяния в больших углах в экваториальном (1) и меридиональном (2) направлении смесей ПЭВП–ПЭО (а) и ПП–ПЭО (б); в – азимутальное распределение интенсивности рефлекса ПЭО (120) в ПП (1) и ПЭВП (2); г – схематичное представление ориентации длинных осей макромолекул ПЭО с М = 4 × 103 в порах полиолефинов, деформированных по механизму крейзинга.
Возникновение ориентации макромолекул ПЭО при кристаллизации в нанопорах, т.е. в ограниченном пространстве, связано с особенностями кристаллизации высокомолекулярных соединений [3, 6–8, 10–16, 19, 37, 38]. Свободная поверхностная энергия грани, образованной складками макромолекул, значительно выше, чем свободная энергия боковых поверхностей: плоскостная ориентация ламелей по отношению к субстрату должна быть предпочтительной. Однако в зависимости от природы взаимодействия кристаллизующегося полимера и субстрата, геометрии ограниченного пространства и условий кристаллизации возможна различная ориентация ламелей. Так, например, авторы работы [28] наблюдали ориентацию ламелей ПЭО c молекулярной массой 20 × 103 параллельно фибриллам ПП и ПЭВП.
Очевидно, что геометрия и размер пор как ограничивающего фактора и взаимодействие кристаллизующегося полимера с поверхностью субстрата влияют на процессы зародышеобразования и роста кристаллов. В смесях ПП–ПЭО и ПЭВП–ПЭО поровое пространство заполнено нанофибриллами, формирующимися при деформации полиолефинов по механизму крейзинга, и зародышеобразование ПЭО, вероятно, происходит на их поверхности. Зародышеобразованию на поверхности фибрилл также может способствовать предшествующая кристаллизации адсорбция ПЭО (см. рис. 2б). При контурной длине 253.2 Å [39] макромолекул ПЭО c молекулярной массой 4 × 103 и размерах кристаллитов по полученным данным 100–210 Å ПЭО способен кристаллизоваться не более чем с одной–двумя складками. Можно предположить, что часть макромолекулы ПЭО остается связанной при адсорбции на поверхности фибрилл ПП и ПЭВП, а оставшаяся часть слишком мала, чтобы образовывать складку, и ПЭО кристаллизуется с выпрямленными цепями перпендикулярно оси вытяжки ПП и ПЭВП.
ЗАКЛЮЧЕНИЕ
Таким образом, установлено, что ПЭО кристаллизуется в нанопорах с ПЭВП и ПП, деформированных по механизму крейзинга, с понижением температуры плавления и степени кристалличности по сравнению со “свободно” закристаллизованным ПЭО. Понижение значений базовых теплофизических характеристик ПЭО связано с пространственными затруднениями в процессе кристаллизации и зависит от размера пор полимерных матриц, который определяется степенью вытяжки и исходной морфологией полиолефина – row-структура ПЭВП и сферолитная структура ПП. Процесс кристаллизации ПЭО в нанопорах сопровождается ориентацией макромолекул перпендикулярно направлению растяжения и, соответственно, плоскостной ориентацией ламелей ПЭО. Таким образом, размеры пор ПЭВП и ПП, деформированных по механизму крейзинга, определяют структурообразование введенного ПЭО. Обнаруженный эффект может представлять практический интерес для получения материалов на основе ПЭО с пониженной степенью кристалличности, используемых в качестве газоразделительных мембран или твердых полиэлектролитов.
Работа выполнена при финансовой поддержке фонда Министерства образования и науки России (соглашение № 075-15-2020-794).
Список литературы
Sangroniz L., Wang B., Su Y., Liu G., Cavallo D., Wang D., Muller A.J. // Progr. Polym. Sci. 2021. P. 101376.
Arutkin M., Raphaël E., Forrest J., Salez T. // Phys. Rev., Amer. Phys. Soc. (APS). 2020. V. 101. P. 032122.
Chengtao Yu, Qing Xie, Yongzhong Bao, Guorong Shan, Pengju Pan // Crystals. 2017. V. 7. № 5. P. 147.
Cangialosi D., Alegría A., Colmenero J. // Progr. Polym. Sci. 2016. V. 54–55. P. 128.
Mijangos C., Hernández R., Martín J. // Progr. Polym. Sci. 2016. V. 54–55. P. 148.
Michell R.M., Müller A.J. // Progr. Polym. Sci. 2016. V. 54–55. P. 183.
Prud’homme R.E. // Progr. Polym. Sci. 2016. V. 54–55. P. 214.
Volynskii A.L., Yarysheva A.Yu., Rukhlya E.G., Yarysheva L.M., Bakeev N.F. // Polymer Science A. 2015. V. 57. № 5. P. 515.
Ediger M.D., Forrest J.A. // Macromolecules. 2014. V. 47. P. 471.
Michell R.M., Blaszczyk-Lezak I., Mijangos C., Muller A.J. // J. Polym. Sci., Polym. Phys. 2014. V. 52. P. 1179.
Michell R.M., Blaszczyk-Lezak I., Mijangos C., Müller A.J. // Polymer. 2013. V. 54. P. 4059.
Lin M.C., Nandan B., Chen H.-L. // Soft Matt. 2012. V. 8. P. 3306.
Carr J.M., Langhe D.S., Ponting M.T., Hiltner A., Baer E. // J. Mater. Res. 2012. V. 27. № 10. P. 1326.
Wu H., Su Z., Takahara A. // Soft Matt. 2012. V. 8. P. 3180.
Martín J., Maiz J., Sacristan J., Mijangos C. // Polymer. 2012. V. 53. P. 1149.
Li H., Yan S. // Macromolecules. 2011. V. 44. P. 417.
Chen R., Huang D. // Front. Chem. China. 2011. V. 6. № 4. P. 332.
Chrissopoulou K., Andrikopoulos K.S., Fotiadou S., Bollas S., Karageorgaki C., Christofilos D., Voyiatzis G.A., Anastasiadis S.H. // Macromolecules. 2011. V. 44. P. 9710.
Adsorption and Phase Behaviour in Nanochannels and Nanotubes / Eds by L.J. Dunne, G. Manos. Springer Science+Business Media B.V., 2010.
Liu Y.-X., Chen E.-Q. // Coordination Chem. Rev. 2010. V. 254. P. 1011.
Grefe A.-K., Kuttich B., Stühn L., Stark R., Stühn B. // Soft Matt. 2019. V. 15. P. 3149.
Yarysheva A.Yu., Arzhakova O.V., Yarysheva L.M., Volynskii A.L. // Polymer. 2018. V. 158. P. 243.
Yarysheva A.Yu., Bagrov D.V., Bakirov A.V., Yarysheva L.M., Chvalun S.N., Volynskii A.L. // Eur. Polym. J. 2018. V. 100. P. 233.
Yarysheva A.Yu., Rukhlya E.G., Yarysheva L.M., Bagrov D.V., Volynskii A.L., Bakeev N.F. // Eur. Polym. J. 2015. V. 66. P. 458.
Yarysheva A., Rukhlya E., Grokhovskaya T., Dolgova A., Arzhakova O.V. // J. Appl. Polym. Sci. 2019. V. 136. P. 48567.
Yarysheva A.Y., Dolgova A.A.,Yarysheva L.M., Arzhakova O.V. // Mendeleev Commun. 2020. V. 30. № 4. P. 507.
Yarysheva A.Y., Bagrov D.V., Yarysheva L.M., Volynskii A.L., Bakeev N.F. // Polymer Science A. 2012. V. 54. № 10. P. 779.
Yarysheva A.Y., Bagrov D.V., Bakirov A.V., Tarasevich B.N., Grohovskaya T.E., Yarysheva L.M., Chvalun S.N., Volynskii A.L. // Macromolecules. 2017. V. 50. № 7. P. 2881.
Casassa E.F., Tagami Y. // Macromolecules. 1969. V. 2. № 1. P. 14.
Caspi Y., Zbaida D., Cohen H., Elbaum M. // Macromolecules. 2009. V. 42. № 3. P. 760.
Grull H., Shaulitch R., Yerushalmi-Rozen R. // Macromolecules. 2001. V. 34. № 23. P. 8315.
Teraoka I., Langley K.H., Karasz F.E. // Macromolecules. 1993. V. 26. № 2. P. 287.
Shin K., Woo E., Jeong Y.G., Kim C., Huh J., Kim K.-W. // Macromolecules. 2007. V. 40. № 18. P. 6617.
Thitisomboon W., Gu Q., Weng Lu-Tao, Gao P. // Polymer. 2021. V. 217. P. 123449.
Maiz J., Martin J., Mijangos C. // Langmuir. 2012. V. 28. № 33. P. 12296.
Takahashi Y., Tadokoro H. // Macromolecules. 1973. V. 6. № 5. P. 672.
Guan Y., Liu G.M., Gao P.Y., Li L., Ding G.Q., Wang D.J. // ACS Macro Lett. 2013. V. 2. № 3. P. 181.
Su Li X., Zhang X., Müller A.J., Wang D., Liu G. // Macromolecules. 2018. V. 51. № 23. P. 9484.
Годовский Ю.К., Слонимский Г.Л., Гарбар Н.М. // Высокомолек. cоед. А. 1973. Т. 15. № 4. С. 813.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия А)